



Biotech Depot 2010 - 500 Beiträge pro Seite
eröffnet am 03.01.10 16:49:57 von
neuester Beitrag 07.01.12 15:22:53 von
neuester Beitrag 07.01.12 15:22:53 von
Beiträge: 334
ID: 1.155.114
ID: 1.155.114
Aufrufe heute: 0
Gesamt: 12.716
Gesamt: 12.716
Aktive User: 0
Top-Diskussionen
| Titel | letzter Beitrag | Aufrufe |
|---|---|---|
| 20.04.24, 12:11 | 677 | |
| vor 1 Stunde | 607 | |
| heute 01:03 | 597 | |
| vor 51 Minuten | 367 | |
| vor 45 Minuten | 291 | |
| gestern 22:37 | 289 | |
| heute 00:58 | 252 | |
| vor 1 Stunde | 226 |
Meistdiskutierte Wertpapiere
| Platz | vorher | Wertpapier | Kurs | Perf. % | Anzahl | ||
|---|---|---|---|---|---|---|---|
| 1. | 1. | 17.963,73 | +0,15 | 210 | |||
| 2. | 2. | 132,56 | -0,53 | 129 | |||
| 3. | 3. | 2.304,33 | -0,98 | 81 | |||
| 4. | 4. | 6,9380 | +0,58 | 72 | |||
| 5. | 5. | 676,90 | +0,49 | 53 | |||
| 6. | 6. | 3,7250 | +0,13 | 40 | |||
| 7. | 8. | 2,3595 | +1,14 | 37 | |||
| 8. | 9. | 45,00 | -0,44 | 35 |
Auf ein neues! 
Während im letzten Jahr Dax, Nasdaq usw große Gewinne einbrachten, hat sich mein Biotech-Depot Thread: Biotech Depot 2009 leider nur sehr durchwachsen entwickelt, was ein wenig entäuschend ist... am Ende blieb es bei einem kleinen Minus. Ein wesentlicher Grund lag in der sehr schlechten Entwicklung von Genmab, die im Laufe des Jahres fast 2/3 an Wert verloren und die ich recht hoch gewichtet hatte.
Dieses Jahr starte ich nun mit folgenden Werten:
11,9% Regeneron http://finance.yahoo.com/q?s=REGN
10,2% Incyte http://finance.yahoo.com/q?s=INCY
8,0% Genmab http://finance.yahoo.com/q?s=GEN.CO
7,3% Cubist http://finance.yahoo.com/q?s=CBST
6,3% Exelixis http://finance.yahoo.com/q?s=EXEL
6,1% Rigel http://finance.yahoo.com/q?s=RIGL
6,0% Onyx http://finance.yahoo.com/q?s=ONXX
5,9% Allos http://finance.yahoo.com/q?s=ALTH
5,4% Isis http://finance.yahoo.com/q?s=ISIS
5,1% Micromet http://finance.yahoo.com/q?s=MITI
3,9% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,8% ArQule http://finance.yahoo.com/q?s=ARQL
3,7% Evotec http://finance.yahoo.com/q?s=EVT.DE
3,6% Medigene http://finance.yahoo.com/q?s=MDG.DE
3,3% Array http://finance.yahoo.com/q?s=ARRY
2,3% Arena http://finance.yahoo.com/q?s=ARNA
2,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
1,8% NicOx http://finance.yahoo.com/q?s=COX.PA
1,2% Progenics http://finance.yahoo.com/q?s=PGNX
1,2% Addex http://finance.yahoo.com/q?s=ADXN.SW
1,0% Sucampo http://finance.yahoo.com/q?s=SCMP
Die letzten Änderungen in 2009 waren:
Aufstockung von ALTH, REGN und ISIS... Reduzierung von Evotec und Kauf von ARQL.
Ob es wohl diesmal eine gute Mischung ergibt?
Mal schauen, was dieses Jahr so bringt... wünsche jedenfalls allen ein gutes und erfolgreiches Jahr 2010 und viel Spaß mit ihren Investments!
mfg ipollit

Während im letzten Jahr Dax, Nasdaq usw große Gewinne einbrachten, hat sich mein Biotech-Depot Thread: Biotech Depot 2009 leider nur sehr durchwachsen entwickelt, was ein wenig entäuschend ist... am Ende blieb es bei einem kleinen Minus. Ein wesentlicher Grund lag in der sehr schlechten Entwicklung von Genmab, die im Laufe des Jahres fast 2/3 an Wert verloren und die ich recht hoch gewichtet hatte.
Dieses Jahr starte ich nun mit folgenden Werten:
11,9% Regeneron http://finance.yahoo.com/q?s=REGN
10,2% Incyte http://finance.yahoo.com/q?s=INCY
8,0% Genmab http://finance.yahoo.com/q?s=GEN.CO
7,3% Cubist http://finance.yahoo.com/q?s=CBST
6,3% Exelixis http://finance.yahoo.com/q?s=EXEL
6,1% Rigel http://finance.yahoo.com/q?s=RIGL
6,0% Onyx http://finance.yahoo.com/q?s=ONXX
5,9% Allos http://finance.yahoo.com/q?s=ALTH
5,4% Isis http://finance.yahoo.com/q?s=ISIS
5,1% Micromet http://finance.yahoo.com/q?s=MITI
3,9% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,8% ArQule http://finance.yahoo.com/q?s=ARQL
3,7% Evotec http://finance.yahoo.com/q?s=EVT.DE
3,6% Medigene http://finance.yahoo.com/q?s=MDG.DE
3,3% Array http://finance.yahoo.com/q?s=ARRY
2,3% Arena http://finance.yahoo.com/q?s=ARNA
2,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
1,8% NicOx http://finance.yahoo.com/q?s=COX.PA
1,2% Progenics http://finance.yahoo.com/q?s=PGNX
1,2% Addex http://finance.yahoo.com/q?s=ADXN.SW
1,0% Sucampo http://finance.yahoo.com/q?s=SCMP
Die letzten Änderungen in 2009 waren:
Aufstockung von ALTH, REGN und ISIS... Reduzierung von Evotec und Kauf von ARQL.
Ob es wohl diesmal eine gute Mischung ergibt?
Mal schauen, was dieses Jahr so bringt... wünsche jedenfalls allen ein gutes und erfolgreiches Jahr 2010 und viel Spaß mit ihren Investments!
mfg ipollit
Antwort auf Beitrag Nr.: 38.656.864 von ipollit am 03.01.10 16:49:57Zum Vergleich der Nasdaq Biotech Index...

Regeneron (REGN) - MK 1,95 Mrd USD (bei 24,18 USD)
http://finance.yahoo.com/q?s=regn
starke Pipeline und Partner, Avastin-ähnliches Aflibercept in PIII mit Sanofi (50% Gewinnanteil), Lucentis-ähnliches VEGF-Trap Eye in PIII mit Bayer (100% US-Rechte, 50% weltweit), Arcalyst zugelassen als Nischenprodukt gegen CAPS, in PIII gegen Gicht (100% weltweit), große AK-Allianz mit Sanofi... Regeneron erhält jährlich 160 Mio USD bis 2017 mit dem Ziel jährlich 4 bis 5 AKs in die PI zu bekommen

Incyte (INCY) - MK 1,08 Mrd USD (bei 9,11 USD)
http://finance.yahoo.com/q?s=incy
die Schuldenkrise scheint überwunden... zuletzt extrem gute Deals für die JAK1/2-Hemmer: Novartis hat die ex-US Rechte am sehr aussichtsreichen INCB018424 gegen Krebs in PIII gekauft (Incyte besitzt 100% der US-Rechte), EliLilly finanziert die teure Entwicklung von INCB28050 gegen Immunkrankheiten wie RA (Incyte bekommt bis zu 20% Royalties)... das brachte zusammen 300 Mio USD Upfront ein. In der Pipeline befinden sich aktuell noch 2 Kandidaten in PII gegen Brustkrebs und Diabetes, die theoretisch auslizensiert werden können

Genmab - MK 710 Mio USD (bei 82 DKK)
recht enttäuschend. zwar haben Genmab/GSK die Zulassung für Arzerra (Rituxan-ähnlich) gegen CLL erhalten, doch ist eine riskante PIII gegen die wichtige Indikation NHL fehlgeschlagen. Damit ist offen, ob Arzerra mit Rituxan konkurrieren kann... direkte, aber riskante Vergleichstudien mit Rituxan wurden begonnen. Zudem wird Arzerra subkutan gegen RA entwickelt. Wichtige PIII-Ergebnisse zum zweiten Kandidaten Zalutumumab (Erbitux-ähnlich) gegen HN-Krebs sollen in Kürze erfolgen... auch hier besteht ein großes Risiko, dass sich Zalutumumab nicht gegen Erbitux absetzen kann. Die Entwicklung von R1507 (Roche) wurde leider gestoppt... wohl aufgrund der Genentech-Übernahme. Zudem ist der (Zwangs-)Verkauf der AK-Produktionsstädten enttäuschend.

Cubist (CBST) - MK 1,1 Mrd USD (bei 18,97 USD)
http://finance.yahoo.com/q?s=cbst
KGV10e von 12,5... das Antibiotikum Cubicin bringt hohe Gewinne ein und entwickelt sich zum Blockbuster. Der relativ niedrige Kurs (und der Einbruch in 2009) ist nur dadurch zu erklären, dass Teva versucht, die Patente (bis 2017/19) auszuhebeln, um Cubicin generisch zu vertreiben. 2011 wird dieser Fall vor Gericht behandelt. In der Pipeline gab es ein paar Rückschläge... ALN-RSV01 in PII wurde vorerst an Alnylam zurückgegeben, Cubist will den präklinischen Nachfolger gegen eine größere Indikation entwickeln. Die PII von Ecallantide gegen Blutungen (lizensiert von Dyax, die zuletzt dafür die Zulassung als Kalbitor gegen akute Schwellungen HAE von der FDA erhalten haben) wurde wegen Unregelmäßigkeiten frühzeitig geschlossen... es besteht das Risiko, dass damit die PII scheitert. Dafür wurde zuletzt mit der Übernahme von Calixa mit CXA-201 ein weiteres ausichtsreiches Antibiotikum erworben, welches 2014 auf den Markt kommen könnte.

Exelixis (EXEL) - MK 791 Mio USD (bei 7,37 USD)
http://finance.yahoo.com/q?s=exel
große Pipline mit 11 Kandidaten in der klinischen Entwicklung, die im wesentlichen von namhaften Pharma-Partnern finanziert wird. In 2009 konnten mehrere gute Deals abgeschlossen werden.

Rigel (RIGL) - MK 493 Mio USD (bei 9,51 USD)
http://finance.yahoo.com/q?s=rigl
Unsicherheit bezügliche R788 als orales Mittel gegen RA. Die PII-Daten zeigten zwar eine gute Wirkung, doch könnten störende Nebenwirkungen wie leicht erhöhter Blutdruck Probleme bereiten. PIII ist in Vorbeitung... es konnte aber immer noch kein Pharma-Partner gefunden werden. R788 ist von der Indikation gegen Autoimmunskrankheiten und Krebs ähnlich wie INCY's JAK-Hemmer... nur handelt es sich mit SYK um ein anderes Target.

Onyx (ONXX) - MK 1,82 Mrd USD (bei 29,34 USD)
http://finance.yahoo.com/q?s=onxx
KGV10e von 38. Nexavar gegen Krebs entwickelt weiter positiv... doch versucht der Partner Bayer an Onyx vorbei eine leicht modifizierte Version zu entwickeln, worauf Onyx Bayer verklagt hat. Nexavar könnte gegen Brustkrebs wirksam sein... dies würde das Potential deutlich erhöhen. Durch die Übernahme von Proteolix wurde zuletzt das Krebsmedikament Carfilzomib in PII erworben, dass ein nebenwirkungsfreieres Velcade sein könnte. Sind die Daten bei MM in 2010 positiv, könnte bereits 2011 ein weiterer potentieller Blockbuster von Onyx auf den Markt kommen.

Allos (ALTH) - MK 684 Mio USD (bei 6,58 USD)
http://finance.yahoo.com/q?s=alth
Ein-Produkt Unternehmen... im September hat Allos die Zulassung von Folotyn (neuartiges Chemotherapeutikum) gegen die seltene Krebsart PTCL erhalten. Es ist zwar eine kleine Indikation, aber es gibt keine Alternativen und der Preis für Folotyn ist entsprechend sehr hoch. Außerdem kann Allos es mit einer kleinen Vertriebsstruktur selber vermarkten. Folotyn wird nun bei größeren Indikationen wie NHL oder NSCLC getestet

Isis (ISIS) - MK 1,09 Mrd USD (bei 11,11 USD)
http://finance.yahoo.com/q?s=isis
Führend in der RNAi-Technologie... umfangreiche Pipeline und Kooperationen, die einiges an Bargeld einbringen (teilweise cashflow positiv), hoher Bargeldbestand mit 600 Mio USD. Wichtigstes Projekt ist Mipomersen in PIII, das zusammen mit Genzyme zur Senkung von Cholesterin, wenn z.B. Statine nicht ausreichend helfen, entwickelt wird. Bis Mitte 2010 sollen Daten von 3 weiteren PIIIs vorliegen... die Zulassung für Hochrisikopatienten soll im Sommer 2011 erfolgen, etwas später als ursprünglich geplant. Zwar konnte in der ersten PIII Mipomersen bei Hochrisiko-Patienten das Cholesterin senken, doch gab es auch bei einem Teil der Patienten erhöhte Leberwerte, ein Anzeichen von möglicher Leberschädigung. Die Frage ist nun, in wie weit Mipomersen dafür verantwortlich ist und ob dies auch bei Patienten, die keine lebensgefährlichen Cholesterinwerte aufweisen, auftritt, denn nur damit kann Mipomersen zu einem Blockbuster werden. Daher ist der Kurs zuletzt gefallen.

Micromet (MITI) - MK 459 Mio USD (bei 6,66 USD)
http://finance.yahoo.com/q?s=miti
hat spezielle BiTE-AKs in der Pipeline, mit denen T-Killerzellen des Immunsystems an bestimmte Targets gebunden werden sollen, damit der Körper diese zerstört. Der CD19-BiTE-AK Blinatumomab hat in PI/II Studien gute Ergebnisse geliefert... so war bei 80% von Patienten mit ALL der Krebs nicht mehr nachweisbar und die Ansprechrate bei NHL, bei denen andere Therapien nicht ansprechen, lag bei 100%. Dies sind zwar unsichere, aber ermutigende Ergebnisse. Der AK ist noch nicht verpartnert und es könnte bald eine Zulassungsstudie begonnen werden.

Seattle Genetics (SGEN) - MK 1,02 Mrd USD (bei 10,16 UDS)
http://finance.yahoo.com/q?s=sgen
Technologie, die AKs mit einer Chemo belädt, um die Wirkung der AKs zu erhöhen. Mit dem CD40-AK SGN-40 gab es einen Rückschlag... eine PII wurde gestoppt und Genentech ist aus der Entwickung ausgestiegen. Vielversprechender CD30 AK SGN-35 gegen HL in PIII konnte mit Takeda/Millennium verpartnert werden... SGEN besitzt noch die vollen US-Rechte. PIII Ergebnisse sollen in Laufe von 2010 vorliegen und die Zulassung in 2011 beantragt werden. Desweiteren sollen in 1H10 PII-Daten zum CD33-AK SGN-33 in AML kommen, die bereits für eine Zulassung ausreichend sein könnten. SGN-33 ist noch nicht verpartnert.

ArQule (ARQL) - MK 165 Mio USD (bei 3,69 USD)
http://finance.yahoo.com/q?s=arql
vielversprechender CMet-Hemmer ARQ-197 in PII gegen Krebs zusammen mit Daiichi... PII-Daten bei NSCLC in Kombination mit Tarceva in 1H10. Mit etwa 160 Mio USD Cash (netto 110) in etwa auf Cash-Niveau.

Evotec - MK 334 Mio USD (bei 1,13 EUR)
Mehrere Rückschläge in 2009 mit Scheitern von u.a. EVT201, EVT302. Umstrukturierung auf mehr Dienstleistung mit dem Ziel, ab 2012 profitabel zu werden. Die Pipeline befindet sich noch in einem sehr frühen Stadium... eine neue Kooperation mit Roche für EVT101/3 in PI gegen Depressionen. Daneben den oralen P2X7-Antagonisten gegen RA in PI.

Medigene - MK 183 Mio USD (bei 3,58 EUR)
Mit Eligard und Veregen 2 Produkte am Markt. Negative Kursentwicklung, da es laufend zu Verzögerungen kommt... Hoffungsträger ist EndoTag-1, liposomales Paclitaxel in PII, auf das sich nun ganz fokussiert wird. Es konnte trotz positiver PII-Daten bei BSDK immer noch kein Partner für eine PIII gefunden werden... in 2010 sollen Daten bei der größeren Indikation Brustkrebs folgen.

Array (ARRY) - MK 139 Mio USD (bei 2,81 USD)
http://finance.yahoo.com/q?s=arry
Ähnlich wie EXEL Konzentration auf Krebs und Autoimmunerkrankungen und unterschiedlichen Targets (7 Kandidaten in der Klinik). Mehrere negative Studienergebnisse in 2009. Finanzierungsprobleme, die mit der letzten Verpartnerung von ARRY-403 gegen Diabetes mit Amgen etwas gelindert werden konnten. In 2010 stehen weitere klinische Daten an.

Arena (ARNA) - MK 329 Mio USD (bei 3,55 USD)
http://finance.yahoo.com/q?s=arna
Arena hängt von Lorcaserin ab, ein Mittel gegen Fettsucht. Zwar gibt es einen großen Bedarf an Mitteln, die das Übergewicht reduzieren, doch dürfen diese keine Nebenwirkungen aufweisen. Lorcaserin scheint kaum Nebenwirkungen zu besitzen, doch ist die Wirkung nur gering, so dass es nicht klar ist, ob es für eine Zulassung ausreicht und ob das Mittel dann auch vom Markt angenommen wird. Es konnte kein Partner gefunden werden... die Zulassung wurde vor Kurzem alleine beantragt. Die Zulassungsentscheidung der FDA dürfte nächstes Jahr fallen... bei einer Zulassung wäre es das erste Mittel dieser Art am Markt.

ViroPharma (VPHM) - MK 650 Mio USD (bei 8,39 USD)
Anfang 2009 gab es mit dem überraschenden Scheitern von Maribavir in PIII einen größeren Rückschlag. Zudem wurde das wichtigste Produkt, das Antibiotikum Vancocin, wie seit langem erwartet generisch. Dafür entwickelt sich das in 2008 erworbene Cinryze als einziges Mittel zur Prophylaxe von HAE gut, so dass Vancocin vielleicht ersetzt werden kann.

NicOx - MK 590 Mio USD (bei 5,68 EUR)
Es konnte immer noch kein Partner für das Schmerzmittel Naproxcinod gefunden werden. Zuletzt wurde alleine die Zulassung beantragt.

Progenics (PGNX) - MK 141 Mio USD (bei 4,44 USD)
http://finance.yahoo.com/q?s=pgnx
Ein zugelassenes Produkt mit Relistor, das Verdauungsprobleme bei Gabe von Morphium verhindern soll. Relistor hat bisher sehr enttäuscht und wurde von Wyeth an PGNX zurückgegeben. PGNX sucht nun nach neuen Vermarktungsstrategien und entwickelt eine orale Version. Mit zuletzt 100 Mio USD Cash relativ hoher Cashbestand.

Addex - MK 120 Mio USD (bei 13,8 CHF)
Allosterische Modulatoren insbesondere für CNS, die u.a. bisherige etablierte Wirkansätze verbessern sollen. Zuletzt gab es mit dem unerwarteten Stopp der Entwicklung von ADX10059 für Langzeittherapien wegen möglicher Leberschäden einen schweren Rückschlag.

Sucampo - MK 169 Mio USD (bei 4,04 USD)
http://finance.yahoo.com/q?s=SCMP
Mit Amitiza ein Produkt am Markt... teilweise in der Gewinnzone

mfg ipollit
Regeneron (REGN) - MK 1,95 Mrd USD (bei 24,18 USD)
http://finance.yahoo.com/q?s=regn
starke Pipeline und Partner, Avastin-ähnliches Aflibercept in PIII mit Sanofi (50% Gewinnanteil), Lucentis-ähnliches VEGF-Trap Eye in PIII mit Bayer (100% US-Rechte, 50% weltweit), Arcalyst zugelassen als Nischenprodukt gegen CAPS, in PIII gegen Gicht (100% weltweit), große AK-Allianz mit Sanofi... Regeneron erhält jährlich 160 Mio USD bis 2017 mit dem Ziel jährlich 4 bis 5 AKs in die PI zu bekommen
Incyte (INCY) - MK 1,08 Mrd USD (bei 9,11 USD)
http://finance.yahoo.com/q?s=incy
die Schuldenkrise scheint überwunden... zuletzt extrem gute Deals für die JAK1/2-Hemmer: Novartis hat die ex-US Rechte am sehr aussichtsreichen INCB018424 gegen Krebs in PIII gekauft (Incyte besitzt 100% der US-Rechte), EliLilly finanziert die teure Entwicklung von INCB28050 gegen Immunkrankheiten wie RA (Incyte bekommt bis zu 20% Royalties)... das brachte zusammen 300 Mio USD Upfront ein. In der Pipeline befinden sich aktuell noch 2 Kandidaten in PII gegen Brustkrebs und Diabetes, die theoretisch auslizensiert werden können
Genmab - MK 710 Mio USD (bei 82 DKK)
recht enttäuschend. zwar haben Genmab/GSK die Zulassung für Arzerra (Rituxan-ähnlich) gegen CLL erhalten, doch ist eine riskante PIII gegen die wichtige Indikation NHL fehlgeschlagen. Damit ist offen, ob Arzerra mit Rituxan konkurrieren kann... direkte, aber riskante Vergleichstudien mit Rituxan wurden begonnen. Zudem wird Arzerra subkutan gegen RA entwickelt. Wichtige PIII-Ergebnisse zum zweiten Kandidaten Zalutumumab (Erbitux-ähnlich) gegen HN-Krebs sollen in Kürze erfolgen... auch hier besteht ein großes Risiko, dass sich Zalutumumab nicht gegen Erbitux absetzen kann. Die Entwicklung von R1507 (Roche) wurde leider gestoppt... wohl aufgrund der Genentech-Übernahme. Zudem ist der (Zwangs-)Verkauf der AK-Produktionsstädten enttäuschend.
Cubist (CBST) - MK 1,1 Mrd USD (bei 18,97 USD)
http://finance.yahoo.com/q?s=cbst
KGV10e von 12,5... das Antibiotikum Cubicin bringt hohe Gewinne ein und entwickelt sich zum Blockbuster. Der relativ niedrige Kurs (und der Einbruch in 2009) ist nur dadurch zu erklären, dass Teva versucht, die Patente (bis 2017/19) auszuhebeln, um Cubicin generisch zu vertreiben. 2011 wird dieser Fall vor Gericht behandelt. In der Pipeline gab es ein paar Rückschläge... ALN-RSV01 in PII wurde vorerst an Alnylam zurückgegeben, Cubist will den präklinischen Nachfolger gegen eine größere Indikation entwickeln. Die PII von Ecallantide gegen Blutungen (lizensiert von Dyax, die zuletzt dafür die Zulassung als Kalbitor gegen akute Schwellungen HAE von der FDA erhalten haben) wurde wegen Unregelmäßigkeiten frühzeitig geschlossen... es besteht das Risiko, dass damit die PII scheitert. Dafür wurde zuletzt mit der Übernahme von Calixa mit CXA-201 ein weiteres ausichtsreiches Antibiotikum erworben, welches 2014 auf den Markt kommen könnte.
Exelixis (EXEL) - MK 791 Mio USD (bei 7,37 USD)
http://finance.yahoo.com/q?s=exel
große Pipline mit 11 Kandidaten in der klinischen Entwicklung, die im wesentlichen von namhaften Pharma-Partnern finanziert wird. In 2009 konnten mehrere gute Deals abgeschlossen werden.
Rigel (RIGL) - MK 493 Mio USD (bei 9,51 USD)
http://finance.yahoo.com/q?s=rigl
Unsicherheit bezügliche R788 als orales Mittel gegen RA. Die PII-Daten zeigten zwar eine gute Wirkung, doch könnten störende Nebenwirkungen wie leicht erhöhter Blutdruck Probleme bereiten. PIII ist in Vorbeitung... es konnte aber immer noch kein Pharma-Partner gefunden werden. R788 ist von der Indikation gegen Autoimmunskrankheiten und Krebs ähnlich wie INCY's JAK-Hemmer... nur handelt es sich mit SYK um ein anderes Target.
Onyx (ONXX) - MK 1,82 Mrd USD (bei 29,34 USD)
http://finance.yahoo.com/q?s=onxx
KGV10e von 38. Nexavar gegen Krebs entwickelt weiter positiv... doch versucht der Partner Bayer an Onyx vorbei eine leicht modifizierte Version zu entwickeln, worauf Onyx Bayer verklagt hat. Nexavar könnte gegen Brustkrebs wirksam sein... dies würde das Potential deutlich erhöhen. Durch die Übernahme von Proteolix wurde zuletzt das Krebsmedikament Carfilzomib in PII erworben, dass ein nebenwirkungsfreieres Velcade sein könnte. Sind die Daten bei MM in 2010 positiv, könnte bereits 2011 ein weiterer potentieller Blockbuster von Onyx auf den Markt kommen.
Allos (ALTH) - MK 684 Mio USD (bei 6,58 USD)
http://finance.yahoo.com/q?s=alth
Ein-Produkt Unternehmen... im September hat Allos die Zulassung von Folotyn (neuartiges Chemotherapeutikum) gegen die seltene Krebsart PTCL erhalten. Es ist zwar eine kleine Indikation, aber es gibt keine Alternativen und der Preis für Folotyn ist entsprechend sehr hoch. Außerdem kann Allos es mit einer kleinen Vertriebsstruktur selber vermarkten. Folotyn wird nun bei größeren Indikationen wie NHL oder NSCLC getestet
Isis (ISIS) - MK 1,09 Mrd USD (bei 11,11 USD)
http://finance.yahoo.com/q?s=isis
Führend in der RNAi-Technologie... umfangreiche Pipeline und Kooperationen, die einiges an Bargeld einbringen (teilweise cashflow positiv), hoher Bargeldbestand mit 600 Mio USD. Wichtigstes Projekt ist Mipomersen in PIII, das zusammen mit Genzyme zur Senkung von Cholesterin, wenn z.B. Statine nicht ausreichend helfen, entwickelt wird. Bis Mitte 2010 sollen Daten von 3 weiteren PIIIs vorliegen... die Zulassung für Hochrisikopatienten soll im Sommer 2011 erfolgen, etwas später als ursprünglich geplant. Zwar konnte in der ersten PIII Mipomersen bei Hochrisiko-Patienten das Cholesterin senken, doch gab es auch bei einem Teil der Patienten erhöhte Leberwerte, ein Anzeichen von möglicher Leberschädigung. Die Frage ist nun, in wie weit Mipomersen dafür verantwortlich ist und ob dies auch bei Patienten, die keine lebensgefährlichen Cholesterinwerte aufweisen, auftritt, denn nur damit kann Mipomersen zu einem Blockbuster werden. Daher ist der Kurs zuletzt gefallen.
Micromet (MITI) - MK 459 Mio USD (bei 6,66 USD)
http://finance.yahoo.com/q?s=miti
hat spezielle BiTE-AKs in der Pipeline, mit denen T-Killerzellen des Immunsystems an bestimmte Targets gebunden werden sollen, damit der Körper diese zerstört. Der CD19-BiTE-AK Blinatumomab hat in PI/II Studien gute Ergebnisse geliefert... so war bei 80% von Patienten mit ALL der Krebs nicht mehr nachweisbar und die Ansprechrate bei NHL, bei denen andere Therapien nicht ansprechen, lag bei 100%. Dies sind zwar unsichere, aber ermutigende Ergebnisse. Der AK ist noch nicht verpartnert und es könnte bald eine Zulassungsstudie begonnen werden.
Seattle Genetics (SGEN) - MK 1,02 Mrd USD (bei 10,16 UDS)
http://finance.yahoo.com/q?s=sgen
Technologie, die AKs mit einer Chemo belädt, um die Wirkung der AKs zu erhöhen. Mit dem CD40-AK SGN-40 gab es einen Rückschlag... eine PII wurde gestoppt und Genentech ist aus der Entwickung ausgestiegen. Vielversprechender CD30 AK SGN-35 gegen HL in PIII konnte mit Takeda/Millennium verpartnert werden... SGEN besitzt noch die vollen US-Rechte. PIII Ergebnisse sollen in Laufe von 2010 vorliegen und die Zulassung in 2011 beantragt werden. Desweiteren sollen in 1H10 PII-Daten zum CD33-AK SGN-33 in AML kommen, die bereits für eine Zulassung ausreichend sein könnten. SGN-33 ist noch nicht verpartnert.
ArQule (ARQL) - MK 165 Mio USD (bei 3,69 USD)
http://finance.yahoo.com/q?s=arql
vielversprechender CMet-Hemmer ARQ-197 in PII gegen Krebs zusammen mit Daiichi... PII-Daten bei NSCLC in Kombination mit Tarceva in 1H10. Mit etwa 160 Mio USD Cash (netto 110) in etwa auf Cash-Niveau.
Evotec - MK 334 Mio USD (bei 1,13 EUR)
Mehrere Rückschläge in 2009 mit Scheitern von u.a. EVT201, EVT302. Umstrukturierung auf mehr Dienstleistung mit dem Ziel, ab 2012 profitabel zu werden. Die Pipeline befindet sich noch in einem sehr frühen Stadium... eine neue Kooperation mit Roche für EVT101/3 in PI gegen Depressionen. Daneben den oralen P2X7-Antagonisten gegen RA in PI.
Medigene - MK 183 Mio USD (bei 3,58 EUR)
Mit Eligard und Veregen 2 Produkte am Markt. Negative Kursentwicklung, da es laufend zu Verzögerungen kommt... Hoffungsträger ist EndoTag-1, liposomales Paclitaxel in PII, auf das sich nun ganz fokussiert wird. Es konnte trotz positiver PII-Daten bei BSDK immer noch kein Partner für eine PIII gefunden werden... in 2010 sollen Daten bei der größeren Indikation Brustkrebs folgen.
Array (ARRY) - MK 139 Mio USD (bei 2,81 USD)
http://finance.yahoo.com/q?s=arry
Ähnlich wie EXEL Konzentration auf Krebs und Autoimmunerkrankungen und unterschiedlichen Targets (7 Kandidaten in der Klinik). Mehrere negative Studienergebnisse in 2009. Finanzierungsprobleme, die mit der letzten Verpartnerung von ARRY-403 gegen Diabetes mit Amgen etwas gelindert werden konnten. In 2010 stehen weitere klinische Daten an.
Arena (ARNA) - MK 329 Mio USD (bei 3,55 USD)
http://finance.yahoo.com/q?s=arna
Arena hängt von Lorcaserin ab, ein Mittel gegen Fettsucht. Zwar gibt es einen großen Bedarf an Mitteln, die das Übergewicht reduzieren, doch dürfen diese keine Nebenwirkungen aufweisen. Lorcaserin scheint kaum Nebenwirkungen zu besitzen, doch ist die Wirkung nur gering, so dass es nicht klar ist, ob es für eine Zulassung ausreicht und ob das Mittel dann auch vom Markt angenommen wird. Es konnte kein Partner gefunden werden... die Zulassung wurde vor Kurzem alleine beantragt. Die Zulassungsentscheidung der FDA dürfte nächstes Jahr fallen... bei einer Zulassung wäre es das erste Mittel dieser Art am Markt.
ViroPharma (VPHM) - MK 650 Mio USD (bei 8,39 USD)
Anfang 2009 gab es mit dem überraschenden Scheitern von Maribavir in PIII einen größeren Rückschlag. Zudem wurde das wichtigste Produkt, das Antibiotikum Vancocin, wie seit langem erwartet generisch. Dafür entwickelt sich das in 2008 erworbene Cinryze als einziges Mittel zur Prophylaxe von HAE gut, so dass Vancocin vielleicht ersetzt werden kann.
NicOx - MK 590 Mio USD (bei 5,68 EUR)
Es konnte immer noch kein Partner für das Schmerzmittel Naproxcinod gefunden werden. Zuletzt wurde alleine die Zulassung beantragt.
Progenics (PGNX) - MK 141 Mio USD (bei 4,44 USD)
http://finance.yahoo.com/q?s=pgnx
Ein zugelassenes Produkt mit Relistor, das Verdauungsprobleme bei Gabe von Morphium verhindern soll. Relistor hat bisher sehr enttäuscht und wurde von Wyeth an PGNX zurückgegeben. PGNX sucht nun nach neuen Vermarktungsstrategien und entwickelt eine orale Version. Mit zuletzt 100 Mio USD Cash relativ hoher Cashbestand.
Addex - MK 120 Mio USD (bei 13,8 CHF)
Allosterische Modulatoren insbesondere für CNS, die u.a. bisherige etablierte Wirkansätze verbessern sollen. Zuletzt gab es mit dem unerwarteten Stopp der Entwicklung von ADX10059 für Langzeittherapien wegen möglicher Leberschäden einen schweren Rückschlag.
Sucampo - MK 169 Mio USD (bei 4,04 USD)
http://finance.yahoo.com/q?s=SCMP
Mit Amitiza ein Produkt am Markt... teilweise in der Gewinnzone
mfg ipollit
5-Tages-Charts...
Nasdaq Biotech Index

Einzelwerte





















mfg ipollit
Nasdaq Biotech Index
Einzelwerte
mfg ipollit
Regeneron - aktuelle Pipeline

mfg ipollit
mfg ipollit
Incyte - aktuelle Pipeline

mfg ipollit
mfg ipollit
Trading Spotlight
Genmab - aktuelle Pipeline

mfg ipollit
mfg ipollit
Vl. kann man heute ja "Decode Genetics"(Island) aufnehmen?
Zwar in Ch11 - Mcap aktuell 4Mio USD. Sehr bekanntes Unternehmen, 300Mio USD Schulden.
Davon mal abgesehen, kennt jemand die Pipeline und den Hintergrund?
Wird morgen von der Nasdaq delistet wegen Kursunterschreitung.
Soll keine Kaufempfehlung sein, nur, um die Hintergründe zu diskutieren, falls bekannt.

Zwar in Ch11 - Mcap aktuell 4Mio USD. Sehr bekanntes Unternehmen, 300Mio USD Schulden.
Davon mal abgesehen, kennt jemand die Pipeline und den Hintergrund?
Wird morgen von der Nasdaq delistet wegen Kursunterschreitung.
Soll keine Kaufempfehlung sein, nur, um die Hintergründe zu diskutieren, falls bekannt.
Cubist - aktuelle Pipeline
noch nicht enthalten ist das durch die Übernahme von Calixa erworbene Antibiotikum CXA-201 in PI (bzw. CXA-101 in PII)...

mfg ipollit
noch nicht enthalten ist das durch die Übernahme von Calixa erworbene Antibiotikum CXA-201 in PI (bzw. CXA-101 in PII)...
mfg ipollit
Antwort auf Beitrag Nr.: 38.670.572 von MrRipley am 05.01.10 19:24:09DCGN... naja, ist ein wenig heiß, sich ein bereits bankrottes Unternehmen ins Depot zu legen mit der Hoffnung, dass es doch noch irgendwie überlebt. Davon lasse ich lieber die Finger. Trotzdem danke für den Hinweis!
mfg ipollit
mfg ipollit
Exelixis - aktuelle Pipeline
mfg ipollit
mfg ipollit
Rigel - aktuelle Pipeline

mfg ipollit
mfg ipollit
Onyx - aktuelle Pipeline

mfg ipollit
mfg ipollit
Ich erlaube mir mal den Hinweis auf ein noch sehr kleines und ziemlich spezielles Unternehmen, nämlich:
NeurogesX
http://www.neurogesx.com/
Sie konzentrieren sich auf die Behandlung von Schmerzen, die durch Nervenschädigungen hervorgerufen werden.
Die Marktkapitalisierung beträgt 130 Millionen Dollar, wobei zum Ende des Q3/2009 ca. 57 Mio. Dollar Cash vorhanden waren.
Hier der Chart:

Der Kursaufschwung hat einen realen Grund, nämlich die Zulassung des Wirkstoffes Qutenza.
Was ist Qutenza?
Qutenza ist ein kutanes Pflaster (ein Pflaster, das ein Arzneimittel durch die Haut abgibt). Es enthält den Wirkstoff Capsaicin (8 %).
Wofür wird Qutenza angewendet?
Qutenza ist für die Behandlung von peripheren neuropathischen Schmerzen (d. h. Schmerzen, die durch Nervenschädigungen hervorgerufen werden) bei erwachsenen Nichtdiabetikern angezeigt. Es kann als Monotherapie oder in Kombination mit anderen Schmerzmitteln angewendet werden.
Das Arzneimittel ist nur auf ärztliche Verschreibung erhältlich.
Hier das File der EMEA:
http://www.emea.europa.eu/humandocs/PDFs/EPAR/Qutenza/H-909-…
Qutenza wird in Europa durch Astellas vertrieben (gegen double-digit royalties), die US-Rechte hält NeurogesX weiterhin selbst und will im 1. HJ 2010 einen eigenen Vertrieb in den USA aufbauen und zu einem Produktunternehmen werden.
Aufgrund der erfolgten Zulassungen werden im Jahre 2010 erstmals relevante Umsätze erzielt werden.
Das ist die Pipeline:

Das Unternehmen hängt also derzeit sehr stark am Wirkstoff capsaicin (Qutenza), was sicher nicht so wirklich schön ist.
Aber: Der Markt für Schmerzmittel in diesem Bereich ist Milliarden-Dollar schwer und Analysten trauen Qutenza langfristig (in einigen Jahren) Umsätze im Bereich mehrerer hundert Millionen Dollar zu.
Außerdem ist es überhaupt eine Leistung, einen Wirkstoff zugelassen und verpartnert zu bekommen. Da müssen andere erst mal "hinriechen", wo NeurogesX schon ist.
Letztlich kostet der ganze Laden nach Abzug des Cash derzeit vielleicht 80 Mio. Dollar oder so. Sollte Qutenza am Markt seine Nische erobern, ist das sicher nicht zu teuer bezahlt.
Kritik willkommen!
Gruß
NeurogesX
http://www.neurogesx.com/
Sie konzentrieren sich auf die Behandlung von Schmerzen, die durch Nervenschädigungen hervorgerufen werden.
Die Marktkapitalisierung beträgt 130 Millionen Dollar, wobei zum Ende des Q3/2009 ca. 57 Mio. Dollar Cash vorhanden waren.
Hier der Chart:
Der Kursaufschwung hat einen realen Grund, nämlich die Zulassung des Wirkstoffes Qutenza.
Was ist Qutenza?
Qutenza ist ein kutanes Pflaster (ein Pflaster, das ein Arzneimittel durch die Haut abgibt). Es enthält den Wirkstoff Capsaicin (8 %).
Wofür wird Qutenza angewendet?
Qutenza ist für die Behandlung von peripheren neuropathischen Schmerzen (d. h. Schmerzen, die durch Nervenschädigungen hervorgerufen werden) bei erwachsenen Nichtdiabetikern angezeigt. Es kann als Monotherapie oder in Kombination mit anderen Schmerzmitteln angewendet werden.
Das Arzneimittel ist nur auf ärztliche Verschreibung erhältlich.
Hier das File der EMEA:
http://www.emea.europa.eu/humandocs/PDFs/EPAR/Qutenza/H-909-…
Qutenza wird in Europa durch Astellas vertrieben (gegen double-digit royalties), die US-Rechte hält NeurogesX weiterhin selbst und will im 1. HJ 2010 einen eigenen Vertrieb in den USA aufbauen und zu einem Produktunternehmen werden.
Aufgrund der erfolgten Zulassungen werden im Jahre 2010 erstmals relevante Umsätze erzielt werden.
Das ist die Pipeline:
Das Unternehmen hängt also derzeit sehr stark am Wirkstoff capsaicin (Qutenza), was sicher nicht so wirklich schön ist.
Aber: Der Markt für Schmerzmittel in diesem Bereich ist Milliarden-Dollar schwer und Analysten trauen Qutenza langfristig (in einigen Jahren) Umsätze im Bereich mehrerer hundert Millionen Dollar zu.
Außerdem ist es überhaupt eine Leistung, einen Wirkstoff zugelassen und verpartnert zu bekommen. Da müssen andere erst mal "hinriechen", wo NeurogesX schon ist.
Letztlich kostet der ganze Laden nach Abzug des Cash derzeit vielleicht 80 Mio. Dollar oder so. Sollte Qutenza am Markt seine Nische erobern, ist das sicher nicht zu teuer bezahlt.
Kritik willkommen!
Gruß
Antwort auf Beitrag Nr.: 38.656.864 von ipollit am 03.01.10 16:49:57warum hast du keine mologen in deinem depot? 

Antwort auf Beitrag Nr.: 38.672.882 von SLGramann am 06.01.10 09:37:06Hallo SLGramann!
Danke für die Infos!... NGSX - hast du die auch aus dem Ohad Hammer Blog. Kritik kann ich da nicht äußern... hört sich schon sehr interessant an. Falls es einen Haken gibt, habe ich ihn bisher nicht entdeckt. Das Marktpotential ist relativ zur MK schon recht hoch... es ist nur ein wenig unsicher, ob NGSX das auf dem US-Markt auch eigenständig gut vermarktet bekommt. Der US-Markt könnte einige 100 Mio Umsatz bringen... und hier in der EU ebenfalls. Könnte mir vorstellen, mir eine paar Stücke ins Depot zu legen. Im Kurs steckt das Potential bisher jedenfalls nicht drin, finde ich.
Ich suche auch immer nach interessanten Werten... was käme sonst noch in Frage? Im Moment habe ich da z.B. Xoma, Theravance, Basilea, Neurosearch und Wilex auf meiner Liste.
mfg ipollit
Danke für die Infos!... NGSX - hast du die auch aus dem Ohad Hammer Blog. Kritik kann ich da nicht äußern... hört sich schon sehr interessant an. Falls es einen Haken gibt, habe ich ihn bisher nicht entdeckt. Das Marktpotential ist relativ zur MK schon recht hoch... es ist nur ein wenig unsicher, ob NGSX das auf dem US-Markt auch eigenständig gut vermarktet bekommt. Der US-Markt könnte einige 100 Mio Umsatz bringen... und hier in der EU ebenfalls. Könnte mir vorstellen, mir eine paar Stücke ins Depot zu legen. Im Kurs steckt das Potential bisher jedenfalls nicht drin, finde ich.
Ich suche auch immer nach interessanten Werten... was käme sonst noch in Frage? Im Moment habe ich da z.B. Xoma, Theravance, Basilea, Neurosearch und Wilex auf meiner Liste.
mfg ipollit
Antwort auf Beitrag Nr.: 38.677.562 von ipollit am 06.01.10 18:37:35
Hi ipollit,
richtig, die Idee kam von Hammer. Er hat die Aktie nach der Zulassung in Europa ins Depot genommen, aber leider nie einen Artikel dazu geschrieben.
Wie die Vermarktung laufen wird, ist wirklich die entscheidende Frage. Ich bin sehr gespannt, wann genau und mit welchem Erfolg das in den USA anläuft.
Es mag auch Leute geben, die den Wirkstoff nicht "sexy" finden. Ist ja nur ne "geraspelte Chilischote", könnte man boshaft (und ziemlich falsch) sagen und kein schicker Antikörper. Aber ganz so simpel ist es dann wohl doch nicht, denn sonst hätte Astellas nicht 42 Millionen upfront überwiesen.
Ansonsten habe ich derzeit nur noch ArQule im Visier, aber die hast Du ja eh schon im Depot.
Von Actelion habe ich mich getrennt und habe jetzt folgendes im Depot:
MorphoSys
Biotest
Seattle Genetics
Immunogen
Micromet
Incyte
Exelixis
NeurogesX
Wir haben also ne Menge Überschneidungen.
Außerdem finde auch ich Regeneron sehr interessant. Aber ich trau mich da nicht ran, weil ich über die P III - Programme nicht genug weiß, an denen aber meiner Meinung nach 30 bis 50% der Marktkapitalisierung hängt.
Gruß
Hi ipollit,
richtig, die Idee kam von Hammer. Er hat die Aktie nach der Zulassung in Europa ins Depot genommen, aber leider nie einen Artikel dazu geschrieben.
Wie die Vermarktung laufen wird, ist wirklich die entscheidende Frage. Ich bin sehr gespannt, wann genau und mit welchem Erfolg das in den USA anläuft.
Es mag auch Leute geben, die den Wirkstoff nicht "sexy" finden. Ist ja nur ne "geraspelte Chilischote", könnte man boshaft (und ziemlich falsch) sagen und kein schicker Antikörper. Aber ganz so simpel ist es dann wohl doch nicht, denn sonst hätte Astellas nicht 42 Millionen upfront überwiesen.
Ansonsten habe ich derzeit nur noch ArQule im Visier, aber die hast Du ja eh schon im Depot.
Von Actelion habe ich mich getrennt und habe jetzt folgendes im Depot:
MorphoSys
Biotest
Seattle Genetics
Immunogen
Micromet
Incyte
Exelixis
NeurogesX
Wir haben also ne Menge Überschneidungen.
Außerdem finde auch ich Regeneron sehr interessant. Aber ich trau mich da nicht ran, weil ich über die P III - Programme nicht genug weiß, an denen aber meiner Meinung nach 30 bis 50% der Marktkapitalisierung hängt.
Gruß
Antwort auf Beitrag Nr.: 38.672.907 von pokemon am 06.01.10 09:42:41warum sollte ich Mologen haben?... nichts über PI hinaus. da gibt es meiner Meinung nach aussichtsreichere Sachen.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.678.515 von SLGramann am 06.01.10 20:21:12"nur ne geraspelte Chilischote"... naja, in Chili befindet sich nur zufällig derselbe Wirkstoff, der, wie man wahrscheinlich selber schon die Erfahrung gemacht hat, wunderbar auf die Schmerzrezeptoren wirkt, denn nichts anderes ist ja die Schärfe. Die Eigenschaften von Qutenza hören sich gut an... anderes als bei den bisherigen Schmerzmitteln sollen die Nebenwirkungen gering sein... außerdem ist die Schmerzfreiheit sehr lange anhaltend: eine Stunde Anwendung sollen zu 3 Monaten schmerzfreiheit führen. Wie gesagt... hört sich interessant an!
Regeneron... eine MK von aktuell 2 Mrd USD ist zwar nicht wenig, doch ist REGN eben nicht nur von einem Produkt abhängig. Quasi zwei Produkte sind bereits auf dem Markt... Arcalyst und Novartis Ilaris (wofür REGN Royalties erhält)... beide wirken auf IL1, was vielversprechend ist (was man z.B. auch an XOMAs 052 sieht). Ist REGN mit Arcalyst gegen Gicht erfolgreich, so könnte wohl alleine das schon ein Blockbuster werden. Arcalyst, Aflibercept und VEGF Trap-Eye sind keine AKs sondern Traps. Traps sind freie Rezeptoren, die wie AKs an den Targets binden, um sie zu neutralisieren. Ob Aflibercept erfolgreich wird, muss man sehen... aber immerhin stellt es eine Alternative zu Avastin dar und die Entwicklung wird zu 100% von Sanofi finanziert. VEGF-Trap Eye scheint vielversprechend zu sein. Nach den bisherigen Ergebnissen scheint es dauerhafter als Lucentis die Sehschärfe zu erhalten und/oder seltener verabreicht werden zu mussen (Spritze durch das Auge hindurch, was ja das Risiko einer Infektion beinhaltet). Zuletzt habe ich zugekauft, weil Sanofi sehr viel in REGN investiert, was Sanofi wohl nur macht, wenn sie in REGN einen wichtigen Partner sehen (vielleicht soetwas wie Roche und Genentech): in den nächsten Jahren erhält REGN weit über 1 Mrd USD von Sanofi für die Entwicklung von AKs... Ziel ist es, 5 neue AKs pro Jahr in die PI zu bringen. Sanofi fianziert dann die komplette klinische Entwicklung und Vermarktung. Dafür erhält REGN am Ende nicht 5% Royalties oder soetwas, sondern die Gewinne werden geteilt (wobei REGN aus dem Anteil dann zunächst die Hälte der Entwicklungskosten an Sanofi zurückzahlt). Das hört sich für mich nach einem sehr guten Deal an. Dadurch konnte REGN zuletzt 400 zusätzliche Mitarbeiter einstellen. 5 Aks pro Jahr... das könnten am Ende insgesamt bis zu 40 neue klinische AKs sein, an denen REGN zur Hälfte beteiligt ist. Naja... wie das zu bewerten ist, ist eine andere Frage... daher ist REGN auch nicht ganz billig.
mfg ipollit
Regeneron... eine MK von aktuell 2 Mrd USD ist zwar nicht wenig, doch ist REGN eben nicht nur von einem Produkt abhängig. Quasi zwei Produkte sind bereits auf dem Markt... Arcalyst und Novartis Ilaris (wofür REGN Royalties erhält)... beide wirken auf IL1, was vielversprechend ist (was man z.B. auch an XOMAs 052 sieht). Ist REGN mit Arcalyst gegen Gicht erfolgreich, so könnte wohl alleine das schon ein Blockbuster werden. Arcalyst, Aflibercept und VEGF Trap-Eye sind keine AKs sondern Traps. Traps sind freie Rezeptoren, die wie AKs an den Targets binden, um sie zu neutralisieren. Ob Aflibercept erfolgreich wird, muss man sehen... aber immerhin stellt es eine Alternative zu Avastin dar und die Entwicklung wird zu 100% von Sanofi finanziert. VEGF-Trap Eye scheint vielversprechend zu sein. Nach den bisherigen Ergebnissen scheint es dauerhafter als Lucentis die Sehschärfe zu erhalten und/oder seltener verabreicht werden zu mussen (Spritze durch das Auge hindurch, was ja das Risiko einer Infektion beinhaltet). Zuletzt habe ich zugekauft, weil Sanofi sehr viel in REGN investiert, was Sanofi wohl nur macht, wenn sie in REGN einen wichtigen Partner sehen (vielleicht soetwas wie Roche und Genentech): in den nächsten Jahren erhält REGN weit über 1 Mrd USD von Sanofi für die Entwicklung von AKs... Ziel ist es, 5 neue AKs pro Jahr in die PI zu bringen. Sanofi fianziert dann die komplette klinische Entwicklung und Vermarktung. Dafür erhält REGN am Ende nicht 5% Royalties oder soetwas, sondern die Gewinne werden geteilt (wobei REGN aus dem Anteil dann zunächst die Hälte der Entwicklungskosten an Sanofi zurückzahlt). Das hört sich für mich nach einem sehr guten Deal an. Dadurch konnte REGN zuletzt 400 zusätzliche Mitarbeiter einstellen. 5 Aks pro Jahr... das könnten am Ende insgesamt bis zu 40 neue klinische AKs sein, an denen REGN zur Hälfte beteiligt ist. Naja... wie das zu bewerten ist, ist eine andere Frage... daher ist REGN auch nicht ganz billig.
mfg ipollit
zu ALTH...
JMP SECURITIES
Allos ($6.65, ALTH, Market Outperform, PT: $20)
Scarcity value makes this hematology company with an approved product an obvious Target. We continue to view Allos as a prime take-out candidate and believe that interim data from a Phase II trial in lung cancer could act as a driving catalyst for acquirers. FOLOTYN is now approved and available in the US for the treatment of PTCL, and we expect a full commercial launch to gain traction early this year. Although early in the launch ramp, we believe that initial sales indications from November and December 2009 may provide upside to Street expectations. We also look forward to learning how quickly and readily reimbursement authorities are adopting the drug’s use.
mfg ipollit
JMP SECURITIES
Allos ($6.65, ALTH, Market Outperform, PT: $20)
Scarcity value makes this hematology company with an approved product an obvious Target. We continue to view Allos as a prime take-out candidate and believe that interim data from a Phase II trial in lung cancer could act as a driving catalyst for acquirers. FOLOTYN is now approved and available in the US for the treatment of PTCL, and we expect a full commercial launch to gain traction early this year. Although early in the launch ramp, we believe that initial sales indications from November and December 2009 may provide upside to Street expectations. We also look forward to learning how quickly and readily reimbursement authorities are adopting the drug’s use.
mfg ipollit
Allos - aktuelle Pipeline

mfg ipollit
mfg ipollit
Isis - aktuelle Pipeline

mfg ipollit
mfg ipollit
Micromet - aktuelle Pipeline

mfg ipollit
mfg ipollit
Seattle Genetics - aktuelle Pipeline

mfg ipollit
mfg ipollit
ArQule - aktuelle Pipeline

mfg ipollit
mfg ipollit
Evotec - aktuelle Pipeline

mfg ipollit
mfg ipollit
Medigene - aktuelle Pipeline

mfg ipollit
mfg ipollit
Array - aktuelle Pipeline
(ARRY-403 ist inzwischen an Amgen verpartnert)


mfg ipollit
(ARRY-403 ist inzwischen an Amgen verpartnert)
mfg ipollit
Arena - aktuelle Pipeline
(die Entwicklung von Mercks Niacin Receptor Agonist ist vor Kurzem gestoppt worden)

mfg ipollit
(die Entwicklung von Mercks Niacin Receptor Agonist ist vor Kurzem gestoppt worden)
mfg ipollit
ViroPharma - aktuelle Pipeline
(Maribavir ist in 2009 in der PIII gescheitert)

mfg ipollit
(Maribavir ist in 2009 in der PIII gescheitert)
mfg ipollit
NicOx - aktuelle Pipeline

mfg ipollit
mfg ipollit
Progenics - aktuelle Pipeline

mfg ipollit
mfg ipollit
Addex - aktuelle Pipeline
ADX10059 scheint leider wegen Nebenwirkungen gescheitert zu sein!
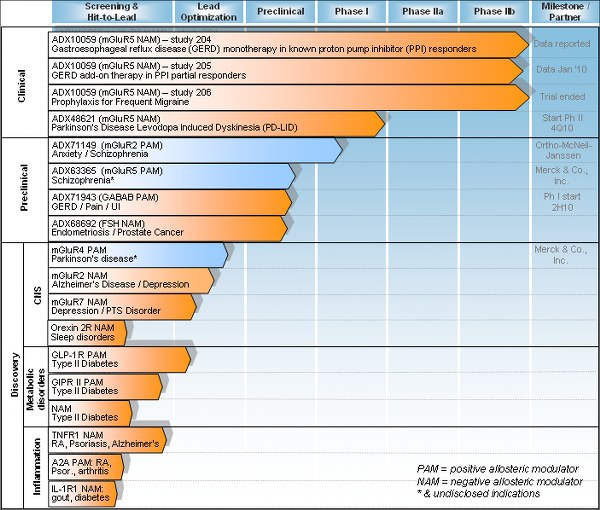
mfg ipollit
ADX10059 scheint leider wegen Nebenwirkungen gescheitert zu sein!
mfg ipollit
und zu guter letzt noch die aktuelle Pipeline von Sucampo


soviel zu den Pipelines Anfang 2010... mal schauen, wie sie sich im Laufe des Jahres entwickeln werden
mfg ipollit
soviel zu den Pipelines Anfang 2010... mal schauen, wie sie sich im Laufe des Jahres entwickeln werden
mfg ipollit
Antwort auf Beitrag Nr.: 38.680.068 von ipollit am 07.01.10 00:42:33Guten Morgen ipollit,
Human Genome ist zwar letztes Jahr schon stark gestiegen, aber da geht noch mehr!
Wenn die endgültige Zulassung für Benlysta kommt geht der Kurs über 40 $.
Danach hat Human Genome so viel Kohle, um auch seine anderen Entwicklungen erfolgreich voranzutreiben, oder es kommt ein Übernahmeangebot von Glaxo o.ä.
Ich habe letztes Jahr bei Human fast den absoluten Tiefpunkt erwischt und konnte meine gesamten Aktienverluste aus den letzten Jahren damit ausgleichen
Viele Grüße
af
Human Genome ist zwar letztes Jahr schon stark gestiegen, aber da geht noch mehr!
Wenn die endgültige Zulassung für Benlysta kommt geht der Kurs über 40 $.
Danach hat Human Genome so viel Kohle, um auch seine anderen Entwicklungen erfolgreich voranzutreiben, oder es kommt ein Übernahmeangebot von Glaxo o.ä.
Ich habe letztes Jahr bei Human fast den absoluten Tiefpunkt erwischt und konnte meine gesamten Aktienverluste aus den letzten Jahren damit ausgleichen
Viele Grüße
af
Antwort auf Beitrag Nr.: 38.680.263 von againstfotsch am 07.01.10 07:41:02Glückwunsch! Soetwas wie bei HGSI findet man nicht oft... ohne Benlysta hätte die Zukunft wohl düster ausgesehen... jetzt stehen die wieder blendend da. Allerdings beträgt die MK nun auch schon mehr als 5 Mrd USD. 40 USD wäre "nur" noch ein bisschen mehr als +20%... wenn dagegen doch noch etwas schief geht, dann gehts wohl wieder steil nach unten. Anfangs war ich nicht dabei gewesen und schaue nun lieber zu, wie es weiter geht.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.686.985 von ipollit am 07.01.10 19:36:50Hi ipollit, schön wieder von dir zu lesen. Drück dir die Daumen bei den US-Biotechs. Werd mich dieses Jahr aber hauptsächlich auf die deutschen konzentrieren. Hab noch paar Vertex im Depot liegen, da geht es ja dieses Jahr um die Wurst.
Grüße
blb
Grüße
blb
Antwort auf Beitrag Nr.: 38.687.140 von blb am 07.01.10 19:50:49Hallo blb!
Danke! - zu den deutschen Bios... wenn ich sie im Vergleich mit beispielsweise den US-Biotechs sehe, dann scheinen sie doch noch ziemlich hinterher zu hängen, so dass ich sie einfach im Vergleich zu wenig interessant finde. Welches deutsche Biotechunternehmen würde denn international Beachtung finden? Da sehe ich eigentlich nur eine Morphosys (Qiagen lasse ich mal außen vor, naja und eine Micromet ist ja eigentlich auch deutsch). Ich habe zwar auch Evotec und Medigene im Depot... aber das sind eigentlich im wesentlichen Spekulationen auf gute Deals für EndoTag-1 und EVT201 gewesen. Letzteres hat nichts ergeben und auch EndoTag-1 zieht sich ewig hin. Interessant finde ich vielleicht noch eine Wilex... vielleicht gelingt denen mal der Durchbruch, zumal die auch eine etwas breitere Pipeline besitzen, wofür sie allerdings recht wenig Geld zur Verfügung haben... immerhin etwas Potential.
Naja, wünsche dir trotzdem viel Glück dabei... z.B. Evotec war letztes Jahr vom Kurs her auch sehr erfolgreich, also warum nicht.
mfg ipollit
Danke! - zu den deutschen Bios... wenn ich sie im Vergleich mit beispielsweise den US-Biotechs sehe, dann scheinen sie doch noch ziemlich hinterher zu hängen, so dass ich sie einfach im Vergleich zu wenig interessant finde. Welches deutsche Biotechunternehmen würde denn international Beachtung finden? Da sehe ich eigentlich nur eine Morphosys (Qiagen lasse ich mal außen vor, naja und eine Micromet ist ja eigentlich auch deutsch). Ich habe zwar auch Evotec und Medigene im Depot... aber das sind eigentlich im wesentlichen Spekulationen auf gute Deals für EndoTag-1 und EVT201 gewesen. Letzteres hat nichts ergeben und auch EndoTag-1 zieht sich ewig hin. Interessant finde ich vielleicht noch eine Wilex... vielleicht gelingt denen mal der Durchbruch, zumal die auch eine etwas breitere Pipeline besitzen, wofür sie allerdings recht wenig Geld zur Verfügung haben... immerhin etwas Potential.
Naja, wünsche dir trotzdem viel Glück dabei... z.B. Evotec war letztes Jahr vom Kurs her auch sehr erfolgreich, also warum nicht.
mfg ipollit
Antwort auf Beitrag Nr.: 38.689.434 von ipollit am 08.01.10 00:00:58du solltest dir ein paar mologene dazulegen.
unbeachtet von den meisten machen die ihren weg! mgn1703 könnte binnen jahresfrist auslizenziert sein, so independent research in deren letzten studie.
unbeachtet von den meisten machen die ihren weg! mgn1703 könnte binnen jahresfrist auslizenziert sein, so independent research in deren letzten studie.
Antwort auf Beitrag Nr.: 38.679.857 von ipollit am 06.01.10 23:21:36
So, habe jetzt auch ArQule und Regeneron ins Depot genommen. Hoffe, dass ich an den Biotech-Werten dieses Jahr nicht mehr rumschrauben muss und gut aufgestellt bin. Entscheidende Daten kommen in 2010 jedenfalls massenweise...
So, habe jetzt auch ArQule und Regeneron ins Depot genommen. Hoffe, dass ich an den Biotech-Werten dieses Jahr nicht mehr rumschrauben muss und gut aufgestellt bin. Entscheidende Daten kommen in 2010 jedenfalls massenweise...
Antwort auf Beitrag Nr.: 38.694.977 von SLGramann am 08.01.10 16:13:42ja, hoffentlich wird dieses Jahr ein gutes für Biotechs. Ich habe jetzt auch meine Evotec in NGSX getauscht. So ein kleines Unternehmen, welches am Markt bisher nur wenig Beachtung findet, macht mich zwar etwas nervös, aber in dem immerhin zugelassenen Produkt steckt auch einiges an Potential, wenn es erfolgreich wird. Im Erfolgsfall könnten hier in Europa alleine Royalties von über 100 Mio USD drin sein. Bei einem Unternehmen, welches sich im Bereich einer 100 Mio MK bewegt, ist das ja schon ein ganze Menge. Naja... das Risiko besteht wohl aber auch, dass sich Qutenza ähnlich schwach wie z.B. Relistor entwickelt. Dann helfen alle tollen Prognosen nicht.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.697.089 von ipollit am 08.01.10 19:28:28Hallo ipollit, kennst du:
IMMUNOMEDICS INC. REGISTERED SHARES DL -,01 (872983)
Was hälst du von der Firma bzw. dem Kursverlauf?
Gruß
af
IMMUNOMEDICS INC. REGISTERED SHARES DL -,01 (872983)
Was hälst du von der Firma bzw. dem Kursverlauf?
Gruß
af
Antwort auf Beitrag Nr.: 38.679.580 von ipollit am 06.01.10 22:31:19heute war MOLOGEN + 11,5 %!
Antwort auf Beitrag Nr.: 38.679.580 von ipollit am 06.01.10 22:31:19sofern TLR 9 behandlungsansatz weiter gute ergebnisse zeigt, dann wird es noch ganz andere unternehmensbewertungen geben.
bei TLR 9 ist mologen international erste liga.
zulassungsrelevate phase II beim metas. darmkrebs nach der erstmaligen chemotheraoie soll in 1/2010 beginnen und bereits in 4/2010 soll erste zwischenauswertung erfolgen.

bei TLR 9 ist mologen international erste liga.
zulassungsrelevate phase II beim metas. darmkrebs nach der erstmaligen chemotheraoie soll in 1/2010 beginnen und bereits in 4/2010 soll erste zwischenauswertung erfolgen.

Antwort auf Beitrag Nr.: 38.712.750 von pokemon am 11.01.10 21:45:28dann Glückwunsch! Trotzdem ist es mir so zu unsicher... vielleicht schaue ich sie mir in ein paar Jahren an.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.712.207 von againstfotsch am 11.01.10 20:52:29mit IMMU kenne ich mich nicht aus, sorry... waren mir nur die Tage aufgefallen, weil es einen größeren Kurssprung gab. Ich weiß nicht recht, ob er gerechtfertigt ist... scheinen etwas vom HGSI-Hype zu profitieren, weil sie auch ein Medikament gegen Lupus in der Pipeline haben. Was mir zum Beispiel nicht gefiel, war der geringe Cash, falls das auf yahoo korrekt ist: 20 Mio USD sind nicht allzu viel. Eine MK von 300 Mio USD ist da auch nicht gerade wenig. Aber wie gesagt, kenne ich mich mit denen nicht aus und kann es somit nicht wirklich einschätzen.
mfg ipollit
mfg ipollit
zu ALTH...
JPM - Allos Therapeutics: Optionality Lies to the Upside Ahead of Folotyn Lung Data; Initiating at Overweight
We are initiating coverage of Allos Therapeutics (NASDAQ: ALTH) with an Overweight rating and a $10 Price Target. We see favorable risk-reward in ALTH shares given mitigated downside risk from one FDA approved indication (PTCL) for key value driver Folotyn, and clear upside potential from Folotyn label-expansion opportunities. The key near-term clinical catalyst is phase 2b data expected in 2nd/3rd-line non-small cell lung cancer (NSCLC) 1H10, which could reveal a sizable NSCLC opportunity but also demonstrate activity in solid tumors. With a valuation that seems anchored on PTCL only, we are Overweight given several free call options in the pipeline.
Folotyn a lower risk asset that could cross the liquid/solid tumor divide. Folotyn is approved in PTCL, a niche market within hematological cancers and could gain compendia listing near term in cutaneous T cell lymphoma (CTCL). However, its mechanism of action is validated in solid tumors as well, and we are focused on the NSCLC data 1H10e given early signs of activity and strong comps vs. Alimta.
PTCL supports valuation. The initial FDA approval is in PTCL, a niche indication of ~5-10k addressable patients. We believe the peak sales opportunity is $500M in PTCL, which in our view, easily supports the current valuation. Concern over potential competition is not a deal breaker, and could assist in expanding the PTCL market.
CTCL up next, but lung cancer the key driver. Folotyn could become compendia-listed by 1H10e in CTCL, a niche indication. However, the real upside near-term driver in our view is the phase 2b data vs. Tarceva in 2nd-/3rd-line NSCLC, which, as we see it, could add $4-5 per share in NPV, assuming activity implies sales in NSCLC and other solid tumors.
Several catalysts in 2010. While we view 1H10e NSCLC data as the key catalyst in 2010, there are other catalysts expected throughout the year. Compendia listing for Folotyn in CTCL could come in 1H10e, while 2H10e data readouts for Folotyn in NHL and CTCL could drive additional upside.
Valuation attractive. Our Dec 2010 PT is $10 (implying 49% upside potential), which is based on a probability-adjusted NPV analysis of the three commercial scenarios for Folotyn. With a risk/reward profile that we view as attractive and not reflected in current levels, we rate ALTH shares Overweight, especially when considering the scarcity value of approved products that are owned exclusively in all indications
mfg ipollit
JPM - Allos Therapeutics: Optionality Lies to the Upside Ahead of Folotyn Lung Data; Initiating at Overweight
We are initiating coverage of Allos Therapeutics (NASDAQ: ALTH) with an Overweight rating and a $10 Price Target. We see favorable risk-reward in ALTH shares given mitigated downside risk from one FDA approved indication (PTCL) for key value driver Folotyn, and clear upside potential from Folotyn label-expansion opportunities. The key near-term clinical catalyst is phase 2b data expected in 2nd/3rd-line non-small cell lung cancer (NSCLC) 1H10, which could reveal a sizable NSCLC opportunity but also demonstrate activity in solid tumors. With a valuation that seems anchored on PTCL only, we are Overweight given several free call options in the pipeline.
Folotyn a lower risk asset that could cross the liquid/solid tumor divide. Folotyn is approved in PTCL, a niche market within hematological cancers and could gain compendia listing near term in cutaneous T cell lymphoma (CTCL). However, its mechanism of action is validated in solid tumors as well, and we are focused on the NSCLC data 1H10e given early signs of activity and strong comps vs. Alimta.
PTCL supports valuation. The initial FDA approval is in PTCL, a niche indication of ~5-10k addressable patients. We believe the peak sales opportunity is $500M in PTCL, which in our view, easily supports the current valuation. Concern over potential competition is not a deal breaker, and could assist in expanding the PTCL market.
CTCL up next, but lung cancer the key driver. Folotyn could become compendia-listed by 1H10e in CTCL, a niche indication. However, the real upside near-term driver in our view is the phase 2b data vs. Tarceva in 2nd-/3rd-line NSCLC, which, as we see it, could add $4-5 per share in NPV, assuming activity implies sales in NSCLC and other solid tumors.
Several catalysts in 2010. While we view 1H10e NSCLC data as the key catalyst in 2010, there are other catalysts expected throughout the year. Compendia listing for Folotyn in CTCL could come in 1H10e, while 2H10e data readouts for Folotyn in NHL and CTCL could drive additional upside.
Valuation attractive. Our Dec 2010 PT is $10 (implying 49% upside potential), which is based on a probability-adjusted NPV analysis of the three commercial scenarios for Folotyn. With a risk/reward profile that we view as attractive and not reflected in current levels, we rate ALTH shares Overweight, especially when considering the scarcity value of approved products that are owned exclusively in all indications
mfg ipollit
zu VPHM... Cinryze entwickelt sich ganz gut
ViroPharma Provides 2010 Cinryze(TM) (C1 Esterase Inhibitor [Human]) Outlook
http://finance.yahoo.com/news/ViroPharma-Provides-2010-prnew…
- Company Also Provides Update to U.S. Hereditary Angioedema (HAE) Peak Year Sales Estimates -
EXTON, Pa., Jan. 11 /PRNewswire/ -- ViroPharma Incorporated (Nasdaq: VPHM) today announced that Vincent Milano, president and chief executive officer of ViroPharma, will provide an overview of the company's business and present a financial update during the 28th Annual J.P. Morgan Healthcare Conference. As previously announced, this presentation will be webcast live at 11:00 A.M. ET (8:00 A.M. PT) on Wednesday, January 13, 2010 and may be accessed via the company's website at www.viropharma.com . The company expects to release full-year 2009 financial results and further discuss 2010 guidance later in the first quarter of 2010.
"The ultimate reward for us in any given year is to achieve our goal of providing solutions for patients with serious diseases and unmet medical needs; 2009 was a remarkable year in that respect," stated Vincent Milano, ViroPharma's chief executive officer. "In 2009 we successfully launched Cinryze™ (C1 esterase inhibitor [human]), the first and only drug approved to prevent HAE attacks. Thanks to meticulous execution by our team, we were able to provide Cinryze during 2009 to over 400 patients who are now actively preventing their attacks. We are pleased to announce that for 2009, we expect our net Cinryze sales will be toward the high end of our previous guidance range of $90 to $95 million, placing Cinryze among the best ever launches of an ultra orphan drug product."
Milano continued, "Our momentum into 2010 is also strong, as we announced this morning an agreement with our partner, Sanquin for the global rights to market and develop Cinryze, and as we continue our execution on our manufacturing scale up efforts to serve HAE patients including the hundreds who are now enrolled in CinryzeSolutions™. We are now producing Cinryze through our scaled-up parallel chromatography process, or PCP, which will begin to enter the trade in the second quarter of this year. Further, we recently conducted a successful meeting with the FDA on our industrial scale initiative where we were able to confirm our previous expectations of the path forward for this significantly expanded manufacturing process. As a result, we are announcing 2010 Cinryze net sales guidance of between $145 and $165 million, which represents tremendous revenue growth over 2009. Although we anticipate that we will be profitable in 2010, we will provide our full guidance later in the first quarter of this year. Finally, I am pleased to announce we have increased our projection of U.S. HAE peak year Cinryze sales to between $350 and $450 million. Our objective continues to be to ensure that every patient who can benefit from Cinryze will have access to this important drug."
***********
ViroPharma expands licensing deal for angioedema drug Cinryze
http://philadelphia.bizjournals.com/philadelphia/stories/201…
ViroPharma Inc. said Monday it has expanded it global licensing deal for Cinryze with Sanquin Blood Supply Foundation in the Netherlands.
The Exton, Pa., specialty pharmaceutical company said the revised agreement “significantly expands ViroPharma's rights to commercialize Cinryze in regions beyond the originally licensed territories of North America, most of the countries in South America, and Israel, and allows for development of potential new indications.”
In October 2008, the Food and Drug Administration approved the use of Cinryze to prevent against angioedema attacks in adolescent and adult patients with hereditary angioedema or HAE. HAE is a rare, severely debilitating, and life-threatening genetic disorder caused by a deficiency of C1 inhibitor, a human plasma protein. The drug is only approved in the United States.
“We have developed an outstanding relationship with Sanquin during the first year of the launch of Cinryze in the United States, and the expansion of our collaboration represents a significant step in our combined efforts to expand the potential markets in which we may commercialize Cinryze,” said Vincent Milano, ViroPharma's president and CEO.
Milano said ViroPharma (NASDAQ:VPHM) will look to getting Cinryze approved in new territories and for other C1-mediated diseases. The company also plans to develop new forms of administration for the drug, which is now delivered intravenously.
Under the terms of the expanded deal, which involved no upfront payments, ViroPharma agreed to modify the existing manufacturing fee, establish minimum purchase requirements, fund research efforts at Sanquin at the rate of 1 million Euros ($1.45 million) a year for five years, and provide an additional loan to Sanquin to fund capacity expansions.
ViroPharma has been granted the exclusive right and license to research, develop, obtain regulatory approvals for and commercialize Cinryze in all countries in Europe and the rest of world, other than certain European and other territories in which Sanquin has existing relationships. ViroPharma also has been granted the right to develop Cinryze for all potential new indications.
mfg ipollit
ViroPharma Provides 2010 Cinryze(TM) (C1 Esterase Inhibitor [Human]) Outlook
http://finance.yahoo.com/news/ViroPharma-Provides-2010-prnew…
- Company Also Provides Update to U.S. Hereditary Angioedema (HAE) Peak Year Sales Estimates -
EXTON, Pa., Jan. 11 /PRNewswire/ -- ViroPharma Incorporated (Nasdaq: VPHM) today announced that Vincent Milano, president and chief executive officer of ViroPharma, will provide an overview of the company's business and present a financial update during the 28th Annual J.P. Morgan Healthcare Conference. As previously announced, this presentation will be webcast live at 11:00 A.M. ET (8:00 A.M. PT) on Wednesday, January 13, 2010 and may be accessed via the company's website at www.viropharma.com . The company expects to release full-year 2009 financial results and further discuss 2010 guidance later in the first quarter of 2010.
"The ultimate reward for us in any given year is to achieve our goal of providing solutions for patients with serious diseases and unmet medical needs; 2009 was a remarkable year in that respect," stated Vincent Milano, ViroPharma's chief executive officer. "In 2009 we successfully launched Cinryze™ (C1 esterase inhibitor [human]), the first and only drug approved to prevent HAE attacks. Thanks to meticulous execution by our team, we were able to provide Cinryze during 2009 to over 400 patients who are now actively preventing their attacks. We are pleased to announce that for 2009, we expect our net Cinryze sales will be toward the high end of our previous guidance range of $90 to $95 million, placing Cinryze among the best ever launches of an ultra orphan drug product."
Milano continued, "Our momentum into 2010 is also strong, as we announced this morning an agreement with our partner, Sanquin for the global rights to market and develop Cinryze, and as we continue our execution on our manufacturing scale up efforts to serve HAE patients including the hundreds who are now enrolled in CinryzeSolutions™. We are now producing Cinryze through our scaled-up parallel chromatography process, or PCP, which will begin to enter the trade in the second quarter of this year. Further, we recently conducted a successful meeting with the FDA on our industrial scale initiative where we were able to confirm our previous expectations of the path forward for this significantly expanded manufacturing process. As a result, we are announcing 2010 Cinryze net sales guidance of between $145 and $165 million, which represents tremendous revenue growth over 2009. Although we anticipate that we will be profitable in 2010, we will provide our full guidance later in the first quarter of this year. Finally, I am pleased to announce we have increased our projection of U.S. HAE peak year Cinryze sales to between $350 and $450 million. Our objective continues to be to ensure that every patient who can benefit from Cinryze will have access to this important drug."
***********
ViroPharma expands licensing deal for angioedema drug Cinryze
http://philadelphia.bizjournals.com/philadelphia/stories/201…
ViroPharma Inc. said Monday it has expanded it global licensing deal for Cinryze with Sanquin Blood Supply Foundation in the Netherlands.
The Exton, Pa., specialty pharmaceutical company said the revised agreement “significantly expands ViroPharma's rights to commercialize Cinryze in regions beyond the originally licensed territories of North America, most of the countries in South America, and Israel, and allows for development of potential new indications.”
In October 2008, the Food and Drug Administration approved the use of Cinryze to prevent against angioedema attacks in adolescent and adult patients with hereditary angioedema or HAE. HAE is a rare, severely debilitating, and life-threatening genetic disorder caused by a deficiency of C1 inhibitor, a human plasma protein. The drug is only approved in the United States.
“We have developed an outstanding relationship with Sanquin during the first year of the launch of Cinryze in the United States, and the expansion of our collaboration represents a significant step in our combined efforts to expand the potential markets in which we may commercialize Cinryze,” said Vincent Milano, ViroPharma's president and CEO.
Milano said ViroPharma (NASDAQ:VPHM) will look to getting Cinryze approved in new territories and for other C1-mediated diseases. The company also plans to develop new forms of administration for the drug, which is now delivered intravenously.
Under the terms of the expanded deal, which involved no upfront payments, ViroPharma agreed to modify the existing manufacturing fee, establish minimum purchase requirements, fund research efforts at Sanquin at the rate of 1 million Euros ($1.45 million) a year for five years, and provide an additional loan to Sanquin to fund capacity expansions.
ViroPharma has been granted the exclusive right and license to research, develop, obtain regulatory approvals for and commercialize Cinryze in all countries in Europe and the rest of world, other than certain European and other territories in which Sanquin has existing relationships. ViroPharma also has been granted the right to develop Cinryze for all potential new indications.
mfg ipollit
zu ONXX...
http://www.thestreet.com/_yahoo/story/10658357/1/more-biotec…
More Biotech Nuggets From the J.P. Morgan Confab
China was a big topic at the Onyx Pharmaceuticals(ONXX Quote) breakout session. Specifically, investors are anxious for some visibility into the Chinese government's plan to reimburse for targeted cancer drugs, which would include Onyx's liver cancer drug Nexavar.
China represents an enormous commercial market opportunity for Nexavar, since about half of the 600,000 liver cancer deaths worldwide occur there. Nexavar is approved in China today but patients have to pay out of pocket for the drug. Sales in China could rocket, however, if the Chinese government decides to pay for the drug.
Onyx executives said the company expects the Chinese government to hold meetings with cancer drug companies over the next year as part of the decision-making process for reimbursement.
Meantime, Onyx expects to report results from the phase III "Nexus" study of Nexavar in non-small cell lung cancer sometime in 2010. A previous phase III study in lung cancer failed, but Onyx has increased the number of patients in the current trial and is using a different chemotherapy backbone, which may help yield more positive results.
mfg ipollit
http://www.thestreet.com/_yahoo/story/10658357/1/more-biotec…
More Biotech Nuggets From the J.P. Morgan Confab
China was a big topic at the Onyx Pharmaceuticals(ONXX Quote) breakout session. Specifically, investors are anxious for some visibility into the Chinese government's plan to reimburse for targeted cancer drugs, which would include Onyx's liver cancer drug Nexavar.
China represents an enormous commercial market opportunity for Nexavar, since about half of the 600,000 liver cancer deaths worldwide occur there. Nexavar is approved in China today but patients have to pay out of pocket for the drug. Sales in China could rocket, however, if the Chinese government decides to pay for the drug.
Onyx executives said the company expects the Chinese government to hold meetings with cancer drug companies over the next year as part of the decision-making process for reimbursement.
Meantime, Onyx expects to report results from the phase III "Nexus" study of Nexavar in non-small cell lung cancer sometime in 2010. A previous phase III study in lung cancer failed, but Onyx has increased the number of patients in the current trial and is using a different chemotherapy backbone, which may help yield more positive results.
mfg ipollit
Antwort auf Beitrag Nr.: 38.713.802 von ipollit am 12.01.10 00:24:15Hallo ipollit,
habe noch eine Frage an dich:
Was hälst du von Affymetrix?
Affymetrix underweight
13.01.2010 - 16:55
Rating-Update:
London (aktiencheck.de AG) - Die Analysten von Barclays Capital stufen die Aktie von Affymetrix (ISIN US00826T1088/ WKN 901198) von "equal weight" auf "underweight" herab. Das Kursziel werde von 6 USD auf 3 USD reduziert. (13.01.2010/ac/a/u)Analyse-Datum: 13.01.2010
Quelle: Finanzen.net
3$ halte ich für untertrieben, was meinst du?
Gruß
af
habe noch eine Frage an dich:
Was hälst du von Affymetrix?
Affymetrix underweight
13.01.2010 - 16:55
Rating-Update:
London (aktiencheck.de AG) - Die Analysten von Barclays Capital stufen die Aktie von Affymetrix (ISIN US00826T1088/ WKN 901198) von "equal weight" auf "underweight" herab. Das Kursziel werde von 6 USD auf 3 USD reduziert. (13.01.2010/ac/a/u)Analyse-Datum: 13.01.2010
Quelle: Finanzen.net
3$ halte ich für untertrieben, was meinst du?
Gruß
af
Antwort auf Beitrag Nr.: 38.689.434 von ipollit am 08.01.10 00:00:58hi ipollit,
ich verstehe nicht ganz, eine morphosys findest du ok, hast sie aber wie's scheint nicht im depot? bitte kurz um aufklärung.
ansonsten halte ich von SGEN und EXEL sehr viel und habe dementsprechend einiges davon.
diese NGSX muss ich mir mal anschaun - klingt interessant.
lg
pf2
ich verstehe nicht ganz, eine morphosys findest du ok, hast sie aber wie's scheint nicht im depot? bitte kurz um aufklärung.
ansonsten halte ich von SGEN und EXEL sehr viel und habe dementsprechend einiges davon.
diese NGSX muss ich mir mal anschaun - klingt interessant.
lg
pf2
@SL
nochmal zu dieser NGSX: in der aktuell in EU und USA zugelassenen indikation dürfte das marktpotenzial doch recht beschränkt sein, oder?!
wann folgen hier weitere indikationsgebiete?
nochmal zu dieser NGSX: in der aktuell in EU und USA zugelassenen indikation dürfte das marktpotenzial doch recht beschränkt sein, oder?!
wann folgen hier weitere indikationsgebiete?
Antwort auf Beitrag Nr.: 38.729.096 von againstfotsch am 13.01.10 17:44:01sorry, ich kenne Affymetrix nicht näher... da sie eher Systeme verkaufen, also weniger klassischer Biotech als mehr Gerätehersteller, sind sie nicht ganz so interessant für mich.
Nur weil irgendwelche Analysten ein Kursziel ausgeben, muss das noch lange nichts bedeuten. Der Kurs ist nachwievor über 6 USD... also keine Auswirkung. Interessant wäre höchstens deren Grund für die Abstufung.
mfg ipollit
Nur weil irgendwelche Analysten ein Kursziel ausgeben, muss das noch lange nichts bedeuten. Der Kurs ist nachwievor über 6 USD... also keine Auswirkung. Interessant wäre höchstens deren Grund für die Abstufung.
mfg ipollit
Antwort auf Beitrag Nr.: 38.731.676 von PathFinder2 am 13.01.10 22:21:12Ich finde, dass Morphoys auch international Beachtung finden kann, da sie recht breit aufgestellt sind und diese klassische AK-Technolgie besitzen, während mehrere Konkurrenten ja schon aufgekauft worden sind. Ich finde die Kooperation mit Novartis interessant und vielversprechend. Die Partner-Pipeline macht langsam Fortschritte und auch aus der eigenen Pipeline könnte etwas werden. Im Depot habe ich sie allerdings nicht... da warte ich noch etwas, bis es mit der Pipeline deutlich Richtung PIII geht. Die MK liegt ja, wenn ich das richtig sehe bereits über 500 Mio USD. Wäre z.B. eine Kursverdopplung d.h. eine MK von mehr als 1 Mrd USD angemessen für ein Unternehmen, dass zwar eine breite Pipeline hat, aber noch weit von möglichen Marktzulassungen entfernt ist? Ich denke, dass MOR im Moment gut bewertet ist... also warte ich erst einmal ab. Stattdessen habe ich bezügl. AK-Technologie z.B. REGN... die haben mehrere eigene PIIIs und eine deutlich bessere AK-Allianz mit Sanofi.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.747.915 von PathFinder2 am 15.01.10 19:05:13zu NGSX...
so schlecht ist die zugelassene Indikation PHN nicht, zumal HIV und PDN offlabel Umsätze einbringen können, auch wenn noch keine Zulassung dafür vorliegt.
Die aktuelle Zulassung+Offlabel soll so ca. 350+ Mio USD peak sales weltweit einbringen, was ja für ein Unternehmen mit einer MK von etwas über 100 Mio USD nicht wenig ist, auch wenn es teilweise nur Royalties usw sind.
http://www.europharmatoday.com/2009/06/neurogesx-brings-aste…
June 25, 2009
NeurogesX Brings Astellas On Board To Launch Pain Patch in Europe
A deal with Astellas to market and distribute the neuropathic pain patch Qutenza in Europe will bolster NeurogesX's cash position, giving the company some breathing room to prepare for regulatory approval and a 2010 launch in the U.S. market.
Qutenza was approved for neuropathic pain in non-diabetic patients by the European Medicines Agency in May and it is pending review at the FDA, with an Aug. 16 user fee date.
NeurogesX is set to get €30 million ($42 million) upfront for Qutenza commercialization rights from the European subsidiary of Japanese drug maker Astellas, according to a deal announced on June 22. In addition to 27 countries in the European Union, the deal covers Iceland, Switzerland, and some countries in the Middle East and Africa. The product will be launched in Europe by the first half of 2010.
Qutenza is a dermal patch packing a high concentration form of synthetic capsaicin (also called trans-capsaicin), which is the substance found in chili peppers. Capsaicin stimulates transient vanilloid 1 receptors in the skin, which in turn subdue overactive pain receptors. The effects are long-lasting, but also reversible, the company says.
As part of the agreement, Astellas also will pay €5 million upfront ($7 million) for a co-development and commercialization option on NGX-1998, a Phase I liquid formulation of the NeurogesX product.
NeurogesX now is eligible for €70 million ($97 million) in sales-based milestone payments and additional option payments for the liquid formulation. Royalty rates start in the high teens and escalate into the mid-20s, the company said during a June 22 investors' call.
Deal gives financial peace of mind
NeurogesX had about $19 million in cash and marketable securities at the close of the first quarter, enough to carry it through 2009, but insufficient to give the company much comfort in making plans for breaking into the U.S. market.
During the call, Chief Financial Officer Stephen Giglieri declined to provide runway guidance, which he says is likely to come in the company's second-quarter earnings report.
"The reason I am hesitant to do that [now] is we will be looking at our development plans and of course that has an impact on our cash runway," he said. "But obviously, putting $49 million into the bank is going to extend our runway fairly significantly."
NeurogesX estimates the potential for each market- the U.S. and Europe- is worth from $300 million to $500 million. Assuming the company receives a 17 percent royalty on net sales in the EU, Lazard analysts project the company could get $110 million in peak sales, including some off-label use in diabetic neuropathy.
In the U.S., Lazard forecasts Qutenza revenues of $40 million in 2010 and $88 million in 2011, with peak sales of at least $400 million in 2015. These figures are based on a treated patient population of 56,000 in HIV-distal sensory polyneuropathy, 256,000 in post-herpetic neuralgia, and 556,000 in diabetic peripheral neuropathy in 2008.
Astellas to foot bill for post-marketing trials
Qutenza, a 179 mg cutaneous patch, was approved in May by the European Medicines Agency for peripheral neuropathic pain in non-diabetic adults. This label includes two of the biggest neuropathic pain conditions- post-herpetic neuralgia, which is caused by shingles, and HIV-DSP, a common neurological problem linked with HIV infection. To get approved in Europe for diabetic neuropathy, the product's third-biggest indication, the company needs to accumulate more data.
Astellas will fund additional studies to support Qutenza marketing and promotion, and will fulfill post-marketing commitments linked to European approval, including a long-term safety study in approved indications.
In the U.S., Qutenza is pending review for post-herpetic neuralgia. Despite the user fee date in August, the company and analysts are expecting delays. After questioning the use of Endo Pharmaceuticals' local pain reliever Lidoderm in the pretreatment phase before Qutenza was applied, the agency requested a small, non-efficacy comparability study of the two treatments. NeurogesX says it has completed enrollment and treated all patients, and hopes to submit the data before the Aug. 19 PDUFA date.
"Although the PDUFA date is Aug. 16, submission of additional data late in a review cycle typically triggers a delay and we would expect this to be the case for Qutenza," wrote Lazard analyst William Tanner in a June 22 note.
During the investors' call CEO Tony DiTonno said the company is hopeful that any delay in the FDA's decision will be relatively short and expressed guarded optimism that an agency action will occur before the end of the year and a launch could happen in the first half of 2010.
Timing of European launch uncertain
Astellas declined to comment on its plans for the European launch, aside from saying that it will take place "as soon as possible." Astellas also is not disclosing its sales and marketing strategy, beyond confirming it will be targeting pain specialists who work in hospitals.
It's possible that that Astellas will launch in Europe in the first half of 2010, not necessarily at the same time of the U.S. rollout, according to NeurogesX executives.
Other compounds approved for neuropathic pain include Lidoderm(lidocaine patch 5 percent), the anti-convulsant gabapentin (Pfizer's Neurontin and generics), Pfizer's Neurontin follow-on compound, Lyrica (pregabalin) and serotonin and Lilly's norepinephrine reuptake inhibitor Cymbalta (duloxetine).
Anti-convulsants and anti-depressants act on the central nervous system and, therefore, have worrisome central side effects including sleepiness, and risk for suicidality, DiTonno pointed out. These medications also require chronic dosing to maintain therapeutic effects.
Qutenza acts locally at the site of pain and is administered for one hour by a doctor, with results lasting 12 weeks. Lidoderm also acts locally but requires much more frequent administration by the patient, raising concerns about compliance, DiTonno maintained. With that product, up to three patches are administered once a day for up to 12 hours (12 hours on and 12 hours off).
Preparing for launch in Europe and U.S.
DiTonno noted that Astellas has pain drugs in its pipeline and will build a pain-focused specialty pharma franchise starting with Qutenza.
Astellas had been working with XenoPort to develop XP13512 (gabapentin enacarbil) in diabetic peripheral neuropathy, but halted a trial in the third quarter of 2008 after an interim analysis showed the drug was unlikely to meet its primary endpoint of pain reduction (1 'The Pink Sheet' DAILY, April 27, 2009).
The company published positive preclinical results for FK1706 in diabetic neuropathy in the journal Neuropharmacology in December 2008. Researchers reported that the compound, a non-immunosuppressive immunophilin ligand, relieved painful neuropathy by changing underlying disease pathology, a different mechanism from gabapentin.
DiTonno declined to comment on which European countries would be targeted first, but he did say that, traditionally, companies focus initially on the UK and Germany because these countries offer the most flexibility on pricing.
In the U.S., NeurogesX plans to build its own sales force of 80 to 100 people to target about 12,000 specialists and will seek a partner to reach about the same number of primary care doctors who prescribe pain medications. "We have already started planning for that and will ramp that up," he said.
http://blog.taragana.com/business/2009/11/17/neurogesx-share…
NeurogesX shares rise as Food and Drug Administration approves Qutenza pain patch
By AP November 17th, 2009
NeurogesX shares rise following FDA approval
NEW YORK — Shares of biotechnology company NeurogesX Inc. jumped Tuesday after the company received Food and Drug Administration approval for its Qutenza pain patch.
The stock rose 51 cents, or 6.2 percent, to $8.75 in afternoon trading. Shares have traded between 89 cents and $9.20 over the last 52 weeks.
The FDA approved Qutenza as a treatment for postherpetic neuralgia, a type of nerve pain condition that results from shingles. The company plans to launch the drug in the U.S. in the first half of 2010. The patch is already approved in Europe for peripheral nerve pain in non-diabetic adults.
Lazard Capital Markets analyst William Tanner reaffirmed a “Buy” rating and $9 price target, saying the approval seemed likely. He forecast sales of $88 million in 2011, which will also likely be the company’s first profitable year. U.S. sales could peak over $350 million annually by 2015.
mfg ipollit
so schlecht ist die zugelassene Indikation PHN nicht, zumal HIV und PDN offlabel Umsätze einbringen können, auch wenn noch keine Zulassung dafür vorliegt.
Die aktuelle Zulassung+Offlabel soll so ca. 350+ Mio USD peak sales weltweit einbringen, was ja für ein Unternehmen mit einer MK von etwas über 100 Mio USD nicht wenig ist, auch wenn es teilweise nur Royalties usw sind.
http://www.europharmatoday.com/2009/06/neurogesx-brings-aste…
June 25, 2009
NeurogesX Brings Astellas On Board To Launch Pain Patch in Europe
A deal with Astellas to market and distribute the neuropathic pain patch Qutenza in Europe will bolster NeurogesX's cash position, giving the company some breathing room to prepare for regulatory approval and a 2010 launch in the U.S. market.
Qutenza was approved for neuropathic pain in non-diabetic patients by the European Medicines Agency in May and it is pending review at the FDA, with an Aug. 16 user fee date.
NeurogesX is set to get €30 million ($42 million) upfront for Qutenza commercialization rights from the European subsidiary of Japanese drug maker Astellas, according to a deal announced on June 22. In addition to 27 countries in the European Union, the deal covers Iceland, Switzerland, and some countries in the Middle East and Africa. The product will be launched in Europe by the first half of 2010.
Qutenza is a dermal patch packing a high concentration form of synthetic capsaicin (also called trans-capsaicin), which is the substance found in chili peppers. Capsaicin stimulates transient vanilloid 1 receptors in the skin, which in turn subdue overactive pain receptors. The effects are long-lasting, but also reversible, the company says.
As part of the agreement, Astellas also will pay €5 million upfront ($7 million) for a co-development and commercialization option on NGX-1998, a Phase I liquid formulation of the NeurogesX product.
NeurogesX now is eligible for €70 million ($97 million) in sales-based milestone payments and additional option payments for the liquid formulation. Royalty rates start in the high teens and escalate into the mid-20s, the company said during a June 22 investors' call.
Deal gives financial peace of mind
NeurogesX had about $19 million in cash and marketable securities at the close of the first quarter, enough to carry it through 2009, but insufficient to give the company much comfort in making plans for breaking into the U.S. market.
During the call, Chief Financial Officer Stephen Giglieri declined to provide runway guidance, which he says is likely to come in the company's second-quarter earnings report.
"The reason I am hesitant to do that [now] is we will be looking at our development plans and of course that has an impact on our cash runway," he said. "But obviously, putting $49 million into the bank is going to extend our runway fairly significantly."
NeurogesX estimates the potential for each market- the U.S. and Europe- is worth from $300 million to $500 million. Assuming the company receives a 17 percent royalty on net sales in the EU, Lazard analysts project the company could get $110 million in peak sales, including some off-label use in diabetic neuropathy.
In the U.S., Lazard forecasts Qutenza revenues of $40 million in 2010 and $88 million in 2011, with peak sales of at least $400 million in 2015. These figures are based on a treated patient population of 56,000 in HIV-distal sensory polyneuropathy, 256,000 in post-herpetic neuralgia, and 556,000 in diabetic peripheral neuropathy in 2008.
Astellas to foot bill for post-marketing trials
Qutenza, a 179 mg cutaneous patch, was approved in May by the European Medicines Agency for peripheral neuropathic pain in non-diabetic adults. This label includes two of the biggest neuropathic pain conditions- post-herpetic neuralgia, which is caused by shingles, and HIV-DSP, a common neurological problem linked with HIV infection. To get approved in Europe for diabetic neuropathy, the product's third-biggest indication, the company needs to accumulate more data.
Astellas will fund additional studies to support Qutenza marketing and promotion, and will fulfill post-marketing commitments linked to European approval, including a long-term safety study in approved indications.
In the U.S., Qutenza is pending review for post-herpetic neuralgia. Despite the user fee date in August, the company and analysts are expecting delays. After questioning the use of Endo Pharmaceuticals' local pain reliever Lidoderm in the pretreatment phase before Qutenza was applied, the agency requested a small, non-efficacy comparability study of the two treatments. NeurogesX says it has completed enrollment and treated all patients, and hopes to submit the data before the Aug. 19 PDUFA date.
"Although the PDUFA date is Aug. 16, submission of additional data late in a review cycle typically triggers a delay and we would expect this to be the case for Qutenza," wrote Lazard analyst William Tanner in a June 22 note.
During the investors' call CEO Tony DiTonno said the company is hopeful that any delay in the FDA's decision will be relatively short and expressed guarded optimism that an agency action will occur before the end of the year and a launch could happen in the first half of 2010.
Timing of European launch uncertain
Astellas declined to comment on its plans for the European launch, aside from saying that it will take place "as soon as possible." Astellas also is not disclosing its sales and marketing strategy, beyond confirming it will be targeting pain specialists who work in hospitals.
It's possible that that Astellas will launch in Europe in the first half of 2010, not necessarily at the same time of the U.S. rollout, according to NeurogesX executives.
Other compounds approved for neuropathic pain include Lidoderm(lidocaine patch 5 percent), the anti-convulsant gabapentin (Pfizer's Neurontin and generics), Pfizer's Neurontin follow-on compound, Lyrica (pregabalin) and serotonin and Lilly's norepinephrine reuptake inhibitor Cymbalta (duloxetine).
Anti-convulsants and anti-depressants act on the central nervous system and, therefore, have worrisome central side effects including sleepiness, and risk for suicidality, DiTonno pointed out. These medications also require chronic dosing to maintain therapeutic effects.
Qutenza acts locally at the site of pain and is administered for one hour by a doctor, with results lasting 12 weeks. Lidoderm also acts locally but requires much more frequent administration by the patient, raising concerns about compliance, DiTonno maintained. With that product, up to three patches are administered once a day for up to 12 hours (12 hours on and 12 hours off).
Preparing for launch in Europe and U.S.
DiTonno noted that Astellas has pain drugs in its pipeline and will build a pain-focused specialty pharma franchise starting with Qutenza.
Astellas had been working with XenoPort to develop XP13512 (gabapentin enacarbil) in diabetic peripheral neuropathy, but halted a trial in the third quarter of 2008 after an interim analysis showed the drug was unlikely to meet its primary endpoint of pain reduction (1 'The Pink Sheet' DAILY, April 27, 2009).
The company published positive preclinical results for FK1706 in diabetic neuropathy in the journal Neuropharmacology in December 2008. Researchers reported that the compound, a non-immunosuppressive immunophilin ligand, relieved painful neuropathy by changing underlying disease pathology, a different mechanism from gabapentin.
DiTonno declined to comment on which European countries would be targeted first, but he did say that, traditionally, companies focus initially on the UK and Germany because these countries offer the most flexibility on pricing.
In the U.S., NeurogesX plans to build its own sales force of 80 to 100 people to target about 12,000 specialists and will seek a partner to reach about the same number of primary care doctors who prescribe pain medications. "We have already started planning for that and will ramp that up," he said.
http://blog.taragana.com/business/2009/11/17/neurogesx-share…
NeurogesX shares rise as Food and Drug Administration approves Qutenza pain patch
By AP November 17th, 2009
NeurogesX shares rise following FDA approval
NEW YORK — Shares of biotechnology company NeurogesX Inc. jumped Tuesday after the company received Food and Drug Administration approval for its Qutenza pain patch.
The stock rose 51 cents, or 6.2 percent, to $8.75 in afternoon trading. Shares have traded between 89 cents and $9.20 over the last 52 weeks.
The FDA approved Qutenza as a treatment for postherpetic neuralgia, a type of nerve pain condition that results from shingles. The company plans to launch the drug in the U.S. in the first half of 2010. The patch is already approved in Europe for peripheral nerve pain in non-diabetic adults.
Lazard Capital Markets analyst William Tanner reaffirmed a “Buy” rating and $9 price target, saying the approval seemed likely. He forecast sales of $88 million in 2011, which will also likely be the company’s first profitable year. U.S. sales could peak over $350 million annually by 2015.
mfg ipollit
Antwort auf Beitrag Nr.: 38.769.508 von ipollit am 20.01.10 00:44:48danke für den artikel - hört sich in der tat interessant an diese firma.
allzulange dürfte der cash nicht reichen, da man wohl zunächst von steigendem cashburn ausgehen kann (sales force etc.). aber dann sollten es ja die ersten produktumsätze wieder richten wenn alles glatt geht.
werd mir wohl demnächst ein paar ins körbchen tun. market cap aktuell USD 125,8 mio.
allzulange dürfte der cash nicht reichen, da man wohl zunächst von steigendem cashburn ausgehen kann (sales force etc.). aber dann sollten es ja die ersten produktumsätze wieder richten wenn alles glatt geht.
werd mir wohl demnächst ein paar ins körbchen tun. market cap aktuell USD 125,8 mio.
Hallo, klasse Thread!
Mein absoluter Favorit ist Isis Pharmaceuticals. Isis sind sozusagen die Pioniere in der Antisense/RNAI-Technologie mit einer marktbeherrschenden Patentlage.
Die Pipeline ist mit aktuell 23 Medikamenten-Kandidaten mehr als vielversprechend. Die Antisense-Technologie macht es möglich die Pipeline jährlich um 3-5 Medikamente zu erweitern(allein in den letzten 6 Wochen um 3 Stück!) und das zu einem Bruchteil der Kosten traditioneller Technologien.
Pipeline
http://www.isispharm.com/Pipeline/index.htm
Isis hat allerdings den Makel mit der 1. Generation von Antisensemedikamenten kläglich gescheitert zu sein!
Mittlerweile wurde die Antisensetechnologie allerdings erheblich weiterentwickelt und befindet sich in Generationen 2 und 2.2 . Die Generation 2.5 ist für 2010 angekündigt mit der die orale Verabreichung möglich sein wird.
Was noch fehlt ist die Validierung der Technologie, d.h. es muss das erste Blockbustermedikament die Zulassung erhalten.
Mit Mipomersen hat Isis dabei ein sehr vielversprechendes Medikament. Dabei handelt es sich um ein Herz-Kreislauf-Mittel, welches helfen soll, den Cholesterinspiegel im Blut zu senken.
Mipomersen wird zurzeit in verschiedenen Patientengruppen (unterschiedliche Risikogruppen) in Phase III getestet. Die riskanteste Phase III-Studie mit 34 Patienten der höchsten Risikogruppe (allerdings kleinsten Marktpotential) wurde mit herausragenden Ergebnissen abgeschlossen. Die Ergebnisse der Phase-III mit 124 Patienten der zweithöchsten Risikogruppe sind noch in diesem (01.Quartal 2010) fällig und zwei weitere Phase-III Ergebnisse (3. und 4.höchste Risikogruppe) sind im Sommer 2010 fällig. Zulassungsanträge in USA und EU sind für Anfang 2011 geplant.
Wer sich für Isis interessiert sollte sich unbedingt folgende Slides vom R&D DAY ansehen !!!
http://phx.corporate-ir.net/External.File?item=UGFyZW50SUQ9M…
Besonders interessant ist auch das Transcript vom R&D DAY welches man allerdings erst nach Anmeldung (anonyme Einträge reichen auch)
herunterladen kann.
http://ir.isispharm.com/phoenix.zhtml?c=222170&p=IROL-Guestb…
Grüße cristrader
Mein absoluter Favorit ist Isis Pharmaceuticals. Isis sind sozusagen die Pioniere in der Antisense/RNAI-Technologie mit einer marktbeherrschenden Patentlage.
Die Pipeline ist mit aktuell 23 Medikamenten-Kandidaten mehr als vielversprechend. Die Antisense-Technologie macht es möglich die Pipeline jährlich um 3-5 Medikamente zu erweitern(allein in den letzten 6 Wochen um 3 Stück!) und das zu einem Bruchteil der Kosten traditioneller Technologien.
Pipeline
http://www.isispharm.com/Pipeline/index.htm
Isis hat allerdings den Makel mit der 1. Generation von Antisensemedikamenten kläglich gescheitert zu sein!
Mittlerweile wurde die Antisensetechnologie allerdings erheblich weiterentwickelt und befindet sich in Generationen 2 und 2.2 . Die Generation 2.5 ist für 2010 angekündigt mit der die orale Verabreichung möglich sein wird.
Was noch fehlt ist die Validierung der Technologie, d.h. es muss das erste Blockbustermedikament die Zulassung erhalten.
Mit Mipomersen hat Isis dabei ein sehr vielversprechendes Medikament. Dabei handelt es sich um ein Herz-Kreislauf-Mittel, welches helfen soll, den Cholesterinspiegel im Blut zu senken.
Mipomersen wird zurzeit in verschiedenen Patientengruppen (unterschiedliche Risikogruppen) in Phase III getestet. Die riskanteste Phase III-Studie mit 34 Patienten der höchsten Risikogruppe (allerdings kleinsten Marktpotential) wurde mit herausragenden Ergebnissen abgeschlossen. Die Ergebnisse der Phase-III mit 124 Patienten der zweithöchsten Risikogruppe sind noch in diesem (01.Quartal 2010) fällig und zwei weitere Phase-III Ergebnisse (3. und 4.höchste Risikogruppe) sind im Sommer 2010 fällig. Zulassungsanträge in USA und EU sind für Anfang 2011 geplant.
Wer sich für Isis interessiert sollte sich unbedingt folgende Slides vom R&D DAY ansehen !!!
http://phx.corporate-ir.net/External.File?item=UGFyZW50SUQ9M…
Besonders interessant ist auch das Transcript vom R&D DAY welches man allerdings erst nach Anmeldung (anonyme Einträge reichen auch)
herunterladen kann.
http://ir.isispharm.com/phoenix.zhtml?c=222170&p=IROL-Guestb…
Grüße cristrader
Antwort auf Beitrag Nr.: 38.778.590 von cristrader am 21.01.10 01:21:27Transcript R&D DAY
http://webcastingplayer.corporate-ir.net/player/SupportMater…
http://webcastingplayer.corporate-ir.net/player/SupportMater…
Antwort auf Beitrag Nr.: 38.778.590 von cristrader am 21.01.10 01:21:27wurde mit herausragenden Ergebnissen abgeschlossen.
das hat der markt aber absolut nicht so gesehen...
ISIS ist auch auf meiner watchlist; was ich vom konzept dieser satellite-companies halten soll, weiß ich nicht so recht. mag vorteile haben, bietet aber auch viel platz für schindluder mmn.
das hat der markt aber absolut nicht so gesehen...
ISIS ist auch auf meiner watchlist; was ich vom konzept dieser satellite-companies halten soll, weiß ich nicht so recht. mag vorteile haben, bietet aber auch viel platz für schindluder mmn.
Antwort auf Beitrag Nr.: 38.786.098 von PathFinder2 am 21.01.10 21:12:39
Was meinst Du mit satellite-companie?
Ich finde ISIS eigentlich auch spannend, aber bisher war ja alles noch nicht so ermutigend und ipollit hat ja auch gleich ganz am Anfang geschrieben:
"Zwar konnte in der ersten PIII Mipomersen bei Hochrisiko-Patienten das Cholesterin senken, doch gab es auch bei einem Teil der Patienten erhöhte Leberwerte, ein Anzeichen von möglicher Leberschädigung. Die Frage ist nun, in wie weit Mipomersen dafür verantwortlich ist und ob dies auch bei Patienten, die keine lebensgefährlichen Cholesterinwerte aufweisen, auftritt, denn nur damit kann Mipomersen zu einem Blockbuster werden. Daher ist der Kurs zuletzt gefallen."
Tja, ich denke, ich bleib zunächst mal an der Seitenlinie, hoffe aber, dass ISIS hier immer wieder diskutiert werden wird.
Was meinst Du mit satellite-companie?
Ich finde ISIS eigentlich auch spannend, aber bisher war ja alles noch nicht so ermutigend und ipollit hat ja auch gleich ganz am Anfang geschrieben:
"Zwar konnte in der ersten PIII Mipomersen bei Hochrisiko-Patienten das Cholesterin senken, doch gab es auch bei einem Teil der Patienten erhöhte Leberwerte, ein Anzeichen von möglicher Leberschädigung. Die Frage ist nun, in wie weit Mipomersen dafür verantwortlich ist und ob dies auch bei Patienten, die keine lebensgefährlichen Cholesterinwerte aufweisen, auftritt, denn nur damit kann Mipomersen zu einem Blockbuster werden. Daher ist der Kurs zuletzt gefallen."
Tja, ich denke, ich bleib zunächst mal an der Seitenlinie, hoffe aber, dass ISIS hier immer wieder diskutiert werden wird.
Antwort auf Beitrag Nr.: 38.778.590 von cristrader am 21.01.10 01:21:27hallo cristrader!
Danke für die Links!
ja, Isis ist recht gut und ich denke, da steckt einiges an Potential drin. Ein Problem gäbe es nur, wenn RNAi insgesamt so nicht funktionieren würde... der große Durchbruch fehlt bisher ja noch. Zudem ist der Cashburn ist relativ gering... und mit 600 Mio USD noch reichlich vorhanden.
Andererseits gibt es eben noch gewisse Unsicherheiten bezügl. Mipomersen (Entwicklungs-Verzögerungen, potentiell gravierende Nebenwirkungen), weshalb wohl schon seit einiger Zeit der Kurs unter Druck ist. In der R&D-Präsentation stellt es Isis ja auch so da, dass Mipomersen von großer Bedeutung ist... mehr als 1/3 des zukünftige Umsatzes soll dadurch generiert werden. Gibt es mit Mipomersen Probleme, dann schlägt es trotz der breiten Pipeline auf den Kurs durch.
Langfristig steht Isis aber dank des Geschäftsmodells wohl gut da.
mfg ipollit
Danke für die Links!
ja, Isis ist recht gut und ich denke, da steckt einiges an Potential drin. Ein Problem gäbe es nur, wenn RNAi insgesamt so nicht funktionieren würde... der große Durchbruch fehlt bisher ja noch. Zudem ist der Cashburn ist relativ gering... und mit 600 Mio USD noch reichlich vorhanden.
Andererseits gibt es eben noch gewisse Unsicherheiten bezügl. Mipomersen (Entwicklungs-Verzögerungen, potentiell gravierende Nebenwirkungen), weshalb wohl schon seit einiger Zeit der Kurs unter Druck ist. In der R&D-Präsentation stellt es Isis ja auch so da, dass Mipomersen von großer Bedeutung ist... mehr als 1/3 des zukünftige Umsatzes soll dadurch generiert werden. Gibt es mit Mipomersen Probleme, dann schlägt es trotz der breiten Pipeline auf den Kurs durch.
Langfristig steht Isis aber dank des Geschäftsmodells wohl gut da.
mfg ipollit
Antwort auf Beitrag Nr.: 38.786.170 von SLGramann am 21.01.10 21:21:55satellite-companies... ich würde sagen Ausgründungen oder Partnerschaften, bei denen andere die Weiterentwicklung übernehmen.
Isis hat ähnlich wie z.B. MOR das Geschäftsmodell, ihre Pipeline alleine maximal bis in PII zu bringen. Danach werden Partnerschaften oder Ausgründungen gesucht, die dann das weitere Risiko und die anfallenden Entwicklungskosten übernehmen. Isis partizipiert dann an den Royalties oder an Unternehmensanteilen... siehe z.B. die R&D-Präsentation. Dies soll den Vorteil haben, dass Isis nicht von einem Medikamentenkandidaten abhängig ist und solange es wenigstens ein paar auf den Markt schaffen, sie irgendwann relativ sicher Gewinne machen. Teilweise hat Isis jetzt schon Cashflow positive Quartale gehabt.
mfg ipollit
Isis hat ähnlich wie z.B. MOR das Geschäftsmodell, ihre Pipeline alleine maximal bis in PII zu bringen. Danach werden Partnerschaften oder Ausgründungen gesucht, die dann das weitere Risiko und die anfallenden Entwicklungskosten übernehmen. Isis partizipiert dann an den Royalties oder an Unternehmensanteilen... siehe z.B. die R&D-Präsentation. Dies soll den Vorteil haben, dass Isis nicht von einem Medikamentenkandidaten abhängig ist und solange es wenigstens ein paar auf den Markt schaffen, sie irgendwann relativ sicher Gewinne machen. Teilweise hat Isis jetzt schon Cashflow positive Quartale gehabt.
mfg ipollit
eine Sache, die ich im Blog http://www.pharmastrategyblog.com/ gefunden habe...
Antisoma hört sich langsam recht interessant an. Anscheinend ist ASA-404, das Novartis von Antisoma lizensiert hat, ein guter Ansatz... eine Mischung aus Chemo und Avastin ("a kind of hybrid cross between an anti-angiogenic such as bevacizumab (Avastin) and a taxane such as paclitaxel or docetaxel.") Bei einem Erfolg stehen Antisoma einiges an Meilensteinzahlungen und Royalties zu. Zu der Höhe der Royalties habe ich nichts definitives gefunden... an einer Stelle wird es allerdings auf 25% geschätzt, was sehr viel wäre, zumal, wenn ASA-404 ein potentieller Blockbuster bei einem BigPharma wäre.
das ganze für ca. 350 Mio USD MK bietet reichlich Kurspotential, oder?

aus http://www.pharmastrategyblog.com/2010/01/aacr-personalized-…
Today's topic is about a new generation flavanoid, ASA-404, from Novartis. Of course, under FTC guidelines, I should disclose that as many of you know, I'm a former employee and have also done consulting work for the company, but not on this particular product.
ASA-404 is an interesting agent in phase III development. It's a vascular disrupting agent (VDA) with a tubulin dependent mechanism of agent. Unlike taxanes, which target the microtubulin in DNA, this agent is also part anti-angiogenic agent because it selectively disrupts the tumour vasculature with both direct and indirect effects. In simple terms, I think of it as a kind of hybrid cross between an anti-angiogenic such as bevacizumab (Avastin) and a taxane such as paclitaxel or docetaxel.
Dr Mark McKeage presentation at the AACR lung cancer meeting was focused on the phase II that has been published to date, including a publication in the British Journal of Cancer (see bottom of article).
The drug was well tolerated in phase I trials, achieving an MTD of 3700 mg/m2.
In a phase II trial, paclitaxel and carboplatin (pc) therapy was used as the reference arm, and ASA-404 added to the standard chemotherapy in the other arm to determine if the agent added any extra benefit for newly diagnosed patients with non-small cell lung cancer (NSCLC) with either a squamous or non-squamous histology. They looked at 2 different doses of ASA-404 - 1200mg/m2 was used in the randomised comparison with carbo-tax and 1800mg/m2 with out the comparator arm but in combination as before. 36 patients were accrued into the reference pc arm, 37 in the lower dose ASA-404 arm and 31 in the higher dose ASA-404 arm. This gave a total of 104 patients in the pooled phase II study.
Overall, similar rates of adverse events (AE's) were seen in the squamous and non-squamous patients, except for a slight increase in AE's related to blood/lymphatic effects. No serious AE's associated with vascular effects of bleeding, hemorrhage or hemoptysis were seen. There was an increase in hematologic toxicities (anemia, neutropenia and thrombocytopenia) but these were not significant and none rose above 20%. There was no apparent increase in neutropenia related sepsis.
In terms of efficacy and median overall survival, McKey reported:
PCA vs. PC
14.0 vs. 8.8 mos for 1200mg/m2
14.9 for 1800mg/m2
Thus we can clearly see that adding ASA-404 to standard carboplatin-paclitaxel therapy improved median overall in frontline NSCLC by 5 months.
Recently, the FDA has approved several therapies in NSCLC in different histologies, due to either issues with side effects (bevacizumab and bleeding issues) or lack of efficacy (pemetrexed), while others such as erlotinib have been shown to be particularly effective in non-smoking female asians with an adenocarcinoma histology, although the approval is not based on histology.
Let's take a look at the ASA-404 histology data:
Squamous: Non-Squamous: Overall:
PCA PC PCA PC PCA PC
ORR (%) 40.0 14.3 31.7 25.0 34.4 22.2
One third of the patients had a squamous histology, but experienced no bleeding issues seen with VEGF agents.
While the results are higher for those with a squamous histology, there was also a trend towards higher efficacy in the non-squamous patients. The positive results provided support and evidence for the drug's effectiveness and rationale for beginning the phase III triala, ATTRACT-1 in first line and ATTRACT-2 in second line NSCLC without restriction on histology.
To put the 5 months improvement in median OS with ASA-404 in context, I checked out the PI for other therapies. Bevacizumab was approved on the basis of 12.3 months vs. 10.3 months, ie a 2 month improvement, while pemetrexed was approved on the basis of non-inferiority when comparing pemetrexed plus cisplatin to gemcitabine plus cisplatin (ie 10.3 months of overall survival in each arm).
The front-line trial has completed accrual and is ongoing, but the ATTRACT-2 trial for patients who have had prior chemotherapy is still enrolling. You can find it here: ATTRACT-2.
Of course, these are early days yet and positive phase II data does not always translate to phase III success, but they are a hopeful sign of good things to come. If the data are repeated, it will be good news for patients with NSCLC.
mfg ipollit
Antisoma hört sich langsam recht interessant an. Anscheinend ist ASA-404, das Novartis von Antisoma lizensiert hat, ein guter Ansatz... eine Mischung aus Chemo und Avastin ("a kind of hybrid cross between an anti-angiogenic such as bevacizumab (Avastin) and a taxane such as paclitaxel or docetaxel.") Bei einem Erfolg stehen Antisoma einiges an Meilensteinzahlungen und Royalties zu. Zu der Höhe der Royalties habe ich nichts definitives gefunden... an einer Stelle wird es allerdings auf 25% geschätzt, was sehr viel wäre, zumal, wenn ASA-404 ein potentieller Blockbuster bei einem BigPharma wäre.
das ganze für ca. 350 Mio USD MK bietet reichlich Kurspotential, oder?
aus http://www.pharmastrategyblog.com/2010/01/aacr-personalized-…
Today's topic is about a new generation flavanoid, ASA-404, from Novartis. Of course, under FTC guidelines, I should disclose that as many of you know, I'm a former employee and have also done consulting work for the company, but not on this particular product.
ASA-404 is an interesting agent in phase III development. It's a vascular disrupting agent (VDA) with a tubulin dependent mechanism of agent. Unlike taxanes, which target the microtubulin in DNA, this agent is also part anti-angiogenic agent because it selectively disrupts the tumour vasculature with both direct and indirect effects. In simple terms, I think of it as a kind of hybrid cross between an anti-angiogenic such as bevacizumab (Avastin) and a taxane such as paclitaxel or docetaxel.
Dr Mark McKeage presentation at the AACR lung cancer meeting was focused on the phase II that has been published to date, including a publication in the British Journal of Cancer (see bottom of article).
The drug was well tolerated in phase I trials, achieving an MTD of 3700 mg/m2.
In a phase II trial, paclitaxel and carboplatin (pc) therapy was used as the reference arm, and ASA-404 added to the standard chemotherapy in the other arm to determine if the agent added any extra benefit for newly diagnosed patients with non-small cell lung cancer (NSCLC) with either a squamous or non-squamous histology. They looked at 2 different doses of ASA-404 - 1200mg/m2 was used in the randomised comparison with carbo-tax and 1800mg/m2 with out the comparator arm but in combination as before. 36 patients were accrued into the reference pc arm, 37 in the lower dose ASA-404 arm and 31 in the higher dose ASA-404 arm. This gave a total of 104 patients in the pooled phase II study.
Overall, similar rates of adverse events (AE's) were seen in the squamous and non-squamous patients, except for a slight increase in AE's related to blood/lymphatic effects. No serious AE's associated with vascular effects of bleeding, hemorrhage or hemoptysis were seen. There was an increase in hematologic toxicities (anemia, neutropenia and thrombocytopenia) but these were not significant and none rose above 20%. There was no apparent increase in neutropenia related sepsis.
In terms of efficacy and median overall survival, McKey reported:
PCA vs. PC
14.0 vs. 8.8 mos for 1200mg/m2
14.9 for 1800mg/m2
Thus we can clearly see that adding ASA-404 to standard carboplatin-paclitaxel therapy improved median overall in frontline NSCLC by 5 months.
Recently, the FDA has approved several therapies in NSCLC in different histologies, due to either issues with side effects (bevacizumab and bleeding issues) or lack of efficacy (pemetrexed), while others such as erlotinib have been shown to be particularly effective in non-smoking female asians with an adenocarcinoma histology, although the approval is not based on histology.
Let's take a look at the ASA-404 histology data:
Squamous: Non-Squamous: Overall:
PCA PC PCA PC PCA PC
ORR (%) 40.0 14.3 31.7 25.0 34.4 22.2
One third of the patients had a squamous histology, but experienced no bleeding issues seen with VEGF agents.
While the results are higher for those with a squamous histology, there was also a trend towards higher efficacy in the non-squamous patients. The positive results provided support and evidence for the drug's effectiveness and rationale for beginning the phase III triala, ATTRACT-1 in first line and ATTRACT-2 in second line NSCLC without restriction on histology.
To put the 5 months improvement in median OS with ASA-404 in context, I checked out the PI for other therapies. Bevacizumab was approved on the basis of 12.3 months vs. 10.3 months, ie a 2 month improvement, while pemetrexed was approved on the basis of non-inferiority when comparing pemetrexed plus cisplatin to gemcitabine plus cisplatin (ie 10.3 months of overall survival in each arm).
The front-line trial has completed accrual and is ongoing, but the ATTRACT-2 trial for patients who have had prior chemotherapy is still enrolling. You can find it here: ATTRACT-2.
Of course, these are early days yet and positive phase II data does not always translate to phase III success, but they are a hopeful sign of good things to come. If the data are repeated, it will be good news for patients with NSCLC.
mfg ipollit
eine zweite Sache ebenfalls aus diesem Blog gezieht sich auf ARQL...
hier stehen genau die im folgenden beschriebenen c-Met + EGFR Daten in Form der PII von ARQ-197 in Kombination mit Tarceva bei NSCLC in diesem Halbjahr an. Theoretisch scheint diese Kombination anscheinend ja sehr sinnvoll und vielversprechend zu sein.
http://www.pharmastrategyblog.com/2010/01/aacr-personalized-…
January 20, 2010
#AACR Personalized therapy in lung cancer - part 4
It's been a bit of a long week on lung cancer articles and while I was planning on talking about something else today, this new article in my database caught my eye:
.
.
.
Part of the reason is nostalgia - it's 20 years ago since I finished my doctorate on early detection of preclinical lung disease and while I was interested in the methods of detecting changes in breathing patterns associated with smoking, part of me wished I'd done research on molecular biology at the time rather than applied physiology.
The reason is that I realised while doing the literature search is that biochemical and physiological changes in the airways would ultimately tell us more about early detection.
In the article above, the researchers suggest a potential mechanism by which the tobacco-specific carcinogen NNK promotes lung tumour formation and development. Now, bearing in mind that most solid tumours take years to develop from hyperplasia to full aggressive carcinoma, finding how it actually happens and why is still an inexact science, as are methods for early detection given not all smokers get lung cancer and non-smokers are not immune from the disease.
Lin et al., suggest that NNK induces the accumulation of a protein known as DNMT1 in the nucleus and that this protein silences genes that suppress tumour formation. They offered evidence to support their hypothesis, including the observation that DNMT1 accumulates in both lung adenomas from NNK-treated mice and tumours from lung cancer patients that were smokers. DNMT1 overexpression in lung cancer patients who smoked continuously correlated with poor prognosis.
However, the interesting part of their abstract to me was:
"We determined that in a human lung cell line, glycogen synthase kinase 3β (GSK3β) phosphorylated DNMT1 to recruit β-transducin repeat–containing protein (βTrCP), resulting in DNMT1 degradation, and that NNK activated AKT, inhibiting GSK3β function and thereby attenuating DNMT1 degradation."
Ah, our friend AKT.
The potential role of AKT in lung cancer came up repeatedly at last week's AACR lung cancer meeting. The researchers there had begun to realise that blocking EGFR or IGF-1R and c-MET or AKT (either directly or indirectly via PI3K inhibition) might cut off an escape route for the cancer cell and reduce drug resistance:

Drs Jeffrey Engelman (MGH) and David Carbone (Vanderbilt) covered excellent quick reviews at AACR on the latest findings related to EGFR inhibition relating to c-MET and proteomics respectively. As our knowledge of various mutations and biologic pathways improves, so does our understanding of how we can better target aberrations and treat patients with NSCLC more effectively.
Engelman's group has just published a paper on c-MET and EGFR inhibition (see references). They noticed that rare MET-amplified cells exist in some EGFR-mutant lung cancers before treatment. What makes the research relevant to this overview is that MET amplification activates ERBB3/PI3K/AKT signaling in EGFR mutant lung cancers and causes resistance to EGFR kinase inhibitors. They demonstrated that MET activation by its ligand, HGF, induces drug resistance through GAB1 signaling. Using high-throughput FISH analyses in both cell lines and in patients with lung cancer, they identified subpopulations of cells with MET amplification prior to drug exposure.
The concept that HGF induces resistance to tyrosine kinase inhibitors in EGFR-addicted cancers is a novel one. They saw that HGF accelerates MET amplification by expanding preexisting MET-amplified cells. What was particularly relevant though was that analysis of pretreatment cancers identified those poised to become MET amplified, thereby offering a way to segment NSCLC patients for more personalised treatment, increasing the chances of better response rates, longer overall survival and improved patient outcomes.
Then came the killer statement:
"EGFR kinase inhibitor resistance, due to either MET amplification or autocrine HGF production, was cured in vivo by combined EGFR and MET inhibition."
Oh my. This leaves us seriously wondering what will happen in practice by combining erlotinib (Tarceva) or gefitinib (Iressa) with a MET inhibitor in patients with NSCLC?
I can't wait for ASCO this year to find out!
mfg ipollit
hier stehen genau die im folgenden beschriebenen c-Met + EGFR Daten in Form der PII von ARQ-197 in Kombination mit Tarceva bei NSCLC in diesem Halbjahr an. Theoretisch scheint diese Kombination anscheinend ja sehr sinnvoll und vielversprechend zu sein.
http://www.pharmastrategyblog.com/2010/01/aacr-personalized-…
January 20, 2010
#AACR Personalized therapy in lung cancer - part 4
It's been a bit of a long week on lung cancer articles and while I was planning on talking about something else today, this new article in my database caught my eye:
.
.
.
Part of the reason is nostalgia - it's 20 years ago since I finished my doctorate on early detection of preclinical lung disease and while I was interested in the methods of detecting changes in breathing patterns associated with smoking, part of me wished I'd done research on molecular biology at the time rather than applied physiology.
The reason is that I realised while doing the literature search is that biochemical and physiological changes in the airways would ultimately tell us more about early detection.
In the article above, the researchers suggest a potential mechanism by which the tobacco-specific carcinogen NNK promotes lung tumour formation and development. Now, bearing in mind that most solid tumours take years to develop from hyperplasia to full aggressive carcinoma, finding how it actually happens and why is still an inexact science, as are methods for early detection given not all smokers get lung cancer and non-smokers are not immune from the disease.
Lin et al., suggest that NNK induces the accumulation of a protein known as DNMT1 in the nucleus and that this protein silences genes that suppress tumour formation. They offered evidence to support their hypothesis, including the observation that DNMT1 accumulates in both lung adenomas from NNK-treated mice and tumours from lung cancer patients that were smokers. DNMT1 overexpression in lung cancer patients who smoked continuously correlated with poor prognosis.
However, the interesting part of their abstract to me was:
"We determined that in a human lung cell line, glycogen synthase kinase 3β (GSK3β) phosphorylated DNMT1 to recruit β-transducin repeat–containing protein (βTrCP), resulting in DNMT1 degradation, and that NNK activated AKT, inhibiting GSK3β function and thereby attenuating DNMT1 degradation."
Ah, our friend AKT.
The potential role of AKT in lung cancer came up repeatedly at last week's AACR lung cancer meeting. The researchers there had begun to realise that blocking EGFR or IGF-1R and c-MET or AKT (either directly or indirectly via PI3K inhibition) might cut off an escape route for the cancer cell and reduce drug resistance:
Drs Jeffrey Engelman (MGH) and David Carbone (Vanderbilt) covered excellent quick reviews at AACR on the latest findings related to EGFR inhibition relating to c-MET and proteomics respectively. As our knowledge of various mutations and biologic pathways improves, so does our understanding of how we can better target aberrations and treat patients with NSCLC more effectively.
Engelman's group has just published a paper on c-MET and EGFR inhibition (see references). They noticed that rare MET-amplified cells exist in some EGFR-mutant lung cancers before treatment. What makes the research relevant to this overview is that MET amplification activates ERBB3/PI3K/AKT signaling in EGFR mutant lung cancers and causes resistance to EGFR kinase inhibitors. They demonstrated that MET activation by its ligand, HGF, induces drug resistance through GAB1 signaling. Using high-throughput FISH analyses in both cell lines and in patients with lung cancer, they identified subpopulations of cells with MET amplification prior to drug exposure.
The concept that HGF induces resistance to tyrosine kinase inhibitors in EGFR-addicted cancers is a novel one. They saw that HGF accelerates MET amplification by expanding preexisting MET-amplified cells. What was particularly relevant though was that analysis of pretreatment cancers identified those poised to become MET amplified, thereby offering a way to segment NSCLC patients for more personalised treatment, increasing the chances of better response rates, longer overall survival and improved patient outcomes.
Then came the killer statement:
"EGFR kinase inhibitor resistance, due to either MET amplification or autocrine HGF production, was cured in vivo by combined EGFR and MET inhibition."
Oh my. This leaves us seriously wondering what will happen in practice by combining erlotinib (Tarceva) or gefitinib (Iressa) with a MET inhibitor in patients with NSCLC?
I can't wait for ASCO this year to find out!
mfg ipollit
CBST... zwar schon etwas her, aber noch zur Calixa-Übernahme
Das dabei erworbene CXA-201 könnte recht aussichtsreich sein... zumindenst waren die Gründer von Calixa zuvor mit Antibiotika bereits sehr erfolgreich.
http://www.pharmacychoice.com/news/article.cfm?Article_ID=36…
3/30/09 - Calixa Pushes Anti-Pseudomonal Antibiotic Toward Phase II for UTI
With $30 million of Series A financing in its coffers, Calixa Therapeutics Inc. is gearing up for a Phase II trial of CXA-101, a broad-spectrum cephalosporin antibiotic that has shown potent anti-pseudomonal activity.
San Diego-based Calixa was founded in late 2007 by antibiotic expert James Ge and serial entrepreneur Eckard Weber. The two have a long history of working together at antibiotic firms funded by Domain Associates, where Weber serves as a partner.
In the early part of the decade, Ge and Weber teamed up at Peninsula Pharmaceuticals Inc., where Ge headed drug development and Weber served as chairman. Peninsula was acquired by Johnson & Johnson in 2005 for $245 million, giving the big pharma control of Peninsula's broad-spectrum carbapenem antibiotic Doribax (doripenem). (See BioWorld Today, April 20, 2005.)
Doribax gained approval for complicated intra-abdominal and urinary tract infections although, like many antibiotics, it has hit regulatory hurdles in hospital-acquired pneumonia. (See BioWorld Today, July 18, 2008.)
While J&J focused on Doribax, the Peninsula team spun out Cerexa Inc. to develop the Phase I broad-spectrum cephalosporin antibiotic ceftaroline, which was licensed from Takeda Chemical Industries Ltd. Again, Ge headed drug development and Weber served as chairman until Cerexa was acquired by Forest Laboratories Inc. in late 2006 for $480 million. (See BioWorld Today, Dec. 15, 2006.)
While Forest moved ceftaroline through Phase III, Ge and Weber had their sights on FR264205, a preclinical broad-spectrum cephalosporin antibiotic in development at Tokyo-based Astellas Pharma Inc. Cerexa had done "extensive due diligence" on the compound, but it hadn't been licensed prior to the Forest acquisition, and Forest wasn't interested in pursuing such an early-stage program, Ge said.
So Ge and Weber founded Calixa, raised $30 million and licensed FR264205 - now known as CXA-101.
The Series A round came from Domain as well as Canaan Partners and Frazier Healthcare Ventures. The money should carry Calixa "into 2010," Ge told BioWorld Today.
What differentiates CXA-101 from other antibiotics on the market or in development is its potent activity against Pseudomonas aeruginosa, Ge explained.
The bacteria ranks alongside Escherichia coli and Streptococcus pneumoniae as one of the most common causes of hospital-acquired infections and can lead to hospital-acquired pneumonia, urinary tract infections, wound infections and sepsis.
P. aeruginosa infections commonly are treated with broad-spectrum antibiotics such as the cephalosporin antibiotics ceftazidime or cefepime, or the carbapenem antibiotic imipenem.
In a paper published in the March 2007 issue of Antimicrobial Agents and Chemotherapy, Astellas' researchers demonstrated that the concentration of CXA-101 needed to inhibit 90 percent of 193 P. aeruginosa isolates was eight times to 16 times lower than that needed for ceftazidime, cefepime or imipenem. CXA-101 also demonstrated activity against strains resistant to the existing drugs and showed evidence that it may be less prone to resistance.
In murine models, CXA-101 showed superior or comparable efficacy to ceftazidime and imipenem.
Calixa filed an investigational new drug application for CXA-101 in mid-2008 and completed two Phase I studies.
CXA-101 was well tolerated with no dose-limiting toxicities and a safety profile similar to that of marketed cephalosporin antibiotics.
With Phase I data in hand, Calixa is now gearing up for a Phase II trial, slated to start in the second quarter. Ge said the double-blind study will compare intravenous CXA-101 to an existing antibiotic for complicated urinary tract infections. Data are expected within a year.
Behind CXA-101, Calixa is developing CXA-201, which combines CXA-101 with a beta-lactamase inhibitor to provide even broader activity. If the data with CXA-201 pan out, Ge said Calixa will focus all of its resources on that program rather than CXA-101.
A similar approach is being pursued by Paris-based Novexel SA, which is conducting a Phase II trial combining its broad-spectrum beta lactamase inhibitor, NXL 104, with ceftazidime for complicated intra-abdominal infections. But Ge noted that CXA-101 has shown better anti-pseudomonal activity than ceftazidime.
He added that CXA-101 has the potential to be "best-in-class" as far as its anti-pseudomonal activity, based on in vitro and in vivo data.
Calixa also is developing an inhaled form of CXA-101 for P. aeruginosa infections in cystic fibrosis patients. The program, dubbed CXA-301, is in preclinical, with formulation work under way.
As of now, the only inhaled antibiotic for CF infections is TOBI (tobramycin solution for inhalation, Novartis AG), but Gilead Sciences Inc. is in regulatory discussions with the FDA regarding its inhaled aztreonam, and Phase II programs include Bayer AG's ciprofloxacin inhaled powder, Aradigm Corp.'s inhaled liposomal ciprofloxacin, Transave Inc.'s Arikace (liposomal amikacin for inhalation) and Mpex Pharmaceuticals Inc.'s MP-376, an aerosol formulation of levofloxacin.
Preclinical data have shown CXA-101 to be more potent than tobramycin and aztreonam, Ge noted
mfg ipollit
Das dabei erworbene CXA-201 könnte recht aussichtsreich sein... zumindenst waren die Gründer von Calixa zuvor mit Antibiotika bereits sehr erfolgreich.
http://www.pharmacychoice.com/news/article.cfm?Article_ID=36…
3/30/09 - Calixa Pushes Anti-Pseudomonal Antibiotic Toward Phase II for UTI
With $30 million of Series A financing in its coffers, Calixa Therapeutics Inc. is gearing up for a Phase II trial of CXA-101, a broad-spectrum cephalosporin antibiotic that has shown potent anti-pseudomonal activity.
San Diego-based Calixa was founded in late 2007 by antibiotic expert James Ge and serial entrepreneur Eckard Weber. The two have a long history of working together at antibiotic firms funded by Domain Associates, where Weber serves as a partner.
In the early part of the decade, Ge and Weber teamed up at Peninsula Pharmaceuticals Inc., where Ge headed drug development and Weber served as chairman. Peninsula was acquired by Johnson & Johnson in 2005 for $245 million, giving the big pharma control of Peninsula's broad-spectrum carbapenem antibiotic Doribax (doripenem). (See BioWorld Today, April 20, 2005.)
Doribax gained approval for complicated intra-abdominal and urinary tract infections although, like many antibiotics, it has hit regulatory hurdles in hospital-acquired pneumonia. (See BioWorld Today, July 18, 2008.)
While J&J focused on Doribax, the Peninsula team spun out Cerexa Inc. to develop the Phase I broad-spectrum cephalosporin antibiotic ceftaroline, which was licensed from Takeda Chemical Industries Ltd. Again, Ge headed drug development and Weber served as chairman until Cerexa was acquired by Forest Laboratories Inc. in late 2006 for $480 million. (See BioWorld Today, Dec. 15, 2006.)
While Forest moved ceftaroline through Phase III, Ge and Weber had their sights on FR264205, a preclinical broad-spectrum cephalosporin antibiotic in development at Tokyo-based Astellas Pharma Inc. Cerexa had done "extensive due diligence" on the compound, but it hadn't been licensed prior to the Forest acquisition, and Forest wasn't interested in pursuing such an early-stage program, Ge said.
So Ge and Weber founded Calixa, raised $30 million and licensed FR264205 - now known as CXA-101.
The Series A round came from Domain as well as Canaan Partners and Frazier Healthcare Ventures. The money should carry Calixa "into 2010," Ge told BioWorld Today.
What differentiates CXA-101 from other antibiotics on the market or in development is its potent activity against Pseudomonas aeruginosa, Ge explained.
The bacteria ranks alongside Escherichia coli and Streptococcus pneumoniae as one of the most common causes of hospital-acquired infections and can lead to hospital-acquired pneumonia, urinary tract infections, wound infections and sepsis.
P. aeruginosa infections commonly are treated with broad-spectrum antibiotics such as the cephalosporin antibiotics ceftazidime or cefepime, or the carbapenem antibiotic imipenem.
In a paper published in the March 2007 issue of Antimicrobial Agents and Chemotherapy, Astellas' researchers demonstrated that the concentration of CXA-101 needed to inhibit 90 percent of 193 P. aeruginosa isolates was eight times to 16 times lower than that needed for ceftazidime, cefepime or imipenem. CXA-101 also demonstrated activity against strains resistant to the existing drugs and showed evidence that it may be less prone to resistance.
In murine models, CXA-101 showed superior or comparable efficacy to ceftazidime and imipenem.
Calixa filed an investigational new drug application for CXA-101 in mid-2008 and completed two Phase I studies.
CXA-101 was well tolerated with no dose-limiting toxicities and a safety profile similar to that of marketed cephalosporin antibiotics.
With Phase I data in hand, Calixa is now gearing up for a Phase II trial, slated to start in the second quarter. Ge said the double-blind study will compare intravenous CXA-101 to an existing antibiotic for complicated urinary tract infections. Data are expected within a year.
Behind CXA-101, Calixa is developing CXA-201, which combines CXA-101 with a beta-lactamase inhibitor to provide even broader activity. If the data with CXA-201 pan out, Ge said Calixa will focus all of its resources on that program rather than CXA-101.
A similar approach is being pursued by Paris-based Novexel SA, which is conducting a Phase II trial combining its broad-spectrum beta lactamase inhibitor, NXL 104, with ceftazidime for complicated intra-abdominal infections. But Ge noted that CXA-101 has shown better anti-pseudomonal activity than ceftazidime.
He added that CXA-101 has the potential to be "best-in-class" as far as its anti-pseudomonal activity, based on in vitro and in vivo data.
Calixa also is developing an inhaled form of CXA-101 for P. aeruginosa infections in cystic fibrosis patients. The program, dubbed CXA-301, is in preclinical, with formulation work under way.
As of now, the only inhaled antibiotic for CF infections is TOBI (tobramycin solution for inhalation, Novartis AG), but Gilead Sciences Inc. is in regulatory discussions with the FDA regarding its inhaled aztreonam, and Phase II programs include Bayer AG's ciprofloxacin inhaled powder, Aradigm Corp.'s inhaled liposomal ciprofloxacin, Transave Inc.'s Arikace (liposomal amikacin for inhalation) and Mpex Pharmaceuticals Inc.'s MP-376, an aerosol formulation of levofloxacin.
Preclinical data have shown CXA-101 to be more potent than tobramycin and aztreonam, Ge noted
mfg ipollit
Antwort auf Beitrag Nr.: 38.786.872 von ipollit am 21.01.10 22:52:08Antisoma...
ein umfangreicherer Analysten-Report:
http://www.oxbio.com/pressreleases/030609_Antisoma.pdf
mfg ipollit
ein umfangreicherer Analysten-Report:
http://www.oxbio.com/pressreleases/030609_Antisoma.pdf
mfg ipollit
Antwort auf Beitrag Nr.: 38.786.637 von ipollit am 21.01.10 22:20:52@SL
siehe hier zu satellite companies:
http://www.isispharm.com/Strategic-Alliances/Satellite-Compa…
also ich find solche konstrukte (von unternehmen A gehören ISIS ein paar prozent, von B ein paar, dort ein Joint Venture mit so und so) u.a. aufgrund transparenzgründen nicht so wahnsinnig toll.
nicht falsch verstehen: ich bin absolut für strategische partnerships. gutes beispiel EXEL: ordentliche partnerdeals, EXEL mit kernfunktion research & frühe entwicklung, big pharma (allem voran BMS) mit kernfunktion späte entwicklung und vermarktung. spielen kann man das in mehreren varianten von voller auslizenzierung in früher phase bis hin zu co-development.
der sinn, warum ein biotech von etlichen anderen biotechs equity-stakes haben sollte, erschließt sich mir nicht ganz, bzw. kann auch risiken bergen. risikostreuung kann man mmn besser mit obigen deals umsetzen (sofern man partner findet...)
siehe hier zu satellite companies:
http://www.isispharm.com/Strategic-Alliances/Satellite-Compa…
also ich find solche konstrukte (von unternehmen A gehören ISIS ein paar prozent, von B ein paar, dort ein Joint Venture mit so und so) u.a. aufgrund transparenzgründen nicht so wahnsinnig toll.
nicht falsch verstehen: ich bin absolut für strategische partnerships. gutes beispiel EXEL: ordentliche partnerdeals, EXEL mit kernfunktion research & frühe entwicklung, big pharma (allem voran BMS) mit kernfunktion späte entwicklung und vermarktung. spielen kann man das in mehreren varianten von voller auslizenzierung in früher phase bis hin zu co-development.
der sinn, warum ein biotech von etlichen anderen biotechs equity-stakes haben sollte, erschließt sich mir nicht ganz, bzw. kann auch risiken bergen. risikostreuung kann man mmn besser mit obigen deals umsetzen (sofern man partner findet...)
Antwort auf Beitrag Nr.: 38.769.508 von ipollit am 20.01.10 00:44:48zu NGSX...
ebenfalls ein umfangreicherer Analysten-Report
(Link zum pdf in google anklicken, sonst klappt der Zugriff nicht)
http://www.google.de/search?hl=de&rlz=1R2GGLL_deDE334&q=zer_…
mfg ipollit
ebenfalls ein umfangreicherer Analysten-Report
(Link zum pdf in google anklicken, sonst klappt der Zugriff nicht)
http://www.google.de/search?hl=de&rlz=1R2GGLL_deDE334&q=zer_…
mfg ipollit
Antwort auf Beitrag Nr.: 38.786.637 von ipollit am 21.01.10 22:20:52Hallo!
Die Mipomersenergebnisse werden sicherlich kurzfristig gesehen entscheidend für den Kursverlauf sein. Bezüglich der Mipomersen- Phase III-Studien bin allerdings mehr als zuversichtlich. An mangelnder Wirksamkeit wird Mipomersen den bisherigen Studien zufolge sicherlich nicht scheitern das Nebenwirkungsprofil wird entscheidend sein. Erhöhte Leberwerte gehören insbesondere bei Beginn der Medikamentenverabreichung auch bei allen anderen Standardtherapien zu den häufigen Nebenwirkungen. Die Nebenwirkungen von Mipomersen bewegen sich bisher im vergleichbaren Rahmen, nur das halt ein völlig neuer Therapieansatz bzw. eine neue Medikamententechnologie angewendet wird, wo es bisher einfach keine langfristigen Erfahrungswerte gibt.
S. Crooke
"Of course, we had three patients in this study that had elevated liver transaminases. We reported this at the AHA. Importantly,
what we've seen as we've developed this drug, and just to let you know, as it was presented by [Professor Rawls], these biochemical
changes in liver enzymes occur with all lipid lowering therapies. What we're seeing is, so far in our safety database, is no liver
toxicity. And that's important, no Hy's Law.We've dosed through a number of -- many of these and they -- and in all cases in this study the transaminases returned to entry
criteria by the end of the planned clinical observation. So, of course additional analysis is being conducted in this, but we're
very comfortable with the safety of this drug and the profile that we're seeing."
Nachfolgend noch ein Auszug eines Research-Berichtes von "Zacks"
Carlsbad, California-based Isis Pharmaceuticals, Inc. is a drug discovery and development company that
focuses on the development of products using ribonucleic acid (RNA)-based technologies, such as
antisense. Antisense technology is a direct route from genomics to drugs. Antisense drugs are the first
class of drugs targeted to control expression of genes through interactions with RNA. Beyond antisense,
Isis scientists have created another technology that exploits their knowledge of RNA. The company has
about 1,600 issued patents worldwide and controls one of the largest antisense and RNA patent estates
in the pharmaceutical industry.
The company discovers new drugs and out-licenses them to partners for license fees, milestone
payments, and royalties. Isis has partnership agreements with leading pharmaceuticals companies like
Bristol-Myers Squibb, Genzyme, Eli Lilly, and Johnson & Johnson among others. The company also has
a collaboration agreement with biotech company, Alnylam Pharmaceuticals. Both companies have joined
hands to form Regulus Therapeutics, Inc., a company that is focused on microRNA (mRNA) therapeutics.
Isis focuses its research and development efforts primarily in cardiovascular, metabolic and
neurodegenerative diseases and cancer while its partners are involved in the development of antisense
drugs in these and other areas, including inflammatory disease.
In 2008, Isis recognized $107 million in total revenues from licensing its intellectual property, satellite
companies, and from forming collaborations with larger partners in developing antisense drug
candidates. Earlier this year, Isis sold its Ibis Biosciences subsidiary to Abbott Molecular, Inc. for $215
million and an earn out on the sales of assay kits and services. As a result, Ibis financial results are
considered discontinuing operations.
Antisense Technology Represents Enormous Potential: We are big fans of antisense technology and
believe that the number of potential therapeutic applications is enormous. Antisense drugs may have
significant potential to treat a number of diseases where small molecules and biologic compounds
have failed. Although still early stage, antisense technology with mechanisms such as small
interfering RNA (siRNA), RNA interference (RNAi), alternate splicing, and microRNA (mRNA) have
the potential to change how we treat disease in the next decade. We see antisense technology
today where biologics were ten years ago. The promise of biologic drugs is finally delivering with
blockbuster compounds such as Rituxan, Herceptin, Avastin, Aranesp, Synagis, and Humira. We
think in ten years the same will be said for antisense.Antisense drugs have several specific advantages over traditional protein or small-molecule derived
therapeutics. Firstly, antisense drugs are highly specific and bind to only the complimentary mRNA
strain that codes for the specific protein being targeted. This has the potential to greatly reduce
unwanted side-effects that occur through secondary mechanisms of action. Secondly, since all
proteins are coded by mRNA, antisense drugs have the potential to work on a broad number of
diseases including cardiovascular, metabolic, inflammatory, ocular, viral, neurodegenerative
disease, and cancer. Finally, because all antisense drugs start with the basic hybridization of mRNA
targets, development timelines are shortened and response is often predictable in the early-stage of
development. Scientists need only choose a specific protein to target and then create a small
antisense molecule to bind to the transcribed mRNA. This creates efficiency in drug discovery and
time to market.
Isis is pioneering the effort into antisense research. The company has already commercialized the
first antisense drug in 1998 with Vitravene and has several proprietary compounds in clinical
development as well as partnered programs with large pharmaceutical companies such as Bristol-
Myers, Eli Lilly, Merck, J&J, Genzyme and Glaxo. We think that by 2012 two to three additional
antisense drugs developed by Isis could be on the market.
Mipomersen - The Blockbuster in Phase III: Isis Pharmaceuticals lead pipeline candidate,
mipomersen (formerly ISIS-301012), is a second generation compound currently in a multitude of
phase III trials for familial hypercholesterolemia (FH). Mipomersen is an antisense drug that targets
apolipoprotein B (apoB) -100, which is critical to the synthesis and transport of low density
lipoprotein (LDL) or bad cholesterol. By potentially knocking out the apoB-100 protein via antisense
technology, Isis scientists believe they can effectively lower the cholesterol of patients and reduce
the risk of serious cardiovascular disease. Given the novel mechanism and narrow initial market
focus of homozygous (Ho) FH, the U.S. Food and Drug Administration (FDA) granted mipomersen
orphan drug status in June 2006.
Meanwhile, highly encouraging phase II data helped Isis strike one of the largest biotechnology
deals in history in early 2008 with Genzyme. As per the terms of the deal, Isis will not only receive
commercialization and development milestones, it will also split net profits with Genzyme on
mipomersen (70% Genzyme, 30% - Isis) until worldwide sales eclipse $2 billion. Thereafter,
Genzyme and Isis will split profits on mipomersen 50/50. This deal is a homerun collaboration for
Isis, in our view. It has not only provided a guaranteed $325 million in up-front funding that allows
Isis the opportunity to grow and expand its internal pipeline, but it provides for the future success of
mipomersen as Genzyme has worldwide blockbuster capabilities.
Isis recently reported positive phase III results on mipomersen from a study that was conducted in
patients with homozygous familial hypercholesterolemia (hoFH). Isis and Genzyme announced that
the trial met its primary endpoint, with a 25% reduction in LDL cholesterol (LDL-C) after 26 weeks of
treatment, vs. only a 3% reduction for placebo (p<0.001). The study also met each of its three
secondary endpoints of reduction in apolipoprotein B, total cholesterol and non-high density
lipoprotein (HDL) cholesterol (all p<0.001). What is even more encouraging is that all patients were
failing maximally tolerated statins and other lipid-lowering therapies the average LDL-C at baseline
was greater than 400 mg/dL so these reductions were in addition to those achieved with the
patients' existing therapeutic regimen. Data from this phase III study in patients with hoFH will form
the basis of Genzyme's initial regulatory filing for marketing approval, which is anticipated in the
second half of 2010. The European filing is scheduled to take place soon thereafter. According to
Genzyme, about 25,000 to 30,000 patients in the U.S. and Europe would be candidates for the use
of mipomersen with this indication.
Management is testing once weekly doses of the drug, but the potential exists to have bi-weekly
doses in future. This would be a big convenience over once daily statins. We believe mipomersen
has blockbuster potential. Cholesterol lowering agents such as statins are the largest selling
pharmaceutical products in the world. The potential to improve the efficacy of these drugs with more
convenient dosing creates a large and exciting opportunity.
Partnering Opportunities with ISIS-113715: Another important proprietary candidate at Isis is ISIS-
113715, a second generation compound for type II diabetes. ISIS-113715 is an antisense compound
that targets protein tyrosine phosphatase-1B (PTP-1B). PTP-1B is an enzyme that has been shown
to play a key role in reducing the ability of insulin to regulate blood sugar levels by inactivating the
insulin receptor. By knocking out PTP-1B, the body s insulin receptors may stay active longer and
thus allow for more glucose uptake. Similar to mipomersen in hyperlipidemia, this is a completely
new mechanism of action for type II diabetes and allows for the combination of therapy with currently
approved medications such as metformin, glitazones, and sulfonylurea.
In June 2006, Isis reported positive phase II results in patients with type II diabetes treated with ISIS-
113715 as a single-agent. An intent-to-treat analysis of all patients enrolled in the trial treated with
200 mg/wk for three months showed statistically significant improvement in multiple measures of glucose control such as lowering HbA1C levels and fasting glucose levels. Additionally, important
safety data was observed in the trial. ISIS-113715 did not cause hypoglycemia (low blood sugar)
and weight gain and was well tolerated throughout.
Isis recently presented positive top-line results from a phase II study evaluating the safety and
efficacy of ISIS-113715 in patients with type II diabetes on stable doses of sulfonylurea. In addition
to lowering blood glucose, ISIS-113715 caused statistically significant and clinically meaningful
reductions in LDL cholesterol. Importantly, a tendency towards weight loss was observed even in
this short-term study without strict dietary control. ISIS-113715 demonstrated a favorable safety
profile with no exacerbation of sulfonylurea-induced hypoglycemia or other clinically significant
adverse effects.
These positive phase II results should help ISIS-113715 strike a lucrative partnership deal for the
candidate. The company s strategy of striking partnership deals has helped strengthen its financial
position. Given the company s cash balance of $637.5 million as of June 30, 2009, we believe Isis
has enough cash to fund operations for at least the next five years. A partnership deal for ISIS-
113715 would solidify the cash position further.
Bezüglich zu den Satelitte Companies:
Die Antisense-Technologie ist so ziemlich in allen medizinischen Indikationen einsetzbar. Isis kann als kleines Unternehmen mit 300 Mitarbeitern nicht das ganze Spektrum abdecken, deshalb werden Medikamente oder Therapiebereiche die nicht zur Kernkompetenz passen auslizensiert. Isis will in allen Bereichen ein Stück vom Kuchen abhaben. In den letzten Jahren hat Isis durch die "Satelitte Companies" alleine 450 Mio $ an Meilensteinen eingenommen und das ohne Risiko und Kosten.
Grüße cristrader
Die Mipomersenergebnisse werden sicherlich kurzfristig gesehen entscheidend für den Kursverlauf sein. Bezüglich der Mipomersen- Phase III-Studien bin allerdings mehr als zuversichtlich. An mangelnder Wirksamkeit wird Mipomersen den bisherigen Studien zufolge sicherlich nicht scheitern das Nebenwirkungsprofil wird entscheidend sein. Erhöhte Leberwerte gehören insbesondere bei Beginn der Medikamentenverabreichung auch bei allen anderen Standardtherapien zu den häufigen Nebenwirkungen. Die Nebenwirkungen von Mipomersen bewegen sich bisher im vergleichbaren Rahmen, nur das halt ein völlig neuer Therapieansatz bzw. eine neue Medikamententechnologie angewendet wird, wo es bisher einfach keine langfristigen Erfahrungswerte gibt.
S. Crooke
"Of course, we had three patients in this study that had elevated liver transaminases. We reported this at the AHA. Importantly,
what we've seen as we've developed this drug, and just to let you know, as it was presented by [Professor Rawls], these biochemical
changes in liver enzymes occur with all lipid lowering therapies. What we're seeing is, so far in our safety database, is no liver
toxicity. And that's important, no Hy's Law.We've dosed through a number of -- many of these and they -- and in all cases in this study the transaminases returned to entry
criteria by the end of the planned clinical observation. So, of course additional analysis is being conducted in this, but we're
very comfortable with the safety of this drug and the profile that we're seeing."
Nachfolgend noch ein Auszug eines Research-Berichtes von "Zacks"
Carlsbad, California-based Isis Pharmaceuticals, Inc. is a drug discovery and development company that
focuses on the development of products using ribonucleic acid (RNA)-based technologies, such as
antisense. Antisense technology is a direct route from genomics to drugs. Antisense drugs are the first
class of drugs targeted to control expression of genes through interactions with RNA. Beyond antisense,
Isis scientists have created another technology that exploits their knowledge of RNA. The company has
about 1,600 issued patents worldwide and controls one of the largest antisense and RNA patent estates
in the pharmaceutical industry.
The company discovers new drugs and out-licenses them to partners for license fees, milestone
payments, and royalties. Isis has partnership agreements with leading pharmaceuticals companies like
Bristol-Myers Squibb, Genzyme, Eli Lilly, and Johnson & Johnson among others. The company also has
a collaboration agreement with biotech company, Alnylam Pharmaceuticals. Both companies have joined
hands to form Regulus Therapeutics, Inc., a company that is focused on microRNA (mRNA) therapeutics.
Isis focuses its research and development efforts primarily in cardiovascular, metabolic and
neurodegenerative diseases and cancer while its partners are involved in the development of antisense
drugs in these and other areas, including inflammatory disease.
In 2008, Isis recognized $107 million in total revenues from licensing its intellectual property, satellite
companies, and from forming collaborations with larger partners in developing antisense drug
candidates. Earlier this year, Isis sold its Ibis Biosciences subsidiary to Abbott Molecular, Inc. for $215
million and an earn out on the sales of assay kits and services. As a result, Ibis financial results are
considered discontinuing operations.
Antisense Technology Represents Enormous Potential: We are big fans of antisense technology and
believe that the number of potential therapeutic applications is enormous. Antisense drugs may have
significant potential to treat a number of diseases where small molecules and biologic compounds
have failed. Although still early stage, antisense technology with mechanisms such as small
interfering RNA (siRNA), RNA interference (RNAi), alternate splicing, and microRNA (mRNA) have
the potential to change how we treat disease in the next decade. We see antisense technology
today where biologics were ten years ago. The promise of biologic drugs is finally delivering with
blockbuster compounds such as Rituxan, Herceptin, Avastin, Aranesp, Synagis, and Humira. We
think in ten years the same will be said for antisense.Antisense drugs have several specific advantages over traditional protein or small-molecule derived
therapeutics. Firstly, antisense drugs are highly specific and bind to only the complimentary mRNA
strain that codes for the specific protein being targeted. This has the potential to greatly reduce
unwanted side-effects that occur through secondary mechanisms of action. Secondly, since all
proteins are coded by mRNA, antisense drugs have the potential to work on a broad number of
diseases including cardiovascular, metabolic, inflammatory, ocular, viral, neurodegenerative
disease, and cancer. Finally, because all antisense drugs start with the basic hybridization of mRNA
targets, development timelines are shortened and response is often predictable in the early-stage of
development. Scientists need only choose a specific protein to target and then create a small
antisense molecule to bind to the transcribed mRNA. This creates efficiency in drug discovery and
time to market.
Isis is pioneering the effort into antisense research. The company has already commercialized the
first antisense drug in 1998 with Vitravene and has several proprietary compounds in clinical
development as well as partnered programs with large pharmaceutical companies such as Bristol-
Myers, Eli Lilly, Merck, J&J, Genzyme and Glaxo. We think that by 2012 two to three additional
antisense drugs developed by Isis could be on the market.
Mipomersen - The Blockbuster in Phase III: Isis Pharmaceuticals lead pipeline candidate,
mipomersen (formerly ISIS-301012), is a second generation compound currently in a multitude of
phase III trials for familial hypercholesterolemia (FH). Mipomersen is an antisense drug that targets
apolipoprotein B (apoB) -100, which is critical to the synthesis and transport of low density
lipoprotein (LDL) or bad cholesterol. By potentially knocking out the apoB-100 protein via antisense
technology, Isis scientists believe they can effectively lower the cholesterol of patients and reduce
the risk of serious cardiovascular disease. Given the novel mechanism and narrow initial market
focus of homozygous (Ho) FH, the U.S. Food and Drug Administration (FDA) granted mipomersen
orphan drug status in June 2006.
Meanwhile, highly encouraging phase II data helped Isis strike one of the largest biotechnology
deals in history in early 2008 with Genzyme. As per the terms of the deal, Isis will not only receive
commercialization and development milestones, it will also split net profits with Genzyme on
mipomersen (70% Genzyme, 30% - Isis) until worldwide sales eclipse $2 billion. Thereafter,
Genzyme and Isis will split profits on mipomersen 50/50. This deal is a homerun collaboration for
Isis, in our view. It has not only provided a guaranteed $325 million in up-front funding that allows
Isis the opportunity to grow and expand its internal pipeline, but it provides for the future success of
mipomersen as Genzyme has worldwide blockbuster capabilities.
Isis recently reported positive phase III results on mipomersen from a study that was conducted in
patients with homozygous familial hypercholesterolemia (hoFH). Isis and Genzyme announced that
the trial met its primary endpoint, with a 25% reduction in LDL cholesterol (LDL-C) after 26 weeks of
treatment, vs. only a 3% reduction for placebo (p<0.001). The study also met each of its three
secondary endpoints of reduction in apolipoprotein B, total cholesterol and non-high density
lipoprotein (HDL) cholesterol (all p<0.001). What is even more encouraging is that all patients were
failing maximally tolerated statins and other lipid-lowering therapies the average LDL-C at baseline
was greater than 400 mg/dL so these reductions were in addition to those achieved with the
patients' existing therapeutic regimen. Data from this phase III study in patients with hoFH will form
the basis of Genzyme's initial regulatory filing for marketing approval, which is anticipated in the
second half of 2010. The European filing is scheduled to take place soon thereafter. According to
Genzyme, about 25,000 to 30,000 patients in the U.S. and Europe would be candidates for the use
of mipomersen with this indication.
Management is testing once weekly doses of the drug, but the potential exists to have bi-weekly
doses in future. This would be a big convenience over once daily statins. We believe mipomersen
has blockbuster potential. Cholesterol lowering agents such as statins are the largest selling
pharmaceutical products in the world. The potential to improve the efficacy of these drugs with more
convenient dosing creates a large and exciting opportunity.
Partnering Opportunities with ISIS-113715: Another important proprietary candidate at Isis is ISIS-
113715, a second generation compound for type II diabetes. ISIS-113715 is an antisense compound
that targets protein tyrosine phosphatase-1B (PTP-1B). PTP-1B is an enzyme that has been shown
to play a key role in reducing the ability of insulin to regulate blood sugar levels by inactivating the
insulin receptor. By knocking out PTP-1B, the body s insulin receptors may stay active longer and
thus allow for more glucose uptake. Similar to mipomersen in hyperlipidemia, this is a completely
new mechanism of action for type II diabetes and allows for the combination of therapy with currently
approved medications such as metformin, glitazones, and sulfonylurea.
In June 2006, Isis reported positive phase II results in patients with type II diabetes treated with ISIS-
113715 as a single-agent. An intent-to-treat analysis of all patients enrolled in the trial treated with
200 mg/wk for three months showed statistically significant improvement in multiple measures of glucose control such as lowering HbA1C levels and fasting glucose levels. Additionally, important
safety data was observed in the trial. ISIS-113715 did not cause hypoglycemia (low blood sugar)
and weight gain and was well tolerated throughout.
Isis recently presented positive top-line results from a phase II study evaluating the safety and
efficacy of ISIS-113715 in patients with type II diabetes on stable doses of sulfonylurea. In addition
to lowering blood glucose, ISIS-113715 caused statistically significant and clinically meaningful
reductions in LDL cholesterol. Importantly, a tendency towards weight loss was observed even in
this short-term study without strict dietary control. ISIS-113715 demonstrated a favorable safety
profile with no exacerbation of sulfonylurea-induced hypoglycemia or other clinically significant
adverse effects.
These positive phase II results should help ISIS-113715 strike a lucrative partnership deal for the
candidate. The company s strategy of striking partnership deals has helped strengthen its financial
position. Given the company s cash balance of $637.5 million as of June 30, 2009, we believe Isis
has enough cash to fund operations for at least the next five years. A partnership deal for ISIS-
113715 would solidify the cash position further.
Bezüglich zu den Satelitte Companies:
Die Antisense-Technologie ist so ziemlich in allen medizinischen Indikationen einsetzbar. Isis kann als kleines Unternehmen mit 300 Mitarbeitern nicht das ganze Spektrum abdecken, deshalb werden Medikamente oder Therapiebereiche die nicht zur Kernkompetenz passen auslizensiert. Isis will in allen Bereichen ein Stück vom Kuchen abhaben. In den letzten Jahren hat Isis durch die "Satelitte Companies" alleine 450 Mio $ an Meilensteinen eingenommen und das ohne Risiko und Kosten.
Grüße cristrader
Antwort auf Beitrag Nr.: 38.787.353 von cristrader am 22.01.10 00:50:28ad satellite companies:
mir ist schon klar, was das unternehmen damit bezwecken will. was mir aber nicht gefällt, sind diese eigentümer-verstrickungen.
wenn zb ein unternehmen A von unternehmen B 20% und unternehmen C 50% hält, dann muss ich mir für eine ordentliche analyse alle unternehmen im detail anschauen (sind unternehmen B und C gut geführt? geht alles mit rechten dingen zu?).
reine projektbedingte auslizenzierungsdeals mit upfront/MS/tantieme fänd ich auch ok.
mir ist schon klar, was das unternehmen damit bezwecken will. was mir aber nicht gefällt, sind diese eigentümer-verstrickungen.
wenn zb ein unternehmen A von unternehmen B 20% und unternehmen C 50% hält, dann muss ich mir für eine ordentliche analyse alle unternehmen im detail anschauen (sind unternehmen B und C gut geführt? geht alles mit rechten dingen zu?).
reine projektbedingte auslizenzierungsdeals mit upfront/MS/tantieme fänd ich auch ok.
Antwort auf Beitrag Nr.: 38.787.033 von ipollit am 21.01.10 23:21:01also, das haar in der suppe hab ich bei der NGSX-story (noch) nicht gefunden, abgesehen davon, dass man das teil vor nicht all zu langer zeit bei 1 USD bekam...
ein härchen in der suppe gibts aber wohl, was die patentlage betrifft - siehe auszug aus dem research paper:
Intellectual Property & Manufacturing
We think it is important to note that Anesiva (ANSV) is also working on a liquid capsaicin product currently in phase
III trials. However, Anesiva s product, Adlea, is an injection or instilled application, not a topical rub, and designed
for post-surgical applications such as total knee or hip arthroscopy (TKA / THA) and bunionectomy. As of now, the
two companies seem content stay within their respective market. However, the mention of Anesiva s Adlea brings
up an important discussion on intellectual property. NeurogesX licensed method of use patents in the U.S. and
Europe that covers the delivery of high-concentration capsaicin for the treatment of neuropathic pain from the
University of California (Cal). The license with Cal is exclusive to NeurogesX. NeurogesX will owe a 5% royalty to
Cal on all up-front and backend milestones, as well as sales of the product.
There were three inventors of the patent however, two of the three inventors of the patent did not assign the rights
of this patent to Cal, so NeurogesX license is non-exclusive with respect to the use of capsaicin to treat pain.
Anesiva has licensed the rights to this same patent for the development and commercialization of pain, including
injection or infiltration of capsaicin for post-surgical pain, osteoarthritis or interdigital neuroma. Anesiva has also
licensed the rights for use of capsaicin from Dr. Pappagallo for neuropathic pain.
das wäre durchaus nähere recherche wert
ein härchen in der suppe gibts aber wohl, was die patentlage betrifft - siehe auszug aus dem research paper:
Intellectual Property & Manufacturing
We think it is important to note that Anesiva (ANSV) is also working on a liquid capsaicin product currently in phase
III trials. However, Anesiva s product, Adlea, is an injection or instilled application, not a topical rub, and designed
for post-surgical applications such as total knee or hip arthroscopy (TKA / THA) and bunionectomy. As of now, the
two companies seem content stay within their respective market. However, the mention of Anesiva s Adlea brings
up an important discussion on intellectual property. NeurogesX licensed method of use patents in the U.S. and
Europe that covers the delivery of high-concentration capsaicin for the treatment of neuropathic pain from the
University of California (Cal). The license with Cal is exclusive to NeurogesX. NeurogesX will owe a 5% royalty to
Cal on all up-front and backend milestones, as well as sales of the product.
There were three inventors of the patent however, two of the three inventors of the patent did not assign the rights
of this patent to Cal, so NeurogesX license is non-exclusive with respect to the use of capsaicin to treat pain.
Anesiva has licensed the rights to this same patent for the development and commercialization of pain, including
injection or infiltration of capsaicin for post-surgical pain, osteoarthritis or interdigital neuroma. Anesiva has also
licensed the rights for use of capsaicin from Dr. Pappagallo for neuropathic pain.
das wäre durchaus nähere recherche wert
Antwort auf Beitrag Nr.: 38.797.282 von PathFinder2 am 23.01.10 12:23:37ich weiß nicht, ob das Patent ein Problem ist... der von dir zitierte Abschnitt geht ja noch etwas weiter. Soweit ich es verstehe, hat NGSX das alleinige Recht, Capsaicin über die Haut anzuwenden, während z.B. das von Anesiva intravenös wäre. Für die Indikationen von Qutenza scheint die direkte Anwendung auf der Haut optimaler zu sein. Im Prinzip ist es ja nur ein Aufkleben einer Art Pflaster unter leichter örtlicher Betäubung für vielleicht eine Stunde... und das bei Bedarf alle 3 Monate, um die Schmerzen dauerhaft zu reduzieren.
Der Research-Report ist ja schon etwas älter... inzwischen ist beispielsweise dieser potentielle Konkurrent Anesiva pleite, was deren Entwicklung von Adlea sicherlich hemmt.
mfg ipollit
Der Research-Report ist ja schon etwas älter... inzwischen ist beispielsweise dieser potentielle Konkurrent Anesiva pleite, was deren Entwicklung von Adlea sicherlich hemmt.
mfg ipollit
Antwort auf Beitrag Nr.: 38.797.282 von PathFinder2 am 23.01.10 12:23:37hier ist auch noch etwas zu den Patenten...
http://www.babybiotechs.com/biotech-investment-reports/ngsx/…
Intellectual Property
Low doses of capsaicin cream have long been used successfully for relief of neuropathic pain. However the first published study to use high doses (5-10%) in combination with regional anesthesia (to numb the normal burning sensation accompanying such high dose) was published in 1998 from UCSF. This study showed that at these levels of capsaicin, 90% of patients (N=10) were relieved from neuropathic pain from 1 to 18 weeks. Interestingly, in 1996, prior to the publication, three of the authors of the study patented the use of high percentage capsaicin with the Regents of the University of California as an assignee on the patent. Subsequently, the first author on the above study, Dr Wendye Robbins, was issued another patent in 1997 (again UC Regents were the assignee) whereby local anesthetic was administered through the use of a transdermal patch containing the high concentration of capsaicin. Dr. Robbins founded NeurogesX in 2000, licensing the patents from the University of California.
That pretty much brings us up to speed on the intellectual property with one final important caveat. The first patent outlining the use of high concentration capsaicin was issued to three authors, and two of the three were not UCSF faculty and so did not assign their patent rights to the University of California. Anesiva, a company focused on the development and commercialization of treatments for pain, including injection or infiltration of a capsaicin derivative for post-surgical pain, osteoarthritis or interdigital neuroma, has licensed from one of the non-assigning inventors the right to use the technology under the method patent.
So NeurogesX has the worldwide exclusive license for a high concentration capsaicin transdermal patch. The use of high-concentration capsaicin itself is not exactly in the public domain but neither does NeurogesX have exclusive worldwide rights. Therefore, it goes without saying any investment in NeurogesX is banking on the utility of the transdermal patch method of capsaicin delivery. As it turns out the only current alternatives to a transdermal patches are a topical creams which of course have the drawback of wildly inaccurate dosing (FDA won’t approve) and injection such as Anesiva is developing for post surgical pain.
mfg ipollit
http://www.babybiotechs.com/biotech-investment-reports/ngsx/…
Intellectual Property
Low doses of capsaicin cream have long been used successfully for relief of neuropathic pain. However the first published study to use high doses (5-10%) in combination with regional anesthesia (to numb the normal burning sensation accompanying such high dose) was published in 1998 from UCSF. This study showed that at these levels of capsaicin, 90% of patients (N=10) were relieved from neuropathic pain from 1 to 18 weeks. Interestingly, in 1996, prior to the publication, three of the authors of the study patented the use of high percentage capsaicin with the Regents of the University of California as an assignee on the patent. Subsequently, the first author on the above study, Dr Wendye Robbins, was issued another patent in 1997 (again UC Regents were the assignee) whereby local anesthetic was administered through the use of a transdermal patch containing the high concentration of capsaicin. Dr. Robbins founded NeurogesX in 2000, licensing the patents from the University of California.
That pretty much brings us up to speed on the intellectual property with one final important caveat. The first patent outlining the use of high concentration capsaicin was issued to three authors, and two of the three were not UCSF faculty and so did not assign their patent rights to the University of California. Anesiva, a company focused on the development and commercialization of treatments for pain, including injection or infiltration of a capsaicin derivative for post-surgical pain, osteoarthritis or interdigital neuroma, has licensed from one of the non-assigning inventors the right to use the technology under the method patent.
So NeurogesX has the worldwide exclusive license for a high concentration capsaicin transdermal patch. The use of high-concentration capsaicin itself is not exactly in the public domain but neither does NeurogesX have exclusive worldwide rights. Therefore, it goes without saying any investment in NeurogesX is banking on the utility of the transdermal patch method of capsaicin delivery. As it turns out the only current alternatives to a transdermal patches are a topical creams which of course have the drawback of wildly inaccurate dosing (FDA won’t approve) and injection such as Anesiva is developing for post surgical pain.
mfg ipollit
ipollit, hast du links zu research reports von SGEN und EXEL?
auf welchen sieten suchst du normal?
danke
auf welchen sieten suchst du normal?
danke
danke - das heißt für mich auf den ersten blick
Qutenza - gute IP-position
NGX-1998 - kaum IP-schutz
was denkst du?
Qutenza - gute IP-position
NGX-1998 - kaum IP-schutz
was denkst du?
bei NGSX stellt sich für mich die hauptfrage, zu welchem zeitpunkt von hier weg ein einstieg sinn machen kann.
rein charttechnisch ist der kurs am unteren ende eines trading-bandes zwischen ca. 7 und 9 USD. von demher ganz ok bei ca. 7.
andererseits, zulassung ist geschehen (da hätt man dabei sein müssen vom niedrigen dollarbereich weg...), und jetzt heißt's auf relevante umsätze warten. 2010 wird's da kaum was geben, 2011, 2012...
was denkt ihr?
rein charttechnisch ist der kurs am unteren ende eines trading-bandes zwischen ca. 7 und 9 USD. von demher ganz ok bei ca. 7.
andererseits, zulassung ist geschehen (da hätt man dabei sein müssen vom niedrigen dollarbereich weg...), und jetzt heißt's auf relevante umsätze warten. 2010 wird's da kaum was geben, 2011, 2012...
was denkt ihr?
Antwort auf Beitrag Nr.: 38.797.527 von PathFinder2 am 23.01.10 13:55:32sorry, richtige Research Reports hab ich auch nicht. Nach den meisten Sachen google ich einfach nur.
zu SGEN und EXEL finden sich einige gute Beiträge im Blog von Ohad Hammer... http://www.hammerstockblog.com/
ansonsten lese ich hin und wieder in den Boards...
ihub http://investorshub.advfn.com/boards/board.aspx?board_id=141…
si http://siliconinvestor.advfn.com/forum.aspx?forumid=62
oder die yahoo-Boards. Viel brauchbares lässt sich da allerdings meist nicht finden.
sonst vielleicht noch
http://www.pharmastrategyblog.com/
http://www.fiercebiotech.com/
http://www.thestreet.com/author/1352996/AdamFeuerstein/all.h…
gibt es sonst noch was?
die letzten SGEN und EXEL JPM-Webcasts sind vielleicht auch interessant...
http://metameetings.com/webcasts/jpmorgan/healthcare10/welco…
mfg ipollit
zu SGEN und EXEL finden sich einige gute Beiträge im Blog von Ohad Hammer... http://www.hammerstockblog.com/
ansonsten lese ich hin und wieder in den Boards...
ihub http://investorshub.advfn.com/boards/board.aspx?board_id=141…
si http://siliconinvestor.advfn.com/forum.aspx?forumid=62
oder die yahoo-Boards. Viel brauchbares lässt sich da allerdings meist nicht finden.
sonst vielleicht noch
http://www.pharmastrategyblog.com/
http://www.fiercebiotech.com/
http://www.thestreet.com/author/1352996/AdamFeuerstein/all.h…
gibt es sonst noch was?
die letzten SGEN und EXEL JPM-Webcasts sind vielleicht auch interessant...
http://metameetings.com/webcasts/jpmorgan/healthcare10/welco…
mfg ipollit
Antwort auf Beitrag Nr.: 38.797.548 von PathFinder2 am 23.01.10 14:00:21NGX-1998 ist zwar flüssig... allerdings ebenfalls nur zur äußeren Anwendung über die Haut. Durch eine bessere Aufnahme soll die Zeit der Anwendung von einer Stunde gesenkt werden.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.797.623 von ipollit am 23.01.10 14:33:58danke!
===
übrigens, wirklich interessant waren die aussagen von Exelixis-CEO Scangos auf deren R&D-day Ende letzten Jahres:
"We know that this goal of building a profitable company we may never get to see because other things might happen, if they do, thats fine, right?"
===
übrigens, wirklich interessant waren die aussagen von Exelixis-CEO Scangos auf deren R&D-day Ende letzten Jahres:
"We know that this goal of building a profitable company we may never get to see because other things might happen, if they do, thats fine, right?"
Antwort auf Beitrag Nr.: 38.797.627 von ipollit am 23.01.10 14:36:09aber NGX-1998 hat nix mit dem patch zu tun, oder? von daher meinte ich kaum IP-schutz. oder hab ich das nicht ganz richtig verstanden?
Antwort auf Beitrag Nr.: 38.797.573 von PathFinder2 am 23.01.10 14:09:43
bei NGSX stellt sich für mich die hauptfrage, zu welchem zeitpunkt von hier weg ein einstieg sinn machen kann.
Hi PathFinder und ipollit,
zunächst mal vielen Dank für eure NGSX-Diskussion, die sehr einsichtsvoll war und mich in meiner Positionierung deutlich bestärkt hat. Besonderen Dank an ipollit für das Ausgraben des Research-Reports.
PathFinder, was Deine Frage angeht: Ich muss bei diesem Investment davon ausgehen, dass Qutenza prinzipiell ein Erfolg wird. Das ist zwar nicht sicher, aber wenn ich davon nicht ausgehe, muss ich eh nicht weiter nachdenken. Was hieße Erfolg konkret? Ich würde es als Erfolg verstehen, wenn sie bis 2015 einen Umsatz aus Royalties und Eigenvertrieb in Höhe von 300 Millionen Dollar generieren würden. Bei "üblichen" Margen und Umsatzmultiplen und ansonsten unfallfrei agierendem Management ließe sich daraus meiner Meinung nach in 2015 eine Marktkapitalisierung irgendwo zwischen 500 und 1.000 Milliarden Dollar ableiten, was also ca. Faktor 4 bis 8 wäre.
Weiteres upside bis 2020 dürfte vorhanden sein.
Ich finde das hinreichend, wenn man das relativ(!) begrenzte Risiko mit berücksichtigt.
So in etwa geht meine Investmentidee und natürlich ist das nur eine Meinung.
bei NGSX stellt sich für mich die hauptfrage, zu welchem zeitpunkt von hier weg ein einstieg sinn machen kann.
Hi PathFinder und ipollit,
zunächst mal vielen Dank für eure NGSX-Diskussion, die sehr einsichtsvoll war und mich in meiner Positionierung deutlich bestärkt hat. Besonderen Dank an ipollit für das Ausgraben des Research-Reports.
PathFinder, was Deine Frage angeht: Ich muss bei diesem Investment davon ausgehen, dass Qutenza prinzipiell ein Erfolg wird. Das ist zwar nicht sicher, aber wenn ich davon nicht ausgehe, muss ich eh nicht weiter nachdenken. Was hieße Erfolg konkret? Ich würde es als Erfolg verstehen, wenn sie bis 2015 einen Umsatz aus Royalties und Eigenvertrieb in Höhe von 300 Millionen Dollar generieren würden. Bei "üblichen" Margen und Umsatzmultiplen und ansonsten unfallfrei agierendem Management ließe sich daraus meiner Meinung nach in 2015 eine Marktkapitalisierung irgendwo zwischen 500 und 1.000 Milliarden Dollar ableiten, was also ca. Faktor 4 bis 8 wäre.
Weiteres upside bis 2020 dürfte vorhanden sein.
Ich finde das hinreichend, wenn man das relativ(!) begrenzte Risiko mit berücksichtigt.
So in etwa geht meine Investmentidee und natürlich ist das nur eine Meinung.
Antwort auf Beitrag Nr.: 38.799.166 von SLGramann am 24.01.10 09:55:34
500 und 1.000 Milliarden Dollar
Millionen bitte, Millionen
500 und 1.000 Milliarden Dollar
Millionen bitte, Millionen

Antwort auf Beitrag Nr.: 38.799.166 von SLGramann am 24.01.10 09:55:34ja ich finde das risiko auch relativ begrenzt - und trotzdem einiges an upside-potenzial.
wieso NGSX allerdings nicht gleich nach zulassung von Qutenza übernommen wurde, ist mir nicht so recht klar (wäre zb für Astellas mit sicherheit wesentlich günstiger gewesen).
wie ist die IR von NGSX? bekommt man antworten; sind sie bemüht?
was ist eigentlich mit Japan, sind für Qutenza neue studien notwendig, oder werden vorliegende studien (teilweise) anerkannt?
wieso NGSX allerdings nicht gleich nach zulassung von Qutenza übernommen wurde, ist mir nicht so recht klar (wäre zb für Astellas mit sicherheit wesentlich günstiger gewesen).
wie ist die IR von NGSX? bekommt man antworten; sind sie bemüht?
was ist eigentlich mit Japan, sind für Qutenza neue studien notwendig, oder werden vorliegende studien (teilweise) anerkannt?
da wir gerade von microcaps sprechen: kennt jemand
http://www.biodeliverysciences.com/
(siehe auch piplinechart)
market cap 80 Mio. USD
http://www.biodeliverysciences.com/
(siehe auch piplinechart)
market cap 80 Mio. USD
Antwort auf Beitrag Nr.: 38.799.377 von PathFinder2 am 24.01.10 11:35:38BioDelivery Sciences (NASDAQ: BDSI) is a specialty pharmaceutical company that is leveraging its novel and proprietary patented drug delivery technologies to develop and commercialize, either on its own or in partnerships with third parties, new applications of proven therapeutics. BDSI is focusing on developing products to meet unmet needs in the areas of pain management and oncology supportive care. BDSI’s pain franchise currently consists of two products utilizing the Company’s patented BEMA buccal soluble film technology: ONSOLIS (fentanyl buccal soluble film) which was approved by FDA on July 16, 2009, for the management of breakthrough pain in patients with cancer, eighteen years of age and older, who are already receiving and who are tolerant to opioid therapy for their underlying persistent cancer pain, and BEMA Buprenorphine, an analgesic in Phase 2 clinical development with at least one potential target indication for the treatment of moderate to severe pain. Additional product candidates are being developed utilizing the BEMA technology for conditions such as nausea/vomiting and migraine. The Company is also working with its patented Bioral cochleate technology to facilitate oral dosing of drugs otherwise given by intravenous administration. The first product under development using the technology is Bioral Amphotericin B. The Company’s headquarters is located in Raleigh, North Carolina. For more information please visit www.biodeliverysciences.com.
Antwort auf Beitrag Nr.: 38.799.382 von PathFinder2 am 24.01.10 11:37:07präsentation, die einen recht guten überblick gibt:
http://www.biodeliverysciences.com/media/documents/BDSIInves…
http://www.biodeliverysciences.com/media/documents/BDSIInves…
noch zu 505(b)(2) falls es jemanden interessiert:
http://www.camargopharma.com/Userfiles/Docs/camargo-505b2.pd…
http://www.camargopharma.com/Userfiles/Docs/camargo-505b2.pd…
Antwort auf Beitrag Nr.: 38.799.377 von PathFinder2 am 24.01.10 11:35:38BDSI kenne ich leider nicht. Wenn ich es aber so überfliege, so geht es ja im Prinzip um eine andere Art der Verabreichung von Medikamenten... z.B. sich so etwas in den Mund zu kleben. D.h. die Medikamente sind nicht von BDSI, sondern von jemand anderen. Dann müssen sie doch bereits generisch sein, wenn BDSI sie für ihr Produkt verwenden will, oder? Sie treten also mit ihren Produkten immer gegen andere etablierte und generische Produkte an mit der Differenzierung einer anderen Art der Verabreichung. Kann man da wirklich vorhersehen, wie der Markt dies dann annimmt? Bei soetwas habe ich ein wenig Bedenken, dass die zwar ihre Produkte auf den Markt bringen, diese sich dann aber kaum verkaufen lassen. Oder verstehe ich da etwas nicht richtig?
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 38.656.864 von ipollit am 03.01.10 16:49:57Während im letzten Jahr Dax, Nasdaq usw große Gewinne einbrachten, hat sich mein Biotech-Depot Thread 1147459 leider nur sehr durchwachsen entwickelt, was ein wenig entäuschend ist... am Ende blieb es bei einem kleinen Minus.
also 2009 mit einem minus abzuschliessen find ich schon
extrem sportlich...2009 war ein extrem gutes biotech jahr.
ich hatte einige misstrades im biotechbereich mit rel.
hohem risikopotential, da sollte ich dran
arbeiten, aber unterm strich war es doch recht
ordentlich in 2009...
damit dein depot mal bessere zeiten sieht:
Labopharm inc. PDUFA date: 11. febr. 2010 - keine kaufempfehlung
aber warum auf irgendwelche p2 daten warten, wenn zulassung
und partner vor der türe stehen
also 2009 mit einem minus abzuschliessen find ich schon
extrem sportlich...2009 war ein extrem gutes biotech jahr.
ich hatte einige misstrades im biotechbereich mit rel.
hohem risikopotential, da sollte ich dran
arbeiten
, aber unterm strich war es doch rechtordentlich in 2009...
damit dein depot mal bessere zeiten sieht:
Labopharm inc. PDUFA date: 11. febr. 2010 - keine kaufempfehlung
aber warum auf irgendwelche p2 daten warten, wenn zulassung
und partner vor der türe stehen
Antwort auf Beitrag Nr.: 38.800.235 von Gustl24 am 24.01.10 16:40:32ja... das war extrem sportlich!
dass es so ein extrem gutes Biotech(!)-Jahr war, fand ich eigentlich nicht. Gemessen an dem, was der Nasdaq oder der gewöhnliche Dax gebracht hat, war es alles andere als gut. Und so ein Jahr (der NBI war währungsbereinigt 13% im Plus) kann man halt auch schonmal im Minus abschließen, wenn man z.B. eine Genmab mit 20% bis 25% gewichtet, die dann fast 2/3 abgibt statt ein kleines Plus zu generieren. Und mussten z.B. CBST oder ONXX in einem extrem guten Biotech-Jahr deutlich zweistellig ins Minus gehen? Und auch so große Biotech-Nrn wie eine AMGN, CELG, GILD oder GENZ haben 2009 nichts abgeworfen, teilweise sogar kräftig abgegeben. Wenn man 100% mit einer HGSI oder VNDA dabei war, kann man natürlich immer lachen... aber auch nur hinterher, denn es hätte für diese beiden auch das Aus kommen können. Dieses Hopp oder Top wollte ich eigentlich nicht mehr spielen.
Daher halte ich mich auch bei Labopharm zurück... die Wahrscheinlichkeit einer glatten Zulassung ist im Schnitt normalerweise deutlich geringer als die Ablehnung. Aber dir viel Spaß damit... vielleicht gehörst du ja zu den Glücklichen und hast dann die Rendite für 2010 bereits im Sack.
mfg ipollit
dass es so ein extrem gutes Biotech(!)-Jahr war, fand ich eigentlich nicht. Gemessen an dem, was der Nasdaq oder der gewöhnliche Dax gebracht hat, war es alles andere als gut. Und so ein Jahr (der NBI war währungsbereinigt 13% im Plus) kann man halt auch schonmal im Minus abschließen, wenn man z.B. eine Genmab mit 20% bis 25% gewichtet, die dann fast 2/3 abgibt statt ein kleines Plus zu generieren. Und mussten z.B. CBST oder ONXX in einem extrem guten Biotech-Jahr deutlich zweistellig ins Minus gehen? Und auch so große Biotech-Nrn wie eine AMGN, CELG, GILD oder GENZ haben 2009 nichts abgeworfen, teilweise sogar kräftig abgegeben. Wenn man 100% mit einer HGSI oder VNDA dabei war, kann man natürlich immer lachen... aber auch nur hinterher, denn es hätte für diese beiden auch das Aus kommen können. Dieses Hopp oder Top wollte ich eigentlich nicht mehr spielen.
Daher halte ich mich auch bei Labopharm zurück... die Wahrscheinlichkeit einer glatten Zulassung ist im Schnitt normalerweise deutlich geringer als die Ablehnung. Aber dir viel Spaß damit... vielleicht gehörst du ja zu den Glücklichen und hast dann die Rendite für 2010 bereits im Sack.
mfg ipollit
naja... zumindest der Start in dieses Jahr war bisher nicht so schlecht:
(währungsbereinigt auf EUR-Basis und mit aktueller Gewichtung)
+5,7% Depot
+4,1% NBI
-4,4% Dax
+2,1% USD
+25,6% ViroPharma ( 2,5% )
+22,0% Genmab ( 9,2% )
+21,6% Micromet ( 5,9% )
+20,3% Allos ( 6,7% )
+9,7% Regeneron ( 12,4% )
+9,6% Cubist ( 7,5% )
+7,2% Seattle Genetics ( 3,9% )
+6,2% Incyte ( 10,3% )
+5,1% Progenics ( 1,2% )
+3,3% Onyx ( 5,9% )
+2,5% Isis ( 5,2% )
+0,3% NicOx ( 1,7% )
+0,3% Medigene ( 3,4% )
-5,3% NeurogesX ( 3,5% )
-6,0% Rigel ( 5,4% )
-6,2% Arena ( 2,0% )
-6,8% Addex ( 1,1% )
-8,4% Array ( 2,9% )
-8,7% Sucampo ( 0,9% )
-11,6% Exelixis ( 5,2% )
-12,0% ArQule ( 3,1% )
wenn es so weitergehen würde, wäre ich leicht zufrieden - dann wird es hoffentlich nicht ganz so sportlich ausfallen wie letztes Jahr
mfg ipollit
(währungsbereinigt auf EUR-Basis und mit aktueller Gewichtung)
+5,7% Depot
+4,1% NBI
-4,4% Dax
+2,1% USD
+25,6% ViroPharma ( 2,5% )
+22,0% Genmab ( 9,2% )
+21,6% Micromet ( 5,9% )
+20,3% Allos ( 6,7% )
+9,7% Regeneron ( 12,4% )
+9,6% Cubist ( 7,5% )
+7,2% Seattle Genetics ( 3,9% )
+6,2% Incyte ( 10,3% )
+5,1% Progenics ( 1,2% )
+3,3% Onyx ( 5,9% )
+2,5% Isis ( 5,2% )
+0,3% NicOx ( 1,7% )
+0,3% Medigene ( 3,4% )
-5,3% NeurogesX ( 3,5% )
-6,0% Rigel ( 5,4% )
-6,2% Arena ( 2,0% )
-6,8% Addex ( 1,1% )
-8,4% Array ( 2,9% )
-8,7% Sucampo ( 0,9% )
-11,6% Exelixis ( 5,2% )
-12,0% ArQule ( 3,1% )
wenn es so weitergehen würde, wäre ich leicht zufrieden - dann wird es hoffentlich nicht ganz so sportlich ausfallen wie letztes Jahr
mfg ipollit
und die Jahres-Charts vielleicht ein wenig schöner als am Anfang vom Thread...





















mfg ipollit
mfg ipollit
Genmab dürfte bald auch die Zulassung von Arzerra gegen CLL in Europa erhalten. Einen großen Einfluss auf den Kurs hat das nicht. Spannender werden die in nächster Zeit erwarteten Zalutumumab PIII-Daten. Offen ist, ob sich Zalutumumab irgendwie von z.B. Erbitux absetzen kann. Die Erwartungen sind eher negativ.
http://www.reuters.com/article/idUSLDE60L0WE20100122
UPDATE 1-Glaxo, Genmab win EU backing for leukaemia drug
* EMEA recommends Arzerra for chronic lymphocytic leukaemia
* Move follows U.S. FDA approval in October
* Genmab shares up 5 pct, Glaxo flat (Updates with details on drug, sales forecast, shares)
LONDON, Jan 22 (Reuters) - GlaxoSmithKline (GSK.L) and Genmab (GEN.CO) won European backing for their leukaemia drug Arzerra on Friday, following a green light for the medicine from U.S. regulators in October.
Arzerra is the first drug to reach the market from the labs of Genmab and shares in the Danish biotech rose 5 percent on the news. Glaxo stock was flat.
The European Medicines Agency said it was recommending Arzerra, or ofatumumab, as a treatment for chronic lymphocytic leukaemia in patients who do not respond to Genzyme's (GENZ.O) Campath or the chemotherapy drug fludarabine.
Recommendations for marketing approval by the agency's Committee for Medicinal Products for Human Use (CHMP) are normally endorsed by the European Commission within a couple of months.
Glaxo bought global rights to Arzerra in December 2006 in a deal worth up to $2.1 billion, a record sum for a biotech product agreement at the time.
The world's second biggest drugmaker is also banking on Arzerra working in other diseases beyond CLL.
But hopes for its use in patients with another blood cancer called non-Hodgkin's lymphoma (NHL) were dealt a blow in August when it failed to help patients as much as expected in a clinical trial.
Current consensus analyst forecasts for Arzerra, which is also being developed for rheumatoid arthritis, suggests annual sales will reach $465 million in 2013, according to Thomson Pharma.
Genmab and Glaxo aim to position Arzerra as a rival to Roche (ROG.VX) and Biogen Idec's (BIIB.O) blockbuster treatment Rituxan. Both Arzerra and Rixtuxan belong to a class of medicines known as anti-CD20 antibodies.
mfg ipollit
http://www.reuters.com/article/idUSLDE60L0WE20100122
UPDATE 1-Glaxo, Genmab win EU backing for leukaemia drug
* EMEA recommends Arzerra for chronic lymphocytic leukaemia
* Move follows U.S. FDA approval in October
* Genmab shares up 5 pct, Glaxo flat (Updates with details on drug, sales forecast, shares)
LONDON, Jan 22 (Reuters) - GlaxoSmithKline (GSK.L) and Genmab (GEN.CO) won European backing for their leukaemia drug Arzerra on Friday, following a green light for the medicine from U.S. regulators in October.
Arzerra is the first drug to reach the market from the labs of Genmab and shares in the Danish biotech rose 5 percent on the news. Glaxo stock was flat.
The European Medicines Agency said it was recommending Arzerra, or ofatumumab, as a treatment for chronic lymphocytic leukaemia in patients who do not respond to Genzyme's (GENZ.O) Campath or the chemotherapy drug fludarabine.
Recommendations for marketing approval by the agency's Committee for Medicinal Products for Human Use (CHMP) are normally endorsed by the European Commission within a couple of months.
Glaxo bought global rights to Arzerra in December 2006 in a deal worth up to $2.1 billion, a record sum for a biotech product agreement at the time.
The world's second biggest drugmaker is also banking on Arzerra working in other diseases beyond CLL.
But hopes for its use in patients with another blood cancer called non-Hodgkin's lymphoma (NHL) were dealt a blow in August when it failed to help patients as much as expected in a clinical trial.
Current consensus analyst forecasts for Arzerra, which is also being developed for rheumatoid arthritis, suggests annual sales will reach $465 million in 2013, according to Thomson Pharma.
Genmab and Glaxo aim to position Arzerra as a rival to Roche (ROG.VX) and Biogen Idec's (BIIB.O) blockbuster treatment Rituxan. Both Arzerra and Rixtuxan belong to a class of medicines known as anti-CD20 antibodies.
mfg ipollit
Genmabs Arzerra ist übrigens mit ca. 100.000 USD pro Behandlungszyklus von 6 Monaten extrem teuer. Entsprechendes gilt auch für Allos Folotyn. Ein wenig liegt es daran, dass die Patientengruppe jeweils relativ klein ist und es keine Alternativen gibt. Irgendwann wird sich aber auch die Frage stellen, ob das ganze noch bezahlbar ist... ich denke aber, dass der Preis automatisch sinkt, wenn diese Therapien breiter eingesetzt werden.
http://www.nytimes.com/2009/12/05/health/05drug.html?pagewan…
Questioning a $30,000-a-Month Cancer Drug
Published: December 4, 2009
A newly approved chemotherapy drug will cost about $30,000 a month, a sign that the prices of cancer medicines are continuing to rise despite growing concern about health care costs.
The price of the new drug, called Folotyn, is at least triple that of other drugs that critics have said are too expensive for the benefits they offer to patients. The colon cancer drug Erbitux, for instance, costs $10,000 a month and the drug Avastin about $8,800 when used to treat lung cancer. The price of Folotyn “seems way higher than I heard of before,” Robert L. Erwin, president of the Marti Nelson Cancer Foundation, a patient advocacy group. “I can’t imagine there not being a backlash against the pricing.”
Drug makers in general have been raising prices sharply in advance of the possible passage of health care overhaul legislation, according to various studies. But the price of cancer drugs has been an issue for several years.
Critics, including many oncologists, say that patients and the health system cannot afford to pay huge prices for drugs that, on average, provide only a few extra months of life at best.
And Folotyn has not even been shown to prolong lives — only to shrink tumors. The drug was approved by the Food and Drug Administration in late September as a treatment for peripheral T-cell lymphoma, a rare and usually aggressive blood cancer that strikes an estimated 5,600 Americans each year. It is available for sale, but its manufacturer, Allos Therapeutics, does not plan to start actively promoting it until January.
Allos defends the price, saying it made a significant investment to develop the first approved drug for this type of cancer.
“It’s a very aggressive disease, and patients right now have no options,” said James V. Caruso, the chief commercial officer for Allos, a 17-year-old publicly traded company based in Westminster, Colo., that has no other drugs on the market.
Mr. Caruso also said the price of Folotyn was not out of line with that of other drugs for rare cancers. Patients, moreover, are likely to use the drug for only a couple of months because the tumor worsens so quickly, he said. So the total cost of using Folotyn will be less than for many other drugs with lower monthly prices.
“We believe we are fairly priced,” he added, “and we’re benchmarked” against other drugs. In a conference call with analysts last month, Mr. Caruso said Allos had “not had pushback of any type at this point” from insurers.
Some drugs for rare cancers are close to Foltyn’s price. Genzyme’s Clolar for pediatric leukemia costs about $34,000 a week, though the company says that only two weeks of treatment are typically needed. Genzyme’s drug Campath, for chronic lymphocytic leukemia, costs about $5,000 a week for several weeks.
GlaxoSmithKline is charging up to $98,000 for a six-month treatment course of Arzerra, a drug approved in late October for chronic lymphocytic leukemia, which strikes about 15,000 Americans a year. About $60,000 of the cost would be incurred in the first eight weeks, when the drug is given more frequently.
Gloucester Pharmaceuticals, which won approval in November for a drug to treat cutaneous T-cell lymphoma, another rare cancer, declined to discuss what it would charge when that treatment, called Istodax, goes on sale in January.
Despite such comparisons, Dr. Lee N. Newcomer, senior vice president for oncology at the big insurer UnitedHealthcare, called the price of Folotyn “unconscionable.” He said that Folotyn alone would cost as much as UnitedHealthcare now typically spends in total to treat a lymphoma patient from diagnosis until death. That median expenditure now, he said, is $87,000 for a little over a year of treatments.
But Dr. Newcomer said insurers would be obligated to pay for Folotyn because there were no alternatives.
Folotyn has not yet shown an effect on longevity. In the clinical trial that led to approval of the drug, 27 percent of the 109 patients experienced a reduction in tumor size. The reductions lasted a median of 9.4 months.
But considering all the patients in the trial, only 12 percent had a reduction in tumor size that lasted for more than 14 weeks. The trial did not compare Folotyn to another drug or a placebo.
“This drug is not a home run,” Dr. Brad S. Kahl, a lymphoma specialist at the University of Wisconsin, said during a meeting of an advisory committee to the F.D.A. on Sept. 2. “It’s not even a double. It’s a single.”
Saying that even a single was helpful, Dr. Kahl was part of a majority on the panel that recommended approval of the drug, 10 to 4.
But after recently learning what Allos planned to charge for Folotyn, Dr. Kahl said he was “disappointed” by the “excessive” price.
“It dampens my enthusiasm for using that drug,” he said. “It creates these huge ethical quandaries about trying a drug that has a modest benefit for the average patient at enormous expense.”
Folotyn is given by a rapid intravenous procedure once a week for six weeks out of every seven. Even to try the drug for the first seven-week cycle to see if it works would cost over $50,000. In the clinical trial, the median duration of use was 70 days, which would cost roughly $70,000 to $80,000. But some patients used the drug for many months.
In a note to clients in October, Joshua Schimmer, an analyst at Leerink Swann, estimated that a typical treatment would last 3.5 months and cost $126,000, or about $36,000 a month.
For investors, a high price is usually a good thing. Mr. Schimmer’s note was entitled “Folotyn Prices High, Reiterate Outperform.” He estimated annual sales of the drug in the United States reaching about $300 million by 2014.
Patient advocacy groups say that while they wish prices were lower, high prices might be needed to encourage companies to develop new drugs.
“It’s a two-edged sword that we have to live with and deal with,” said Louis J. DeGennaro, chief scientific officer of the Leukemia and Lymphoma Society, which has received donations from Allos and other companies. “A peripheral T-cell lymphoma patient,” he said, “at first blush will see this therapy as a very good thing.”
Allos, which is still unprofitable, has lost $350 million since its founding in 1992 and failed to win approval of a previous drug.
“Every dime that goes into the company supports Folotyn,” Mr. Caruso said.
At the time Folotyn was approved in September, stock in Allos briefly peaked above $8.50 but has slipped since then, closing up 16 cents at $6.62, or an increase of nearly 2.5 percent, on Friday.
After the approval, Allos raised $93 million in a secondary stock offering. In the prospectus for that offering, the company said that one of the risks for investors was “the relative price of Folotyn as compared to alternate treatment options.” It said there was a risk it might have to lower the price or offer discounts to successfully market Folotyn.
Like many other companies with high-priced drugs, Allos has established a program to help patients arrange insurance reimbursement. It says it will give the drug free to uninsured patients who cannot pay for it any other way.
And because a patient’s out-of-pocket co-payments alone — Medicare’s is 20 percent — could be thousands of dollars a month for Folotyn, Allos is financing a co-payment assistance program run by the National Organization for Rare Disorders, a patient advocacy group.
While this helps patients, it also helps the company sell more of its drug. If the 20 percent Medicare co-payment is made, then Medicare will pay the other 80 percent of the drug’s price — or about $24,000 a month.
mfg ipollit
http://www.nytimes.com/2009/12/05/health/05drug.html?pagewan…
Questioning a $30,000-a-Month Cancer Drug
Published: December 4, 2009
A newly approved chemotherapy drug will cost about $30,000 a month, a sign that the prices of cancer medicines are continuing to rise despite growing concern about health care costs.
The price of the new drug, called Folotyn, is at least triple that of other drugs that critics have said are too expensive for the benefits they offer to patients. The colon cancer drug Erbitux, for instance, costs $10,000 a month and the drug Avastin about $8,800 when used to treat lung cancer. The price of Folotyn “seems way higher than I heard of before,” Robert L. Erwin, president of the Marti Nelson Cancer Foundation, a patient advocacy group. “I can’t imagine there not being a backlash against the pricing.”
Drug makers in general have been raising prices sharply in advance of the possible passage of health care overhaul legislation, according to various studies. But the price of cancer drugs has been an issue for several years.
Critics, including many oncologists, say that patients and the health system cannot afford to pay huge prices for drugs that, on average, provide only a few extra months of life at best.
And Folotyn has not even been shown to prolong lives — only to shrink tumors. The drug was approved by the Food and Drug Administration in late September as a treatment for peripheral T-cell lymphoma, a rare and usually aggressive blood cancer that strikes an estimated 5,600 Americans each year. It is available for sale, but its manufacturer, Allos Therapeutics, does not plan to start actively promoting it until January.
Allos defends the price, saying it made a significant investment to develop the first approved drug for this type of cancer.
“It’s a very aggressive disease, and patients right now have no options,” said James V. Caruso, the chief commercial officer for Allos, a 17-year-old publicly traded company based in Westminster, Colo., that has no other drugs on the market.
Mr. Caruso also said the price of Folotyn was not out of line with that of other drugs for rare cancers. Patients, moreover, are likely to use the drug for only a couple of months because the tumor worsens so quickly, he said. So the total cost of using Folotyn will be less than for many other drugs with lower monthly prices.
“We believe we are fairly priced,” he added, “and we’re benchmarked” against other drugs. In a conference call with analysts last month, Mr. Caruso said Allos had “not had pushback of any type at this point” from insurers.
Some drugs for rare cancers are close to Foltyn’s price. Genzyme’s Clolar for pediatric leukemia costs about $34,000 a week, though the company says that only two weeks of treatment are typically needed. Genzyme’s drug Campath, for chronic lymphocytic leukemia, costs about $5,000 a week for several weeks.
GlaxoSmithKline is charging up to $98,000 for a six-month treatment course of Arzerra, a drug approved in late October for chronic lymphocytic leukemia, which strikes about 15,000 Americans a year. About $60,000 of the cost would be incurred in the first eight weeks, when the drug is given more frequently.
Gloucester Pharmaceuticals, which won approval in November for a drug to treat cutaneous T-cell lymphoma, another rare cancer, declined to discuss what it would charge when that treatment, called Istodax, goes on sale in January.
Despite such comparisons, Dr. Lee N. Newcomer, senior vice president for oncology at the big insurer UnitedHealthcare, called the price of Folotyn “unconscionable.” He said that Folotyn alone would cost as much as UnitedHealthcare now typically spends in total to treat a lymphoma patient from diagnosis until death. That median expenditure now, he said, is $87,000 for a little over a year of treatments.
But Dr. Newcomer said insurers would be obligated to pay for Folotyn because there were no alternatives.
Folotyn has not yet shown an effect on longevity. In the clinical trial that led to approval of the drug, 27 percent of the 109 patients experienced a reduction in tumor size. The reductions lasted a median of 9.4 months.
But considering all the patients in the trial, only 12 percent had a reduction in tumor size that lasted for more than 14 weeks. The trial did not compare Folotyn to another drug or a placebo.
“This drug is not a home run,” Dr. Brad S. Kahl, a lymphoma specialist at the University of Wisconsin, said during a meeting of an advisory committee to the F.D.A. on Sept. 2. “It’s not even a double. It’s a single.”
Saying that even a single was helpful, Dr. Kahl was part of a majority on the panel that recommended approval of the drug, 10 to 4.
But after recently learning what Allos planned to charge for Folotyn, Dr. Kahl said he was “disappointed” by the “excessive” price.
“It dampens my enthusiasm for using that drug,” he said. “It creates these huge ethical quandaries about trying a drug that has a modest benefit for the average patient at enormous expense.”
Folotyn is given by a rapid intravenous procedure once a week for six weeks out of every seven. Even to try the drug for the first seven-week cycle to see if it works would cost over $50,000. In the clinical trial, the median duration of use was 70 days, which would cost roughly $70,000 to $80,000. But some patients used the drug for many months.
In a note to clients in October, Joshua Schimmer, an analyst at Leerink Swann, estimated that a typical treatment would last 3.5 months and cost $126,000, or about $36,000 a month.
For investors, a high price is usually a good thing. Mr. Schimmer’s note was entitled “Folotyn Prices High, Reiterate Outperform.” He estimated annual sales of the drug in the United States reaching about $300 million by 2014.
Patient advocacy groups say that while they wish prices were lower, high prices might be needed to encourage companies to develop new drugs.
“It’s a two-edged sword that we have to live with and deal with,” said Louis J. DeGennaro, chief scientific officer of the Leukemia and Lymphoma Society, which has received donations from Allos and other companies. “A peripheral T-cell lymphoma patient,” he said, “at first blush will see this therapy as a very good thing.”
Allos, which is still unprofitable, has lost $350 million since its founding in 1992 and failed to win approval of a previous drug.
“Every dime that goes into the company supports Folotyn,” Mr. Caruso said.
At the time Folotyn was approved in September, stock in Allos briefly peaked above $8.50 but has slipped since then, closing up 16 cents at $6.62, or an increase of nearly 2.5 percent, on Friday.
After the approval, Allos raised $93 million in a secondary stock offering. In the prospectus for that offering, the company said that one of the risks for investors was “the relative price of Folotyn as compared to alternate treatment options.” It said there was a risk it might have to lower the price or offer discounts to successfully market Folotyn.
Like many other companies with high-priced drugs, Allos has established a program to help patients arrange insurance reimbursement. It says it will give the drug free to uninsured patients who cannot pay for it any other way.
And because a patient’s out-of-pocket co-payments alone — Medicare’s is 20 percent — could be thousands of dollars a month for Folotyn, Allos is financing a co-payment assistance program run by the National Organization for Rare Disorders, a patient advocacy group.
While this helps patients, it also helps the company sell more of its drug. If the 20 percent Medicare co-payment is made, then Medicare will pay the other 80 percent of the drug’s price — or about $24,000 a month.
mfg ipollit
Antwort auf Beitrag Nr.: 38.679.580 von ipollit am 06.01.10 22:31:19nochmals zu mologen: die kursentwicklung nach der bekanntgabe der erfolgreich plazierten ke ist beeindruckend.
ich bin bei inlands biotechwerten bei mologen wilex und 4 sc investiert.
ich bin bei inlands biotechwerten bei mologen wilex und 4 sc investiert.
Antwort auf Beitrag Nr.: 38.800.950 von pokemon am 24.01.10 20:01:42
Kurzfristige Kursentwicklungen sind irrelevant. Bei Biotechs schon gar.
Wie bist Du auf Mologen gekommen? Hast Du bspw. eine Interpretation folgender Aussage:
"Die im Rahmen der aktuellen klinischen Studie der Phase 1b durchgeführte vorläufige Bewertung der Wirksamkeit sowie das positive Sicherheitsprofil lassen das hohe Potenzial von MGN1703 erkennen. Selbst bei Patienten mit weit fortgeschrittenen metastasierten Tumor-Erkrankungen, wie sie für die Phase 1b Studie ausgewählt wurden, konnte zumindest eine Stabilisierung der Erkrankung erreicht werden. Insgesamt wurden 15 Patienten in verschiedenen Dosierungsgruppen über einen Zeitraum von 6 Wochen mit MGN1703 behandelt.
Bei sechs Patienten wurde nach Abschluss der 6-wöchigen Therapie ein stabiler Erkrankungszustand festgestellt. Zudem zeigten bislang zwei Patienten dieser sechs Patienten auch nach 12 Wochen Behandlungsdauer einen stabilen Krankheitsverlauf."
Ich kann das nicht interpretieren, weil ich ein Laie bin, aber es liest sich zunächst nicht so wahnsinnig aufregend. Eine stable disease von 40% nach 6 Wochen bzw. 13% nach 12 Wochen dürfte doch mehr oder weniger im Rahmen des "normalen" Krankheitsverlaufs liegen. Das sage ich jetzt allerdings mit ganz großem Vorbehalt, weil mir da die wirklichen Kenntnisse fehlen.
Auf die Schnelle war bspw. das zu finden:
"Beim ersten planmäßigen Arztbesuch (Woche 8) betrug die
progressionsfreie Überlebensrate 45,5% bei Vectibix plus BSC (BSC = Best Supportive Care, ohne Chemotherapie) und 24,6% bei alleiniger BSC; ein Unterschied von 20,9% (95% KI: 12,4; 29,4). Es wurde kein Unterschied im Gesamtüberleben
beobachtet."
oder das:
"Eine retrospektive Analyse zeigte, dass das progressionsfreie Überleben bei Patienten mit nicht-mutiertem KRAS-Gen unter Panitumumab durchschnittlich bei 12,3 Wochen lag, im Vergleich zu 7,3 Wochen bei alleiniger BSC. Patienten mit KRAS-Mutationen hatten keinen Vorteil von der Antikörpertherapie."
Vectibix gilt offensichtlich nicht gerade als Erfolg, hat aber bessere Daten als der Kandidat von Mologen gezeigt. PFS nach 12 Wochen lag bspw. bei über 40%.
Siehe dazu hier auf Seite 10:
http://www.ema.europa.eu/humandocs/PDFs/EPAR/vectibix/H-741-…
Jetzt will ich mal die Daten dieser 15-Patienten-1b-Studie nicht überbewerten. Allein da Prozente auszurechnen ist schon etwas komisch. Aber so richtig positiv liest sich das für mich nicht.
Oder sehe ich das falsch, was sehr gut sein kann? Dann bitte ich um Richtigstellung.
Kurzfristige Kursentwicklungen sind irrelevant. Bei Biotechs schon gar.
Wie bist Du auf Mologen gekommen? Hast Du bspw. eine Interpretation folgender Aussage:
"Die im Rahmen der aktuellen klinischen Studie der Phase 1b durchgeführte vorläufige Bewertung der Wirksamkeit sowie das positive Sicherheitsprofil lassen das hohe Potenzial von MGN1703 erkennen. Selbst bei Patienten mit weit fortgeschrittenen metastasierten Tumor-Erkrankungen, wie sie für die Phase 1b Studie ausgewählt wurden, konnte zumindest eine Stabilisierung der Erkrankung erreicht werden. Insgesamt wurden 15 Patienten in verschiedenen Dosierungsgruppen über einen Zeitraum von 6 Wochen mit MGN1703 behandelt.
Bei sechs Patienten wurde nach Abschluss der 6-wöchigen Therapie ein stabiler Erkrankungszustand festgestellt. Zudem zeigten bislang zwei Patienten dieser sechs Patienten auch nach 12 Wochen Behandlungsdauer einen stabilen Krankheitsverlauf."
Ich kann das nicht interpretieren, weil ich ein Laie bin, aber es liest sich zunächst nicht so wahnsinnig aufregend. Eine stable disease von 40% nach 6 Wochen bzw. 13% nach 12 Wochen dürfte doch mehr oder weniger im Rahmen des "normalen" Krankheitsverlaufs liegen. Das sage ich jetzt allerdings mit ganz großem Vorbehalt, weil mir da die wirklichen Kenntnisse fehlen.
Auf die Schnelle war bspw. das zu finden:
"Beim ersten planmäßigen Arztbesuch (Woche 8) betrug die
progressionsfreie Überlebensrate 45,5% bei Vectibix plus BSC (BSC = Best Supportive Care, ohne Chemotherapie) und 24,6% bei alleiniger BSC; ein Unterschied von 20,9% (95% KI: 12,4; 29,4). Es wurde kein Unterschied im Gesamtüberleben
beobachtet."
oder das:
"Eine retrospektive Analyse zeigte, dass das progressionsfreie Überleben bei Patienten mit nicht-mutiertem KRAS-Gen unter Panitumumab durchschnittlich bei 12,3 Wochen lag, im Vergleich zu 7,3 Wochen bei alleiniger BSC. Patienten mit KRAS-Mutationen hatten keinen Vorteil von der Antikörpertherapie."
Vectibix gilt offensichtlich nicht gerade als Erfolg, hat aber bessere Daten als der Kandidat von Mologen gezeigt. PFS nach 12 Wochen lag bspw. bei über 40%.
Siehe dazu hier auf Seite 10:
http://www.ema.europa.eu/humandocs/PDFs/EPAR/vectibix/H-741-…
Jetzt will ich mal die Daten dieser 15-Patienten-1b-Studie nicht überbewerten. Allein da Prozente auszurechnen ist schon etwas komisch. Aber so richtig positiv liest sich das für mich nicht.
Oder sehe ich das falsch, was sehr gut sein kann? Dann bitte ich um Richtigstellung.
ISIS - Barclays assessment:
ISIS Pharmaceuticals
Key Issues
"Mipomersen opportunity in homozygous FH - positive phase III data establish statistically significant 25% LDL reduction in most difficult to treat population failing statins and multi-drug combinations. Liver enzyme elevations without bilirubin rise is acceptable in very high risk population. Unmet need should support approval. Actual clinical use by lipidologists and reimbursement by payers unlikely to be constrained
by genotype and more likely to follow phenotype and actual CV risk.
Additional phase III data in heterozygous FH and high risk populations should be even better - LDL reduction with mipomersen in phase II was greatest in patients with lower baseline LDL levels. Responsiveness of HeFH and high risk patients to ApoB100 inhibition is greater than for HoFH patients and expect more significant LDL reductions in upcoming phase III trials
Theoretical concerns regarding fatty liver with mipomersen appear overdone - initial liver imaging study suggests no excess
accumulation of liver fat. Preclinical primate models suggest actual regression of fatty liver. Alternate pathways for lipid oxidation activated along with ApoB100 inhibition. No Hy's law criteria met in very sick HoFH patients in phase III. Further liver imaging data expected in 2010
and should better establish liver safety profile.
Antisense Platform Supports Substantial Pipeline opportunity – Complete ownership of antisense platform and royalty rights to RNAi therapeutics should support platform beyond $1.1B purchase price for 2nd tier RNAi player SIRNA by Merck. Phase II PTP-1B inhibitor ISIS- 113715 for type II diabetes has demonstrated statistically significant reductions in fasting blood sugar (FBS), weekly average fasting blood
sugar, glycated albumin and modest weight loss over a short period of 13 weeks with 6 weeks required to achieve steady state drug levels.
Phase III clusterin inhibitor OGX-011 has demonstrated a clinically meaningful 6.9 month improvement in overall survival (OS) in a randomized phase II study in castrate resistant prostate cancer (CRPC) with 39% reduction in risk of death (HR=0.61). Feedback on OGX- 011 from prostate CA experts has been highly positive. Phase II VLA-4 inhibitor ATL1102 has demonstrated Tysabri-like efficacy in RRMS.
Survivin inhibitor LY2181308 could yield phase II data in leukemia for partner LLY in 2010."
The stock has not recovered from the drop when the hoFH results were released, but that should change when heFH results are reported in 1st qtr.
Grüße cristrader
ISIS Pharmaceuticals
Key Issues
"Mipomersen opportunity in homozygous FH - positive phase III data establish statistically significant 25% LDL reduction in most difficult to treat population failing statins and multi-drug combinations. Liver enzyme elevations without bilirubin rise is acceptable in very high risk population. Unmet need should support approval. Actual clinical use by lipidologists and reimbursement by payers unlikely to be constrained
by genotype and more likely to follow phenotype and actual CV risk.
Additional phase III data in heterozygous FH and high risk populations should be even better - LDL reduction with mipomersen in phase II was greatest in patients with lower baseline LDL levels. Responsiveness of HeFH and high risk patients to ApoB100 inhibition is greater than for HoFH patients and expect more significant LDL reductions in upcoming phase III trials
Theoretical concerns regarding fatty liver with mipomersen appear overdone - initial liver imaging study suggests no excess
accumulation of liver fat. Preclinical primate models suggest actual regression of fatty liver. Alternate pathways for lipid oxidation activated along with ApoB100 inhibition. No Hy's law criteria met in very sick HoFH patients in phase III. Further liver imaging data expected in 2010
and should better establish liver safety profile.
Antisense Platform Supports Substantial Pipeline opportunity – Complete ownership of antisense platform and royalty rights to RNAi therapeutics should support platform beyond $1.1B purchase price for 2nd tier RNAi player SIRNA by Merck. Phase II PTP-1B inhibitor ISIS- 113715 for type II diabetes has demonstrated statistically significant reductions in fasting blood sugar (FBS), weekly average fasting blood
sugar, glycated albumin and modest weight loss over a short period of 13 weeks with 6 weeks required to achieve steady state drug levels.
Phase III clusterin inhibitor OGX-011 has demonstrated a clinically meaningful 6.9 month improvement in overall survival (OS) in a randomized phase II study in castrate resistant prostate cancer (CRPC) with 39% reduction in risk of death (HR=0.61). Feedback on OGX- 011 from prostate CA experts has been highly positive. Phase II VLA-4 inhibitor ATL1102 has demonstrated Tysabri-like efficacy in RRMS.
Survivin inhibitor LY2181308 could yield phase II data in leukemia for partner LLY in 2010."
The stock has not recovered from the drop when the hoFH results were released, but that should change when heFH results are reported in 1st qtr.
Grüße cristrader
Antwort auf Beitrag Nr.: 38.801.191 von SLGramann am 24.01.10 21:06:57wenn ich das richtig verstanden habe, waren die patienten bei der mologen studie austherapiert, d.h. sämtliche bestehenden möglichkeiten der behandlung halfen nicht mehr. da sind diese ergebnisse doch ausser gewöhnlich gut. oder? wird das bestätigt in der phase II, so hat das medikament großes potential? habe ich da einen denkfehler?
war das bei den erwähnten bsp. auch solche patienten, für die es keine behandlungsmöglichkeit mehr gab?
war das bei den erwähnten bsp. auch solche patienten, für die es keine behandlungsmöglichkeit mehr gab?
Vectibix ist als Monotherapie indiziert zur Behandlung des metastasierten, EGFR-exprimierenden, kolorektalen Karzinoms mit nicht-mutiertem (Wildtyp-) KRAS-Gen bei Patienten, bei denen Fluoropyrimidin-, Oxaliplatin- und Irinotecan-haltige Chemotherapieregime versagt haben.
In der bescheibung wird auch von massiven nebenwirkungen gewarnt. das mgn1703 ist laut ad hoc nebenwirkungsfrei.
beide produkte sind aber auch ganz unterschiedlich im ansatz.
im gegensatz zu dem antikörper ist das mgn1703 ein dna basierter wirkstoff im bereich der tlr9 therapeutik.
siehe dazu mologen.com
In der bescheibung wird auch von massiven nebenwirkungen gewarnt. das mgn1703 ist laut ad hoc nebenwirkungsfrei.
beide produkte sind aber auch ganz unterschiedlich im ansatz.
im gegensatz zu dem antikörper ist das mgn1703 ein dna basierter wirkstoff im bereich der tlr9 therapeutik.
siehe dazu mologen.com
Antwort auf Beitrag Nr.: 38.801.475 von pokemon am 24.01.10 22:42:42"mgn1703 ist laut ad hoc nebenwirkungsfrei"
es gibt kein Medikament ohne Nebenwirkungen... selbst Placebos haben Nebenwirkungen. Die Nebenwirkung kommt automatisch mit der Wirkung... keine Nebenwirkung bedeutet eigentlich auch keine Wirkung. Aber vielleicht meinst du keine gravierende Nebenwirkung.
mfg ipollit
es gibt kein Medikament ohne Nebenwirkungen... selbst Placebos haben Nebenwirkungen. Die Nebenwirkung kommt automatisch mit der Wirkung... keine Nebenwirkung bedeutet eigentlich auch keine Wirkung. Aber vielleicht meinst du keine gravierende Nebenwirkung.
mfg ipollit
Antwort auf Beitrag Nr.: 38.801.475 von pokemon am 24.01.10 22:42:42
beide produkte sind aber auch ganz unterschiedlich im ansatz.
Ich weiß. Mir ging es um die Frage, ob die bekannt gegebenen stable disease-Daten wirklich ein brauchbarer Hinweis auf Wirksamkeit sind. Ich habe da Zweifel, kann es aber letztlich nicht beurteilen. Deshalb war die Frage, ob Du zu diesem Punkt Einschätzungen hast oder kennst, die nicht vom Unternehmen stammen.
Aber immerhin wird es ja in ca. einem Jahr bereits Zwischendaten der P II geben. Ich bin sehr skeptisch, Du bist optimistisch. Ahnung haben wir - unterstelle ich mal - beide nicht wirklich. Warten wirs einfach bis dahin ab.
beide produkte sind aber auch ganz unterschiedlich im ansatz.
Ich weiß. Mir ging es um die Frage, ob die bekannt gegebenen stable disease-Daten wirklich ein brauchbarer Hinweis auf Wirksamkeit sind. Ich habe da Zweifel, kann es aber letztlich nicht beurteilen. Deshalb war die Frage, ob Du zu diesem Punkt Einschätzungen hast oder kennst, die nicht vom Unternehmen stammen.
Aber immerhin wird es ja in ca. einem Jahr bereits Zwischendaten der P II geben. Ich bin sehr skeptisch, Du bist optimistisch. Ahnung haben wir - unterstelle ich mal - beide nicht wirklich. Warten wirs einfach bis dahin ab.
Antwort auf Beitrag Nr.: 38.801.558 von ipollit am 24.01.10 23:02:53ja
seit langem habe ich NeuroSearch auf meiner Watchliste... jetzt versuche ich es mal mit einer kleinen Position. REGN habe ich dagegen leicht reduziert.
Die NeuroSearch Position ist recht spekulativ und riskant... in Kürze stehen die ersten PIII-Daten zu Huntexil (ACR16) gegen Huntington an. Huntexil sieht eigentlich recht vielversprechend aus, weshalb ich mal auf positive Daten setze... im negativen Fall ist der Schaden einigermaßen begrenzt. Bisher gibt es nichts derartiges bei Huntington, eine Erkrankung, die schwerwiegend und eher selten ist mit jeweils ca. 35.000 Patienten in Europa und den USA. Die PII sah gut aus und das Mittel basiert immerhin auf der Arbeit des Nobelpreis-Trägers Arvid Carlsson (aus der Übernahme von Carlsson Research).


http://www.jyskebank.dk/_Jb/ASP/Apps/redirect.asp?ShadowID=2…
Huntexil bodes well
We expect that NeuroSearch will publish results from the MermaiHD and HART studies in the course of 2010, and the first results from the six-monthly blinded treatment process under the MermaiHD study are anticipated in early 2010. The results from the HART study are expected to be accessible around mid-2010 and will be followed by the results from the six-month extension study to MermaiHD. In previous clinical studies, Huntexil has shown promising effects against a number of serious symptoms associated with Huntington's disease. Particularly, improvement has been seen in connection with patients’ ability to perform voluntary movements and of their way of walking so they do not experience as many incidents of falling, resulting in improved functionality and quality of life. In phase II, Huntexil managed to demonstrate significant improvement of the voluntary movement by -2.8 points measured as a change from the baseline in modified Motor Score (mMS) against -0.8 point for the placebo group. Also, in phase II, Huntexil demonstrated a significant effect on soft parameters such as depression and anxiety. If NeuroSearch is able to repeat the same effect in the MermaiHD study, the primary endpoint will be reached, and this will pave the way for a product launch in 2011.
...
The disease occurs at a rate of about one in every 10,000 of European descent, and therefore it is estimated that the total number of patients with Huntington’s disease in North America amounts to about 34,000 and in Europe to about 37,000. We estimate that about 33.3% of these will experience a mild degree of the disease, 33.3% a moderate degree and 33.3% a severe degree of the disease, and therefore they will be difficult to treat. We assess that only patients with a mild and moderate degree of the disease will benefit from treatment with Huntexil, i.e. two out of three patients. This results in a total target group of 47,333 patients of which 22,668 are in North America and 24,665 in Europe.
...
mfg ipollit
Die NeuroSearch Position ist recht spekulativ und riskant... in Kürze stehen die ersten PIII-Daten zu Huntexil (ACR16) gegen Huntington an. Huntexil sieht eigentlich recht vielversprechend aus, weshalb ich mal auf positive Daten setze... im negativen Fall ist der Schaden einigermaßen begrenzt. Bisher gibt es nichts derartiges bei Huntington, eine Erkrankung, die schwerwiegend und eher selten ist mit jeweils ca. 35.000 Patienten in Europa und den USA. Die PII sah gut aus und das Mittel basiert immerhin auf der Arbeit des Nobelpreis-Trägers Arvid Carlsson (aus der Übernahme von Carlsson Research).
http://www.jyskebank.dk/_Jb/ASP/Apps/redirect.asp?ShadowID=2…
Huntexil bodes well
We expect that NeuroSearch will publish results from the MermaiHD and HART studies in the course of 2010, and the first results from the six-monthly blinded treatment process under the MermaiHD study are anticipated in early 2010. The results from the HART study are expected to be accessible around mid-2010 and will be followed by the results from the six-month extension study to MermaiHD. In previous clinical studies, Huntexil has shown promising effects against a number of serious symptoms associated with Huntington's disease. Particularly, improvement has been seen in connection with patients’ ability to perform voluntary movements and of their way of walking so they do not experience as many incidents of falling, resulting in improved functionality and quality of life. In phase II, Huntexil managed to demonstrate significant improvement of the voluntary movement by -2.8 points measured as a change from the baseline in modified Motor Score (mMS) against -0.8 point for the placebo group. Also, in phase II, Huntexil demonstrated a significant effect on soft parameters such as depression and anxiety. If NeuroSearch is able to repeat the same effect in the MermaiHD study, the primary endpoint will be reached, and this will pave the way for a product launch in 2011.
...
The disease occurs at a rate of about one in every 10,000 of European descent, and therefore it is estimated that the total number of patients with Huntington’s disease in North America amounts to about 34,000 and in Europe to about 37,000. We estimate that about 33.3% of these will experience a mild degree of the disease, 33.3% a moderate degree and 33.3% a severe degree of the disease, and therefore they will be difficult to treat. We assess that only patients with a mild and moderate degree of the disease will benefit from treatment with Huntexil, i.e. two out of three patients. This results in a total target group of 47,333 patients of which 22,668 are in North America and 24,665 in Europe.
...
mfg ipollit
Antwort auf Beitrag Nr.: 38.800.415 von ipollit am 24.01.10 17:39:17würde an deiner stelle schnell mal die medigene gegen mologen umstauschen.
medigene wird den tec dax bald verlassen müssen, mologen wird bald im tech dax sein dürfen!
SCHAU DIR DEN TREND AN!!
schau dir nur mal beide charts an!
1 jahr und
5 jahre
mologen war vor 5 jahren bei 3! jetzt 9!
medigene war vor 5 jahren bei über 11! und jetzt bei unter 4!
mologen war vor einem jahr bei 6 jetzt bei 9!!!!!!
medigene war vor einem jahr bei 4,2 jetzt bei 3,8!!!!!!
Börsenkapitalisierung bei Mologen knapp 100 mio
Börsenwert bei Medigene knapp 130 mio
was meinst du?
medigene wird den tec dax bald verlassen müssen, mologen wird bald im tech dax sein dürfen!
SCHAU DIR DEN TREND AN!!
schau dir nur mal beide charts an!
1 jahr und
5 jahre
mologen war vor 5 jahren bei 3! jetzt 9!
medigene war vor 5 jahren bei über 11! und jetzt bei unter 4!
mologen war vor einem jahr bei 6 jetzt bei 9!!!!!!
medigene war vor einem jahr bei 4,2 jetzt bei 3,8!!!!!!
Börsenkapitalisierung bei Mologen knapp 100 mio
Börsenwert bei Medigene knapp 130 mio
was meinst du?
Antwort auf Beitrag Nr.: 38.818.463 von ipollit am 26.01.10 23:14:03http://www.investindk.com/visNyhed.asp?artikelID=22787
NeuroSearch prepares to manufacture and sell its first drug
(2009.10.21)
Danish biopharmaceutical company NeuroSearch is changing its business to handle production and sales of its first drug Huntexil
Danish biopharmaceutical company NeuroSearch is changing its business to handle production and sales of its first drug Huntexil, which is currently in phase III development, reports financial daily newspaper Børsen. Huntexil is for the treatment of Huntington's disease, a neurodegenerative genetic disorder that affects muscle coordination and cognitive functions. NeuroSearch expects the final approval of the drug to be given in about one year.
Carsten Lønborg Madsen, analyst at investment bank Carnegie comments: "It is a very interesting drug which has the potential to make a transformation of NeuroSearch, but one should not underestimate the risk in late stage studies of diseases in the central nervous system. There are still some obstacles that need to be cleared."
CEO of NeuroSearch Flemming Pedersen says that forecasts from analysts indicate the market value of Huntexil to be around DKK 5bn (USD 1bn). The company will need between 30 and 50 sales representatives to cover both the European and the US markets. Pedersen expects sales of the drug to quickly reach its maximum potential since doctors and patients are showing great interest in the drug.
Huntexil was adopted into the pipeline a few years ago in association with a company acquisition. When the final stages have been completed, NeuroSearch will have invested approx. DKK 500m (USD 100m) in the drug.
Headquartered in Ballerup on the outskirts of Copenhagen, NeuroSearch’s core business covers the development of novel drugs, based on a discovery platform focusing on ion channels and CNS disorders. A substantial share of the activities is partner-financed through strategic alliances with Eli Lilly and Company, Janssen Pharmaceutica and GlaxoSmithKline (GSK), and a license collaboration with Abbott. The company is listed on Nasdaq OMX Copenhagen and has approximately 220 employees.
mfg ipollit
NeuroSearch prepares to manufacture and sell its first drug
(2009.10.21)
Danish biopharmaceutical company NeuroSearch is changing its business to handle production and sales of its first drug Huntexil
Danish biopharmaceutical company NeuroSearch is changing its business to handle production and sales of its first drug Huntexil, which is currently in phase III development, reports financial daily newspaper Børsen. Huntexil is for the treatment of Huntington's disease, a neurodegenerative genetic disorder that affects muscle coordination and cognitive functions. NeuroSearch expects the final approval of the drug to be given in about one year.
Carsten Lønborg Madsen, analyst at investment bank Carnegie comments: "It is a very interesting drug which has the potential to make a transformation of NeuroSearch, but one should not underestimate the risk in late stage studies of diseases in the central nervous system. There are still some obstacles that need to be cleared."
CEO of NeuroSearch Flemming Pedersen says that forecasts from analysts indicate the market value of Huntexil to be around DKK 5bn (USD 1bn). The company will need between 30 and 50 sales representatives to cover both the European and the US markets. Pedersen expects sales of the drug to quickly reach its maximum potential since doctors and patients are showing great interest in the drug.
Huntexil was adopted into the pipeline a few years ago in association with a company acquisition. When the final stages have been completed, NeuroSearch will have invested approx. DKK 500m (USD 100m) in the drug.
Headquartered in Ballerup on the outskirts of Copenhagen, NeuroSearch’s core business covers the development of novel drugs, based on a discovery platform focusing on ion channels and CNS disorders. A substantial share of the activities is partner-financed through strategic alliances with Eli Lilly and Company, Janssen Pharmaceutica and GlaxoSmithKline (GSK), and a license collaboration with Abbott. The company is listed on Nasdaq OMX Copenhagen and has approximately 220 employees.
mfg ipollit
Hallo, hast Du eine Meinung(egal welche) zu Repros Therapeut.?
Antwort auf Beitrag Nr.: 38.826.700 von MrRipley am 27.01.10 21:36:48Repros? Sorry, kenne ich nicht. Die sind zwar heute ziemlich gestiegen, aber von der MK mit 23 Mio und dem Cash mit 2 Mio USD (wenn das richtig ist) sehr klein. Bei dieser Größe kann es natürlich spekulativ zu großen Kursbewegungen kommen, aber dazu braucht es schon recht viel Glück, denke ich. Genauso gut können da in absehbarer Zeit die Lichter ausgehen. Aber im Prinzip kann ich nichts dazu sagen, da ich den Wert wie gesagt nicht kenne.
mfg ipollit
mfg ipollit
die 26 Neuzulassungen der FDA in 2009...
http://www.fiercebiotech.com/slideshows/fda-approvals-2009
1. Savella - Forest Labs
2. Uloric - Takeda
3. Afinitor - Novartis
4. Coartem - Novartis
5. Ulesfia - Sciele Pharma
6. Simponi - Johnson & Johnson
7. Dysport - Ipsen, Medicis
8. Fanapt - Vanda Pharma
9. Samsca - Otsuka Pharma
10. Besivance - Bausch & Lomb
11. Ilaris - Novartis
12. Multaq - Sanofi-Aventis
13. Effient - Eli Lilly, Daiichi Sankyo
14. Onglyza - AstraZeneca, BMS
15. Livalo - Kowa Research
16. Saphris - Merck's Organon USA
17. Extavia - Novartis
18. Sabril - Lundbeck
19. Bepreve - Ista Pharma
20. Vibativ - Theravance, Astellas
21. Folotyn - Allos Therapeutics
22. Stelara - Johnson & Johnson
23. Votrient - GSK
24. Arzerra - GSK
25. Istodax - Gloucester Pharma
26. Kalbitor - Dyax Corp
zwei davon sind hier direkt relevant... Folotyn von Allos und Arzerra von Genmab

Drug: Folotyn
Indication: Relapsed peripheral T-cell lymphoma
Company: Allos Therapeutics
Date Approved: Sept. 24
Scoop: Folotyn is the first and only drug approved for PTCL, a rare form of blood cancer that has a poor prognosis and a high relapse rate. An expert panel backed the treatment despite questions about the drugmaker's clinical data and the safety profile. Allos has faced some criticism of the drug due to it's $30,000-per-month price tag, but the company says it's priced similarly to other rare disease drugs and adds that patients only receive Folotyn for a few months.

Drug: Arzerra
Indication: chronic lymphocytic leukemia
Company: GlaxoSmithKline
Date Approved: Oct. 26
Scoop: Arzerra is designed to attach to the CD20 molecule found on B cells, which are vulnerable to leukemia. It flags the cells for destruction, much like Rituxan does, though it uses a different mechanism. Arzerra is also undergoing clinical trials for rheumatoid arthritis. It was also in testing for non-Hodgkin's lymphoma, but produced underwhelming results for that indication. GSK licensed the drug, also known as ofatumumab or HuMax CD-20, in 2007 for $2.1 billion.
und zwei weitere sind indirekt relevant... Ilaris wurde zusammen mit REGN entwickelt... REGN erhält Royalties. Und CBST besitzt die Rechte zur Entwicklkung von Kalbitor gegen Blutungen.

Drug: Ilaris (canakinumab)
Indication: CAPS
Company: Novartis
Approval Date: June 17
Scoop: The FDA approved Ilaris for the treatment of children and adults with cryopyrin-associated periodic syndrome (CAPS), which includes a number of rare but life-long auto-inflammatory disorders, last June. Only 300 cases of CAPS have been diagnosed in the U.S., but there are potentially many more undiagnosed patients with the condition due to poor disease recognition. The approval marks one of the first solid commercial successes for CEO Daniel Vasella's long quest to overhaul the pharma company's pipeline.

Drug: Kalbitor (ecallantide)
Indication: Sudden attacks of hereditary angioedema
Companies: Dyax
Approval Date: Nov. 27
Scoop: Last November the FDA approved Kalbitor for patients with hereditary angioedema, a rare and often lethal genetic disease characterized by pain and swelling in the face, lungs and upper airway. The approval marked a big win for Dyax, which has gone $335 million in the red as it pursued clinical development work. Kalbitor will be the 14-year-old biotech company's first marketed therapy. Dyax also licenses out its phage display technology to researchers.
mfg ipollit
http://www.fiercebiotech.com/slideshows/fda-approvals-2009
1. Savella - Forest Labs
2. Uloric - Takeda
3. Afinitor - Novartis
4. Coartem - Novartis
5. Ulesfia - Sciele Pharma
6. Simponi - Johnson & Johnson
7. Dysport - Ipsen, Medicis
8. Fanapt - Vanda Pharma
9. Samsca - Otsuka Pharma
10. Besivance - Bausch & Lomb
11. Ilaris - Novartis
12. Multaq - Sanofi-Aventis
13. Effient - Eli Lilly, Daiichi Sankyo
14. Onglyza - AstraZeneca, BMS
15. Livalo - Kowa Research
16. Saphris - Merck's Organon USA
17. Extavia - Novartis
18. Sabril - Lundbeck
19. Bepreve - Ista Pharma
20. Vibativ - Theravance, Astellas
21. Folotyn - Allos Therapeutics
22. Stelara - Johnson & Johnson
23. Votrient - GSK
24. Arzerra - GSK
25. Istodax - Gloucester Pharma
26. Kalbitor - Dyax Corp
zwei davon sind hier direkt relevant... Folotyn von Allos und Arzerra von Genmab
Drug: Folotyn
Indication: Relapsed peripheral T-cell lymphoma
Company: Allos Therapeutics
Date Approved: Sept. 24
Scoop: Folotyn is the first and only drug approved for PTCL, a rare form of blood cancer that has a poor prognosis and a high relapse rate. An expert panel backed the treatment despite questions about the drugmaker's clinical data and the safety profile. Allos has faced some criticism of the drug due to it's $30,000-per-month price tag, but the company says it's priced similarly to other rare disease drugs and adds that patients only receive Folotyn for a few months.
Drug: Arzerra
Indication: chronic lymphocytic leukemia
Company: GlaxoSmithKline
Date Approved: Oct. 26
Scoop: Arzerra is designed to attach to the CD20 molecule found on B cells, which are vulnerable to leukemia. It flags the cells for destruction, much like Rituxan does, though it uses a different mechanism. Arzerra is also undergoing clinical trials for rheumatoid arthritis. It was also in testing for non-Hodgkin's lymphoma, but produced underwhelming results for that indication. GSK licensed the drug, also known as ofatumumab or HuMax CD-20, in 2007 for $2.1 billion.
und zwei weitere sind indirekt relevant... Ilaris wurde zusammen mit REGN entwickelt... REGN erhält Royalties. Und CBST besitzt die Rechte zur Entwicklkung von Kalbitor gegen Blutungen.
Drug: Ilaris (canakinumab)
Indication: CAPS
Company: Novartis
Approval Date: June 17
Scoop: The FDA approved Ilaris for the treatment of children and adults with cryopyrin-associated periodic syndrome (CAPS), which includes a number of rare but life-long auto-inflammatory disorders, last June. Only 300 cases of CAPS have been diagnosed in the U.S., but there are potentially many more undiagnosed patients with the condition due to poor disease recognition. The approval marks one of the first solid commercial successes for CEO Daniel Vasella's long quest to overhaul the pharma company's pipeline.
Drug: Kalbitor (ecallantide)
Indication: Sudden attacks of hereditary angioedema
Companies: Dyax
Approval Date: Nov. 27
Scoop: Last November the FDA approved Kalbitor for patients with hereditary angioedema, a rare and often lethal genetic disease characterized by pain and swelling in the face, lungs and upper airway. The approval marked a big win for Dyax, which has gone $335 million in the red as it pursued clinical development work. Kalbitor will be the 14-year-old biotech company's first marketed therapy. Dyax also licenses out its phage display technology to researchers.
mfg ipollit
eine neue Studie zu Micromet als pdf (mit Verkaufsempfehlung, die aber schlecht begründet ist imho):
http://www.zacks.com/ZER/zer_get_pdf.php?r=Z566702&t=MITI&id…
http://www.zacks.com/ZER/zer_get_pdf.php?r=Z566702&t=MITI&id…
ein paar infos rund um den SGEN/Millenium-deal für brentuximab-v.
Millennium, In a New Role, Flexes Global Muscle to Cut Deal With Seattle Genetics
http://www.xconomy.com/boston/2010/01/26/millennium-in-a-new…
Millennium, In a New Role, Flexes Global Muscle to Cut Deal With Seattle Genetics
http://www.xconomy.com/boston/2010/01/26/millennium-in-a-new…
ARNA... trotz der nur schwachen Einzel-Wirkung könnte der Pluspunkt von Lorcaserin das niedrige Risiko von Nebenwirkungen und die relativ große Datenmenge aus der PIII sein. Bei einer Zulassung dürfte hier mittelfristig auch die klassische Fen-Phen Kombi - also Locaserin+Phentermine ausprobiert werden, die vielleicht dann auch zu einer höheren Effektivität führt.
http://www.signonsandiego.com/news/2010/jan/31/dash-for-diet…
Dash for diet drug
If the FDA approves, three biotech firms — two in San Diego — say they will change how obesity is treated
By Thomas Kupper, UNION-TRIBUNE STAFF WRITER
Sunday, January 31, 2010 at 1:04 a.m.
For decades, the pharmaceutical industry has chased an elusive target, a drug that will make you lose weight without putting your health at risk.
For a brief period in the 1990s, the two-drug combo known as Fen-phen achieved blockbuster status — before potentially fatal side effects knocked it off the market. More recently, the risk-benefit profile of the leading diet drug Meridia has come under scrutiny, with European authorities deciding to ban it this month.
Now, three California biotechnology companies are trying to convince the U.S. Food and Drug Administration that they’ve found drugs that are effective and safe. Not only that, they say the drugs can help with conditions that often accompany obesity, such as high cholesterol or blood pressure.
If the companies are right, it could bring about a sweeping change in the treatment of obesity while also bringing in blockbuster profits for the companies — San Diego’s Arena Pharmaceuticals and Orexigen Therapeutics, along with Vivus in the San Francisco Bay Area.
“The stage is set for obesity, to finally make some paradigm changes in the way the condition is thought about and treated,” said Michael Narachi, chief executive of Orexigen.
Narachi pointed to new drugs that have dramatically improved the treatment of conditions like high cholesterol or hypertension while creating multibillion-dollar markets for drug companies. Obesity, he said, offers the same kind of potential for advance.
Nothing will alter the traditional prescription for weight loss — diet and exercise. But the companies say their drugs could give physicians an additional tool to treat excess weight as a medical condition with potentially serious consequences.
All three announced study data last year that showed significant numbers of patients losing 5 percent or more of their weight. Arena and Vivus have since filed with the FDA for clearance to sell their drugs, while Orexigen says it will do so in the coming months.
Donna Ryan, a researcher who leads the Obesity Society in Maryland, said the data suggest all three are effective enough to win the FDA’s blessing when it rules late this year or in 2011. Ryan said safety, the other factor the agency looks at, is harder to judge.
While there are a few drugs doctors prescribe for weight loss, Ryan said it is important to have new options that might prove more effective.
“It’s a great thing,” Ryan said of the potential for multiple drugs to win approval. “In terms of what tools physicians have to manage weight loss, we haven’t gotten any drugs out to help us in a decade.”
Of the three drugs, the only truly new one comes from Arena. Its approach was to design a drug, known as lorcaserin, that would hit the same molecular “receptor” the fenfluramine component of Fen-phen targeted for weight loss, without hitting other receptors that caused side effects.
The other two companies have combination drugs. Orexigen’s Contrave combines the antidepressant Wellbutrin, known generically as bupropion, with a sustained-release version of naltrexone, which is used to treat alcoholism and other addictions.
Vivus’ Qnexa combines a generic form of the appetite suppressant phentermine, the other component of Fen-phen, with the anticonvulsant drug topiramate.
Other companies, including San Diego’s Amylin Pharmaceuticals, have weight-loss drugs in earlier stages of development. Orexigen also has a second drug in development.
With two-thirds of Americans qualifying as overweight, according to the Obesity Society, there appears to be room for multiple drugs in the market.
“There’s room for 13, or 30, new drugs,” Arena chief executive Jack Lief said. “I’d like to see all three of them be approved, because I think that will open up the market very significantly. Patients will go to their physicians and want to do something about their weight.”
Ted Tenthoff, a biotech analyst who follows Arena for investment bank Piper Jaffray & Co., said the drugs could end up addressing distinct segments of the market.
Tenthoff said Vivus’ Qnexa, which showed the most dramatic weight loss in trials, would likely would end up being prescribed for the heaviest patients — though the company also has lower-dose formulations it says will address a broad range of patients.
Tenthoff said a better safety profile could provide a niche for Arena’s lorcaserin with less-obese patients, despite the drug’s weaker data on weight loss. The company believes this also could make it the drug doctors try first.
Orexigen, whose drug Tenthoff described as falling somewhere between the other two, plays up Contrave’s potential to help patients with other conditions that often accompany obesity. For example, Narachi said one-third of obesity patients also suffer from depression and therefore might want a weight-loss drug that includes an antidepressant.
Many patients also might try more than one approach.
“What typically happens in markets like this is that there’s quite a bit of churn,” Narachi said. “There might be somebody who tries a product and decides to stop, either because it’s not effective for them or because some patients are more sensitive to certain tolerability aspects than others. Or people may lose a certain amount of weight on one product and then switch to another one and see if they can lose a little bit more.”
As the FDA considers whether to approve the drugs, a big question is likely to be safety. Past problems with obesity drugs could be one reason investors have been slow to embrace the three companies’ stocks, all of which trade in single digits.
Inevitably, regulators will think back to the Fen-phen fiasco, which led drugmaker Wyeth to set aside $21 billion to resolve litigation related to Fen-phen’s links to heart damage and lung disease.
Tenthoff said more recent experience with Meridia could make the FDA that much more conservative. Data that linked that drug to strokes, heart attacks and other cardiovascular problems led European regulators to suspend sales of the drug this month while the FDA asked for a stronger warning label.
“These drugs are going to be used in millions of patients,” Tenthoff said. “That is going to be on the mind of the FDA.”
Questions about safety also could make some patients hesitant, even if the FDA approves all three drugs.
The companies point to substantial pent-up demand in the market, given the lack of new weight-loss drugs in recent years. When Fen-phen was on the market, prescriptions tripled in one year to hit 20.6 million in 1996.
Right now, a major activity for all three companies is seeking large pharmaceutical company partners with sales forces to help market the drugs if they get on the market. Narachi said multiple potential product launches around the same time also could help speed acceptance.
“There will be three companies, perhaps with three partners, all trying to convince people that pharmacotherapy approaches should be more heavily considered as a treatment for obesity,” Narachi said.
The Obesity Society’s Ryan said doctors and patients are eager for new tools.
“They are clamoring for aids to help with their patients,” Ryan said. “Just telling people to eat less and exercise more is not going to slow the obesity epidemic.”
mfg ipollit
http://www.signonsandiego.com/news/2010/jan/31/dash-for-diet…
Dash for diet drug
If the FDA approves, three biotech firms — two in San Diego — say they will change how obesity is treated
By Thomas Kupper, UNION-TRIBUNE STAFF WRITER
Sunday, January 31, 2010 at 1:04 a.m.
For decades, the pharmaceutical industry has chased an elusive target, a drug that will make you lose weight without putting your health at risk.
For a brief period in the 1990s, the two-drug combo known as Fen-phen achieved blockbuster status — before potentially fatal side effects knocked it off the market. More recently, the risk-benefit profile of the leading diet drug Meridia has come under scrutiny, with European authorities deciding to ban it this month.
Now, three California biotechnology companies are trying to convince the U.S. Food and Drug Administration that they’ve found drugs that are effective and safe. Not only that, they say the drugs can help with conditions that often accompany obesity, such as high cholesterol or blood pressure.
If the companies are right, it could bring about a sweeping change in the treatment of obesity while also bringing in blockbuster profits for the companies — San Diego’s Arena Pharmaceuticals and Orexigen Therapeutics, along with Vivus in the San Francisco Bay Area.
“The stage is set for obesity, to finally make some paradigm changes in the way the condition is thought about and treated,” said Michael Narachi, chief executive of Orexigen.
Narachi pointed to new drugs that have dramatically improved the treatment of conditions like high cholesterol or hypertension while creating multibillion-dollar markets for drug companies. Obesity, he said, offers the same kind of potential for advance.
Nothing will alter the traditional prescription for weight loss — diet and exercise. But the companies say their drugs could give physicians an additional tool to treat excess weight as a medical condition with potentially serious consequences.
All three announced study data last year that showed significant numbers of patients losing 5 percent or more of their weight. Arena and Vivus have since filed with the FDA for clearance to sell their drugs, while Orexigen says it will do so in the coming months.
Donna Ryan, a researcher who leads the Obesity Society in Maryland, said the data suggest all three are effective enough to win the FDA’s blessing when it rules late this year or in 2011. Ryan said safety, the other factor the agency looks at, is harder to judge.
While there are a few drugs doctors prescribe for weight loss, Ryan said it is important to have new options that might prove more effective.
“It’s a great thing,” Ryan said of the potential for multiple drugs to win approval. “In terms of what tools physicians have to manage weight loss, we haven’t gotten any drugs out to help us in a decade.”
Of the three drugs, the only truly new one comes from Arena. Its approach was to design a drug, known as lorcaserin, that would hit the same molecular “receptor” the fenfluramine component of Fen-phen targeted for weight loss, without hitting other receptors that caused side effects.
The other two companies have combination drugs. Orexigen’s Contrave combines the antidepressant Wellbutrin, known generically as bupropion, with a sustained-release version of naltrexone, which is used to treat alcoholism and other addictions.
Vivus’ Qnexa combines a generic form of the appetite suppressant phentermine, the other component of Fen-phen, with the anticonvulsant drug topiramate.
Other companies, including San Diego’s Amylin Pharmaceuticals, have weight-loss drugs in earlier stages of development. Orexigen also has a second drug in development.
With two-thirds of Americans qualifying as overweight, according to the Obesity Society, there appears to be room for multiple drugs in the market.
“There’s room for 13, or 30, new drugs,” Arena chief executive Jack Lief said. “I’d like to see all three of them be approved, because I think that will open up the market very significantly. Patients will go to their physicians and want to do something about their weight.”
Ted Tenthoff, a biotech analyst who follows Arena for investment bank Piper Jaffray & Co., said the drugs could end up addressing distinct segments of the market.
Tenthoff said Vivus’ Qnexa, which showed the most dramatic weight loss in trials, would likely would end up being prescribed for the heaviest patients — though the company also has lower-dose formulations it says will address a broad range of patients.
Tenthoff said a better safety profile could provide a niche for Arena’s lorcaserin with less-obese patients, despite the drug’s weaker data on weight loss. The company believes this also could make it the drug doctors try first.
Orexigen, whose drug Tenthoff described as falling somewhere between the other two, plays up Contrave’s potential to help patients with other conditions that often accompany obesity. For example, Narachi said one-third of obesity patients also suffer from depression and therefore might want a weight-loss drug that includes an antidepressant.
Many patients also might try more than one approach.
“What typically happens in markets like this is that there’s quite a bit of churn,” Narachi said. “There might be somebody who tries a product and decides to stop, either because it’s not effective for them or because some patients are more sensitive to certain tolerability aspects than others. Or people may lose a certain amount of weight on one product and then switch to another one and see if they can lose a little bit more.”
As the FDA considers whether to approve the drugs, a big question is likely to be safety. Past problems with obesity drugs could be one reason investors have been slow to embrace the three companies’ stocks, all of which trade in single digits.
Inevitably, regulators will think back to the Fen-phen fiasco, which led drugmaker Wyeth to set aside $21 billion to resolve litigation related to Fen-phen’s links to heart damage and lung disease.
Tenthoff said more recent experience with Meridia could make the FDA that much more conservative. Data that linked that drug to strokes, heart attacks and other cardiovascular problems led European regulators to suspend sales of the drug this month while the FDA asked for a stronger warning label.
“These drugs are going to be used in millions of patients,” Tenthoff said. “That is going to be on the mind of the FDA.”
Questions about safety also could make some patients hesitant, even if the FDA approves all three drugs.
The companies point to substantial pent-up demand in the market, given the lack of new weight-loss drugs in recent years. When Fen-phen was on the market, prescriptions tripled in one year to hit 20.6 million in 1996.
Right now, a major activity for all three companies is seeking large pharmaceutical company partners with sales forces to help market the drugs if they get on the market. Narachi said multiple potential product launches around the same time also could help speed acceptance.
“There will be three companies, perhaps with three partners, all trying to convince people that pharmacotherapy approaches should be more heavily considered as a treatment for obesity,” Narachi said.
The Obesity Society’s Ryan said doctors and patients are eager for new tools.
“They are clamoring for aids to help with their patients,” Ryan said. “Just telling people to eat less and exercise more is not going to slow the obesity epidemic.”
mfg ipollit
Hi ipollit
Ich sehe du hast hauptsächlich nur Mid-Large Caps Biotechs im Depot du solltest auch mal in Kleine Unternehmen investieren da steckt das große geld .
Ich sehe du hast hauptsächlich nur Mid-Large Caps Biotechs im Depot du solltest auch mal in Kleine Unternehmen investieren da steckt das große geld .
Antwort auf Beitrag Nr.: 38.850.447 von BrauchGeld am 31.01.10 12:30:58Mid-Large Caps? Was verstehst du darunter. Ich denke, ich habe quer von allem etwas zwischen Mid- und Smallcaps. Aber warum soll die größe des Unternehmens etwas mit den Erfolg der Anlage zutun haben? Je kleiner das Unternehmen, umso riskanter ist es in der Regel, da es einfach nicht so breit aufgestellt sein kann und auch nicht soviel Geld zur Verfügung hat, um etwas zu bewegen. Bei den Positionen in meinem Depot ist es eher so, dass die Erfolgreichen steigen und damit teuer aussehen und die Flops fallen und damit billig aussehen. Zum Beispiel eine Addex muss wieder ganz neu beginnen... das spiegelt sich in der niedrigen Bewertung wieder und man sollte dies nicht mit "billig" oder "preiswert" verwechseln. Ich fand die Mittelgroßen bisher jedenfalls nicht schlecht.
die MKs in Mio USD...
2.150 Regeneron
1.799 Onyx
1.270 Incyte
1.190 Cubist
1.100 Isis
1.040 Seattle Genetics
825 Genmab
765 ViroPharma
755 Allos
712 Exelixis
565 NicOx
536 Micromet
425 Rigel
378 NeuroSearch
289 Arena
178 Medigene
151 Sucampo
144 ArQule
142 Progenics
125 NeurogesX
118 Array
69 Addex
mfg ipollit
die MKs in Mio USD...
2.150 Regeneron
1.799 Onyx
1.270 Incyte
1.190 Cubist
1.100 Isis
1.040 Seattle Genetics
825 Genmab
765 ViroPharma
755 Allos
712 Exelixis
565 NicOx
536 Micromet
425 Rigel
378 NeuroSearch
289 Arena
178 Medigene
151 Sucampo
144 ArQule
142 Progenics
125 NeurogesX
118 Array
69 Addex
mfg ipollit
Was haltet ihr von ISIS?
Die zweite Generation von Antisensemolekülen scheint zu funktionieren!?! Oder?
Die zweite Generation von Antisensemolekülen scheint zu funktionieren!?! Oder?
zu CBST...
http://www.xconomy.com/boston/2010/01/25/cubist-maintains-gr…
Cubist Maintains Growth Streak, As Investors Fear Generic Threat, Thin Pipeline
Ryan McBride 1/25/10
Cubist Pharmaceuticals has grown into one of the big success stories in biotech industry of the past few years, based almost entirely on the sales of a single product. The Lexington, MA-based company’s big hit is an intravenous antibiotic for deadly infections called daptomycin (Cubicin). Even though this has propelled Cubist into profitable territory, the company’s (NASDAQ:CBST) stock price has been flat for more than four years amid concerns on Wall Street about potential threats to its antibiotics franchise.
Last week, I visited the company’s headquarters and met with CEO Michael Bonney and discussed the successes and challenges he faces at Cubist. This month the company announced 2009 revenue of $562.1 million, a 30-percent jump from the year before. Revenues have grown rapidly every year since the market debut of daptomycin in 2003. But Bonney was clear that the company doesn’t plan to rest on its laurels, and the firm is taking more aggressive measures than in previous years to bring a second commercial product to market. (The company also sells an antibiotic called meropenem on behalf of AstraZeneca in the U.S., but that agreement brought Cubist only $22.5 million in revenue last year.)
Indeed, analysts have criticized the company’s lack of an encore to its success with daptomycin. Bonney acknowledged that the company’s critics have a point, and that he’s tackling it now.
“I think we could have been a little more aggressive at pipeline building earlier than we started to,” Bonney said. “It’s always a fine balancing act between [profitability] and how much you are going to spend, and there’s no formula that I’ve found in any textbook that says this is how you do it. But I do think, with the benefit of hindsight, that is something we could have done differently.”
Cubist has more than just its pipeline to worry about. It generates 93 percent of its revenue from daptomycin, a compound used in hospitals to treat lethal MRSA (Methicillin-resistant Staphylococcus aureus) infections and other bugs. And while analysts say daptomycin has potential to reach $1 billion in peak annual sales, that is no sure thing. Generic drug maker Teva Pharmaceutical has plans to market a cheaper generic copy of the drug before Cubist’s patents for the product expire between 2016 and 2019. Cubist plans to prove in its pending lawsuit against Teva that its patents protect its lead antibiotic from generic competition.
Cubist’s problem isn’t exactly unique in the biotech game; there are a number of mid-sized drug developers whose success hinges largely on one product. A couple of those companies include Cheshire, CT-based Alexion Pharmaceuticals (NASDAQ:ALXN), which gets all of its sales revenue from one product, eculizumab (Soliris), a treatment for a rare blood disorder called paroxysmal nocturnal hemoglobinuria, as well as Emeryville, CA-based Onyx Pharmaceuticals (NASDAQ:ONXX), which has an anti-tumor drug called sorafenib (Nexavar) that it develops and markets with the help of Bayer .
Though Cubist may not have another drug in its pipeline to bring to the market within the next two years, the company completed several deals in 2009 to broaden its portfolio of drug candidates. Last month, the firm bought San Diego-based Calixa Therapeutics for $92.5 million up front and an additional $310 million in potential payments. The buyout gave Cubist most of the commercial rights to Calixa’slead antibiotic treatment, a combination of anti-bacterial agents, that has the potential of being a top seller on par with daptomycin. That’s because it may be able about to treat a life-threatening bug called P. aeruginosa better than existing antibiotics on the market, Bonney says. That promising treatment is in mid-stage clinical trials, and Cubist predicts that it could be seeking U.S. approval for the product in 2013.
Cubist also revealed separate deals last year with a trio of Massachusetts-based biotechs with products in various stages of development: Alnylam Pharmaceuticals, Forma Therapeutics, and Hydra Biosciences. Perhaps the most high profile of those deals was with the RNA-interference (RNAi) drug developer Alnylam (NASDAQ:ALNY), from which Cubist licensed rights to Alnylam’s gene-silencing technology for treating respiratory syncytial virus (RSV), which sends about 125,000 children to the hospital in the U.S. every year, according to Alnylam. Alnylam is in mid-stage clinical development of a first-generation RNAi therapy for RSV. Bonney told me that his company is keenly interested in a newer version of the treatment specifically for children. Yet the therapy hasn’t reached human clinical trials, meaning that its potential commercial impact on the firm is both many years away and highly uncertain.
Another big question mark in Cubist’s pipeline is the future of its experimental drug ecallantide. The company put the brakes on a mid-stage clinical trial of the drug, which it is developing as an anti-bleeding agent for heart surgeries, after more patients who were treated with the drug died compared with those who didn’t take it. (Ecallantide is a protein drug that was discovered by Cambridge, MA-based Dyax (NASDAQ YAX), which has licensed the drug to Cubist for use in heart surgeries.) Cubist is expected to provide an update on what happened in its trial with ecallantide sometime in the first half of this year.
YAX), which has licensed the drug to Cubist for use in heart surgeries.) Cubist is expected to provide an update on what happened in its trial with ecallantide sometime in the first half of this year.
Still, what perhaps keeps the company’s stockholders up at night is the near-term challenge Teva has brought to the daptomycin patents. Cubist mounted a lawsuit against Teva early last year in response to Teva’s notice that it planned to start seek FDA approval of generic version of the antibiotic, before the expiration of Cubist’s patents on the treatment between 2016 and 2019. Bonney said that his company had been ready for such a case since 2007, expecting that Teva or another generic drug-maker would challenge its patents.
Cubist filed two of the three patents in question in the case, Bonney said, after the company acquired rights to daptomycin from the company that discovered the molecule, Indianapolis-based drug giant Eli Lilly (NYSE:LLY). Daptomycin is used in hospitals mostly to treat infections in the skin and tissue just beneath the skin. Cubist is credited with discovering how best to administer the antibiotic to patients at certain dose levels, something that Eli Lilly wasn’t able to figure out. (In fact, Bonney told me that the agreement that brought daptomycin to Cubist required the company to pay Eli Lilly an upfront fee of about $1 million and royalty payments to Lilly—an amazingly small amount considering that the antibiotic now generates more than half a billion dollars in annual revenue.)
Despite the rapid growth in Cubist’s sales of daptomycin, the price of the company’s common stock has been lingering in the $15 to $25 range since mid-2005. Wall Street analysts have cited the potential approval of a generic version of daptomycin, competition from new antibiotics such as South San Francisco-based Theravance’s telavancin, and Cubist’s lack of a late-stage product candidate in its pipeline, as reasons to be concerned about the company’s future.
“I think it’s generally recognized by everybody that the key thing for Cubist is to continue to build the pipeline and find a follow on to Cubicin. That’s relevant whether or not Teva is successful,” says Alan Carr, a biotech analyst for the investment bank Needham & Company in New York. Carr, who is a bit more optimistic about Cubist’s prospects than some of his fellow analysts, has a “buy” rating for the company’s stock.
Bonney said that building the company’s pipeline, including staying in the market for acquisitions that would give the company a new product to bring to market, is one of his top three priorities. However, he said that he isn’t willing to spend too much to acquire new products, especially at the expense of his other big priorities, which are to continue boosting daptomycin sales and to increase the company’s operating profits.
Massachusetts officials are also rooting for Cubist to continue growing in the state. As part of state’s 10-year plan to invest about $1 billion to boost its life sciences industry, Cubist garnered $1.74 million in tax incentives last year tied to its commitment to add jobs in the commonwealth. Cubist employed 362 people in Lexington and 227 more workers outside of the town as of late last month, company spokesman Francis McLoughlin says. He noted that the firm has budgeted for 87 new hires in 2010.
The company is also planning an 110,000-square-foot expansion to its facilities in Lexington. There are still some permitting requirements before construction begins, McLoughlin says, but the project could break ground before the end of this year.
mfg ipollit
http://www.xconomy.com/boston/2010/01/25/cubist-maintains-gr…
Cubist Maintains Growth Streak, As Investors Fear Generic Threat, Thin Pipeline
Ryan McBride 1/25/10
Cubist Pharmaceuticals has grown into one of the big success stories in biotech industry of the past few years, based almost entirely on the sales of a single product. The Lexington, MA-based company’s big hit is an intravenous antibiotic for deadly infections called daptomycin (Cubicin). Even though this has propelled Cubist into profitable territory, the company’s (NASDAQ:CBST) stock price has been flat for more than four years amid concerns on Wall Street about potential threats to its antibiotics franchise.
Last week, I visited the company’s headquarters and met with CEO Michael Bonney and discussed the successes and challenges he faces at Cubist. This month the company announced 2009 revenue of $562.1 million, a 30-percent jump from the year before. Revenues have grown rapidly every year since the market debut of daptomycin in 2003. But Bonney was clear that the company doesn’t plan to rest on its laurels, and the firm is taking more aggressive measures than in previous years to bring a second commercial product to market. (The company also sells an antibiotic called meropenem on behalf of AstraZeneca in the U.S., but that agreement brought Cubist only $22.5 million in revenue last year.)
Indeed, analysts have criticized the company’s lack of an encore to its success with daptomycin. Bonney acknowledged that the company’s critics have a point, and that he’s tackling it now.
“I think we could have been a little more aggressive at pipeline building earlier than we started to,” Bonney said. “It’s always a fine balancing act between [profitability] and how much you are going to spend, and there’s no formula that I’ve found in any textbook that says this is how you do it. But I do think, with the benefit of hindsight, that is something we could have done differently.”
Cubist has more than just its pipeline to worry about. It generates 93 percent of its revenue from daptomycin, a compound used in hospitals to treat lethal MRSA (Methicillin-resistant Staphylococcus aureus) infections and other bugs. And while analysts say daptomycin has potential to reach $1 billion in peak annual sales, that is no sure thing. Generic drug maker Teva Pharmaceutical has plans to market a cheaper generic copy of the drug before Cubist’s patents for the product expire between 2016 and 2019. Cubist plans to prove in its pending lawsuit against Teva that its patents protect its lead antibiotic from generic competition.
Cubist’s problem isn’t exactly unique in the biotech game; there are a number of mid-sized drug developers whose success hinges largely on one product. A couple of those companies include Cheshire, CT-based Alexion Pharmaceuticals (NASDAQ:ALXN), which gets all of its sales revenue from one product, eculizumab (Soliris), a treatment for a rare blood disorder called paroxysmal nocturnal hemoglobinuria, as well as Emeryville, CA-based Onyx Pharmaceuticals (NASDAQ:ONXX), which has an anti-tumor drug called sorafenib (Nexavar) that it develops and markets with the help of Bayer .
Though Cubist may not have another drug in its pipeline to bring to the market within the next two years, the company completed several deals in 2009 to broaden its portfolio of drug candidates. Last month, the firm bought San Diego-based Calixa Therapeutics for $92.5 million up front and an additional $310 million in potential payments. The buyout gave Cubist most of the commercial rights to Calixa’slead antibiotic treatment, a combination of anti-bacterial agents, that has the potential of being a top seller on par with daptomycin. That’s because it may be able about to treat a life-threatening bug called P. aeruginosa better than existing antibiotics on the market, Bonney says. That promising treatment is in mid-stage clinical trials, and Cubist predicts that it could be seeking U.S. approval for the product in 2013.
Cubist also revealed separate deals last year with a trio of Massachusetts-based biotechs with products in various stages of development: Alnylam Pharmaceuticals, Forma Therapeutics, and Hydra Biosciences. Perhaps the most high profile of those deals was with the RNA-interference (RNAi) drug developer Alnylam (NASDAQ:ALNY), from which Cubist licensed rights to Alnylam’s gene-silencing technology for treating respiratory syncytial virus (RSV), which sends about 125,000 children to the hospital in the U.S. every year, according to Alnylam. Alnylam is in mid-stage clinical development of a first-generation RNAi therapy for RSV. Bonney told me that his company is keenly interested in a newer version of the treatment specifically for children. Yet the therapy hasn’t reached human clinical trials, meaning that its potential commercial impact on the firm is both many years away and highly uncertain.
Another big question mark in Cubist’s pipeline is the future of its experimental drug ecallantide. The company put the brakes on a mid-stage clinical trial of the drug, which it is developing as an anti-bleeding agent for heart surgeries, after more patients who were treated with the drug died compared with those who didn’t take it. (Ecallantide is a protein drug that was discovered by Cambridge, MA-based Dyax (NASDAQ
 YAX), which has licensed the drug to Cubist for use in heart surgeries.) Cubist is expected to provide an update on what happened in its trial with ecallantide sometime in the first half of this year.
YAX), which has licensed the drug to Cubist for use in heart surgeries.) Cubist is expected to provide an update on what happened in its trial with ecallantide sometime in the first half of this year.Still, what perhaps keeps the company’s stockholders up at night is the near-term challenge Teva has brought to the daptomycin patents. Cubist mounted a lawsuit against Teva early last year in response to Teva’s notice that it planned to start seek FDA approval of generic version of the antibiotic, before the expiration of Cubist’s patents on the treatment between 2016 and 2019. Bonney said that his company had been ready for such a case since 2007, expecting that Teva or another generic drug-maker would challenge its patents.
Cubist filed two of the three patents in question in the case, Bonney said, after the company acquired rights to daptomycin from the company that discovered the molecule, Indianapolis-based drug giant Eli Lilly (NYSE:LLY). Daptomycin is used in hospitals mostly to treat infections in the skin and tissue just beneath the skin. Cubist is credited with discovering how best to administer the antibiotic to patients at certain dose levels, something that Eli Lilly wasn’t able to figure out. (In fact, Bonney told me that the agreement that brought daptomycin to Cubist required the company to pay Eli Lilly an upfront fee of about $1 million and royalty payments to Lilly—an amazingly small amount considering that the antibiotic now generates more than half a billion dollars in annual revenue.)
Despite the rapid growth in Cubist’s sales of daptomycin, the price of the company’s common stock has been lingering in the $15 to $25 range since mid-2005. Wall Street analysts have cited the potential approval of a generic version of daptomycin, competition from new antibiotics such as South San Francisco-based Theravance’s telavancin, and Cubist’s lack of a late-stage product candidate in its pipeline, as reasons to be concerned about the company’s future.
“I think it’s generally recognized by everybody that the key thing for Cubist is to continue to build the pipeline and find a follow on to Cubicin. That’s relevant whether or not Teva is successful,” says Alan Carr, a biotech analyst for the investment bank Needham & Company in New York. Carr, who is a bit more optimistic about Cubist’s prospects than some of his fellow analysts, has a “buy” rating for the company’s stock.
Bonney said that building the company’s pipeline, including staying in the market for acquisitions that would give the company a new product to bring to market, is one of his top three priorities. However, he said that he isn’t willing to spend too much to acquire new products, especially at the expense of his other big priorities, which are to continue boosting daptomycin sales and to increase the company’s operating profits.
Massachusetts officials are also rooting for Cubist to continue growing in the state. As part of state’s 10-year plan to invest about $1 billion to boost its life sciences industry, Cubist garnered $1.74 million in tax incentives last year tied to its commitment to add jobs in the commonwealth. Cubist employed 362 people in Lexington and 227 more workers outside of the town as of late last month, company spokesman Francis McLoughlin says. He noted that the firm has budgeted for 87 new hires in 2010.
The company is also planning an 110,000-square-foot expansion to its facilities in Lexington. There are still some permitting requirements before construction begins, McLoughlin says, but the project could break ground before the end of this year.
mfg ipollit
Antwort auf Beitrag Nr.: 38.851.355 von bussibaer12 am 31.01.10 17:46:09zu ISIS wurde doch hier schon ein wenig gepostet...
mfg ipollit
mfg ipollit
ONXX - neben der möglichen Zulassung von Carfilzomib aufgrund der ausstehenden PII-Daten wird bald auch eine PIII gestartet
http://www.prnewswire.com/news-releases/onyx-pharmaceuticals…
Onyx Pharmaceuticals Announces Agreement with the FDA on a Special Protocol Assessment for Planned Phase 3 Carfilzomib Combination Trial in Relapsed Multiple Myeloma
EMERYVILLE, Calif., Feb. 2 /PRNewswire-FirstCall/ -- Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX) today announced that the company reached agreement with the U.S. Food and Drug Administration (FDA) on a Special Protocol Assessment (SPA) for the Phase 3 international randomized trial. This pivotal trial, which is expected to begin in the first half of 2010, will enroll patients with relapsed multiple myeloma following treatment with one to three prior regimens. It is designed to evaluate the efficacy of carfilzomib in combination with lenalidomide and low dose dexamethasone, versus lenalidomide and low dose dexamethasone alone.
"The SPA enables us to initiate this carfilzomib Phase 3 combination trial in the first half of this year with increased clarity on the full approval pathway," said Michael Kauffman, M.D., Ph.D., chief medical officer at Onyx. "In the Phase 2b data (from the "006" trial) reported at the American Society of Hematology meeting last December, carfilzomib showed promising response rates and good tolerability with this three-drug combination in patients with relapsed or refractory myeloma."
Dr. Kauffman continued, "This planned Phase 3 study complements and builds upon the encouraging single-agent carfilzomib data reported earlier in heavily pretreated patients with relapsed and refractory multiple myeloma. Despite the established use of existing therapies, over time virtually all multiple myeloma patients relapse and succumb to their disease, meaning new innovative agents are needed. As a medical community, we have made important strides in extending the life of myeloma patients and need to continue to identify new ways we can benefit those who have this disease."
Carfilzomib Development Strategy
Based on carfilzomib's efficacy and tolerability profile demonstrated to-date, Onyx is pursuing a broad approach to advancing this therapy for multiple myeloma patients. In the advanced relapsed refractory setting, carfilzomib is currently being evaluated in a Phase 2b study that could support a new drug application filing by the end of 2010. In addition to this potential accelerated approval pathway and for patients earlier in the course of their disease, Onyx is evaluating the combination of carfilzomib, Revlimid® and low-dose dexamethasone in a Phase 3 study, as covered by this SPA, which is expected to begin in the first half of 2010.
About Special Protocol Assessments
A Special Protocol Assessment is a written agreement with the FDA on the design and planned analysis for a clinical trial. It is intended to form the basis for a marketing application and may only be changed through a written agreement between the sponsor and the FDA, or if the FDA becomes aware of new public health concerns.
About the Phase 3 Trial
The Phase 3 ("009") trial is a 700 patient, randomized, open-label, global multi-center study comparing two treatment regimens for patients with relapsed multiple myeloma and will be conducted in approximately 200 centers worldwide. This study is designed to demonstrate the clinical benefit of carfilzomib in combination with lenalidomide and dexamethasone based on a primary endpoint of progression-free survival. Patients will be randomized to receive carfilzomib (20mg/m2 on days 1 and 2 of cycle 1 only, then 27mg/m2 subsequently) or matching placebo, in addition to a standard dosing schedule of lenalidomide (25mg qd 21 days on, 7 days off) and low-dose dexamethasone (40mg q week).
About Carfilzomib
Carfilzomib is a selective, next-generation proteasome inhibitor that has shown encouraging results in a broad clinical trial program in multiple myeloma. Carfilzomib is currently undergoing evaluation as a single agent in multiple Phase 2 and Phase 1 clinical trials in relapsed or refractory multiple myeloma. These trials include a Phase 2b monotherapy study (known as "003-A1") in patients with relapsed, refractory multiple myeloma, which could support a new drug application (NDA) filing by the end of 2010. Carfilzomib is also being evaluated in advanced solid tumors.
About Multiple Myeloma
Multiple myeloma (MM) is the second most common hematologic cancer and results from an abnormality of plasma cells, usually in the bone marrow. In the United States, more than 50,000 people are living with MM and approximately 20,000 new cases are diagnosed annually(i). Worldwide, more than 180,000 people are living with MM and approximately 86,000 new cases are diagnosed annually(ii).
mfg ipollit
http://www.prnewswire.com/news-releases/onyx-pharmaceuticals…
Onyx Pharmaceuticals Announces Agreement with the FDA on a Special Protocol Assessment for Planned Phase 3 Carfilzomib Combination Trial in Relapsed Multiple Myeloma
EMERYVILLE, Calif., Feb. 2 /PRNewswire-FirstCall/ -- Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX) today announced that the company reached agreement with the U.S. Food and Drug Administration (FDA) on a Special Protocol Assessment (SPA) for the Phase 3 international randomized trial. This pivotal trial, which is expected to begin in the first half of 2010, will enroll patients with relapsed multiple myeloma following treatment with one to three prior regimens. It is designed to evaluate the efficacy of carfilzomib in combination with lenalidomide and low dose dexamethasone, versus lenalidomide and low dose dexamethasone alone.
"The SPA enables us to initiate this carfilzomib Phase 3 combination trial in the first half of this year with increased clarity on the full approval pathway," said Michael Kauffman, M.D., Ph.D., chief medical officer at Onyx. "In the Phase 2b data (from the "006" trial) reported at the American Society of Hematology meeting last December, carfilzomib showed promising response rates and good tolerability with this three-drug combination in patients with relapsed or refractory myeloma."
Dr. Kauffman continued, "This planned Phase 3 study complements and builds upon the encouraging single-agent carfilzomib data reported earlier in heavily pretreated patients with relapsed and refractory multiple myeloma. Despite the established use of existing therapies, over time virtually all multiple myeloma patients relapse and succumb to their disease, meaning new innovative agents are needed. As a medical community, we have made important strides in extending the life of myeloma patients and need to continue to identify new ways we can benefit those who have this disease."
Carfilzomib Development Strategy
Based on carfilzomib's efficacy and tolerability profile demonstrated to-date, Onyx is pursuing a broad approach to advancing this therapy for multiple myeloma patients. In the advanced relapsed refractory setting, carfilzomib is currently being evaluated in a Phase 2b study that could support a new drug application filing by the end of 2010. In addition to this potential accelerated approval pathway and for patients earlier in the course of their disease, Onyx is evaluating the combination of carfilzomib, Revlimid® and low-dose dexamethasone in a Phase 3 study, as covered by this SPA, which is expected to begin in the first half of 2010.
About Special Protocol Assessments
A Special Protocol Assessment is a written agreement with the FDA on the design and planned analysis for a clinical trial. It is intended to form the basis for a marketing application and may only be changed through a written agreement between the sponsor and the FDA, or if the FDA becomes aware of new public health concerns.
About the Phase 3 Trial
The Phase 3 ("009") trial is a 700 patient, randomized, open-label, global multi-center study comparing two treatment regimens for patients with relapsed multiple myeloma and will be conducted in approximately 200 centers worldwide. This study is designed to demonstrate the clinical benefit of carfilzomib in combination with lenalidomide and dexamethasone based on a primary endpoint of progression-free survival. Patients will be randomized to receive carfilzomib (20mg/m2 on days 1 and 2 of cycle 1 only, then 27mg/m2 subsequently) or matching placebo, in addition to a standard dosing schedule of lenalidomide (25mg qd 21 days on, 7 days off) and low-dose dexamethasone (40mg q week).
About Carfilzomib
Carfilzomib is a selective, next-generation proteasome inhibitor that has shown encouraging results in a broad clinical trial program in multiple myeloma. Carfilzomib is currently undergoing evaluation as a single agent in multiple Phase 2 and Phase 1 clinical trials in relapsed or refractory multiple myeloma. These trials include a Phase 2b monotherapy study (known as "003-A1") in patients with relapsed, refractory multiple myeloma, which could support a new drug application (NDA) filing by the end of 2010. Carfilzomib is also being evaluated in advanced solid tumors.
About Multiple Myeloma
Multiple myeloma (MM) is the second most common hematologic cancer and results from an abnormality of plasma cells, usually in the bone marrow. In the United States, more than 50,000 people are living with MM and approximately 20,000 new cases are diagnosed annually(i). Worldwide, more than 180,000 people are living with MM and approximately 86,000 new cases are diagnosed annually(ii).
mfg ipollit
Neurosearch - puh... noch rechzeitig dabei und Glück gehabt!
erste Huntexil PIII war erfolgreich... somit ist der Weg frei. Wegen fehlender Alternativen bei Huntington ist sogar ein potentieller Blockbuster drin, den Neurosearch wegen der relativ geringen Anzahl an Patienten auch selber vermarkten kann.
03.02.2010 10:18
UPDATE 1-Neurosearch shares leap on Huntington's drug study
COPENHAGEN, Feb 3 (Reuters) - Shares in Danish biopharmaceuticals firm Neurosearch A/S surged as much as 74 percent after it announced positive results from a Phase III trial with a candidate Huntington's disease drug.
If approved, the drug, Huntexil, could become Neurosearch's first product on the market.
'This could pave the way for a launch in the near future, possibly in early 2011 or even late 2010, and turn Neurosearch into a profitable company,' Jyske Bank analyst Frank Andersen said.
The shares hit a one-year high to trade up 49 percent at 126.50 crowns by 0850 GMT, against the trend of a 0.9 percent fall in the DJ Stoxx European healthcare index.
'Huntexil significantly improves motor functions in Huntington patients,' Neurosearch said in a statement.
It said positive effects were observed in both voluntary and involuntary motor symptoms and Huntexil was very well tolerated with adverse effects similar to placebo.
Neurosearch said it is now starting talks with scientific advisors and regulatory agencies in Europe and the United States to discuss the outcome of the study and plans for submitting applications for market authorisation for for Huntexil.
'This supports our 'buy' recommendation on the shares. This is very good news,' Andersen said.
He said the drug would likely be launched in Europe first since the Phase III study was conducted in Europe.
Huntington's disease is a highly disabling, hereditary neurodegenerative genetic disorder which leads to damage of the nerve cells in certain areas of the brain.
mfg ipollit
erste Huntexil PIII war erfolgreich... somit ist der Weg frei. Wegen fehlender Alternativen bei Huntington ist sogar ein potentieller Blockbuster drin, den Neurosearch wegen der relativ geringen Anzahl an Patienten auch selber vermarkten kann.
03.02.2010 10:18
UPDATE 1-Neurosearch shares leap on Huntington's drug study
COPENHAGEN, Feb 3 (Reuters) - Shares in Danish biopharmaceuticals firm Neurosearch A/S surged as much as 74 percent after it announced positive results from a Phase III trial with a candidate Huntington's disease drug.
If approved, the drug, Huntexil, could become Neurosearch's first product on the market.
'This could pave the way for a launch in the near future, possibly in early 2011 or even late 2010, and turn Neurosearch into a profitable company,' Jyske Bank analyst Frank Andersen said.
The shares hit a one-year high to trade up 49 percent at 126.50 crowns by 0850 GMT, against the trend of a 0.9 percent fall in the DJ Stoxx European healthcare index.
'Huntexil significantly improves motor functions in Huntington patients,' Neurosearch said in a statement.
It said positive effects were observed in both voluntary and involuntary motor symptoms and Huntexil was very well tolerated with adverse effects similar to placebo.
Neurosearch said it is now starting talks with scientific advisors and regulatory agencies in Europe and the United States to discuss the outcome of the study and plans for submitting applications for market authorisation for for Huntexil.
'This supports our 'buy' recommendation on the shares. This is very good news,' Andersen said.
He said the drug would likely be launched in Europe first since the Phase III study was conducted in Europe.
Huntington's disease is a highly disabling, hereditary neurodegenerative genetic disorder which leads to damage of the nerve cells in certain areas of the brain.
mfg ipollit
Antwort auf Beitrag Nr.: 38.869.634 von ipollit am 03.02.10 10:29:48dieser Jyske Bank Analyst ist echt eine ziemliche Pfeife... ich verstehe manchmal nicht, was die so treiben, wenn sie ihre Analysen schreiben. Die Zahlen von denen sind kaum etwas wert. 
z.B. heute: Jyske Bank analyst Frank Andersen raised his target price on Neurosearch twice in one day, first to 260 crowns from 165 crowns and then to 380 crowns, and repeated a "buy" recommendation on the back of the news.
...
He said he had raised his target price on Neurosearch shares for the second time in one day because the treatment price with Huntexil should be higher than his previous estimate and in line with the treatment price for patients using Danish drug maker Lundbeck's (LUN.CO) Huntington's drug Xenazine.
http://www.reuters.com/article/idUSLDE6120GC20100203?type=ma…
in seiner letzten Analyse schrieb er noch vor wenigen Monaten: "We assess that Huntexil will be priced in line with other Orphan drugs, and hence we will in the US see a treatment price of DKK 75,000, while we assess that in Europe the price will come to DKK 60,000." 75k DDK sind 14k USD
dabei schreibt Neurosearch in ihrer letzten Präsentation selber, dass Huntexil als Orphan Drug mindestens vergleichbar mit Xenazine im Preis liegen sollte, den sie mit 40k USD angeben: "Attractive value increment in orphan pricing (Xenazine (chorea symptoms only); ~ 40000 $ p.a."
und heute fällt diesem Analysten plötzlich auf, dass Huntexil ja vielleicht genauso teuer sein könnte... und muss sich innerhalb eines Tages auch nochmal korrigieren
woanders stand ein möglicher Preis im Bereich von 40-45k EUR, was schon eher hinkommen dürfte... also ca. 60k USD für die Therapie pro Jahr.
An Huntington leiden angeblich in den USA und in Europa zusammen etwa 70.000 Menschen... 2/3 davon sollen für Huntexil in Frage kommen. In der Summe steckt in dem Mittel, das eigentlich in der Form keine Konkurrenz hat und bei dem sehr hoher Bedarf besteht, also eine Menge Potential. Vor allen Dingen wird die Vermarktung von NeuroSearch alleine bewältigt werden können... und NeuroSearch ist dagegen noch ein relativ kleines Unternehmen.
Zwar soll die Zulassung erst Ende des Jahres beantragt werden, doch soll vorher schon Huntexil einem begrenzten Patientenkreis zugänglich gemacht werden.
mfg ipollit

z.B. heute: Jyske Bank analyst Frank Andersen raised his target price on Neurosearch twice in one day, first to 260 crowns from 165 crowns and then to 380 crowns, and repeated a "buy" recommendation on the back of the news.
...
He said he had raised his target price on Neurosearch shares for the second time in one day because the treatment price with Huntexil should be higher than his previous estimate and in line with the treatment price for patients using Danish drug maker Lundbeck's (LUN.CO) Huntington's drug Xenazine.
http://www.reuters.com/article/idUSLDE6120GC20100203?type=ma…
in seiner letzten Analyse schrieb er noch vor wenigen Monaten: "We assess that Huntexil will be priced in line with other Orphan drugs, and hence we will in the US see a treatment price of DKK 75,000, while we assess that in Europe the price will come to DKK 60,000." 75k DDK sind 14k USD
dabei schreibt Neurosearch in ihrer letzten Präsentation selber, dass Huntexil als Orphan Drug mindestens vergleichbar mit Xenazine im Preis liegen sollte, den sie mit 40k USD angeben: "Attractive value increment in orphan pricing (Xenazine (chorea symptoms only); ~ 40000 $ p.a."
und heute fällt diesem Analysten plötzlich auf, dass Huntexil ja vielleicht genauso teuer sein könnte... und muss sich innerhalb eines Tages auch nochmal korrigieren
woanders stand ein möglicher Preis im Bereich von 40-45k EUR, was schon eher hinkommen dürfte... also ca. 60k USD für die Therapie pro Jahr.
An Huntington leiden angeblich in den USA und in Europa zusammen etwa 70.000 Menschen... 2/3 davon sollen für Huntexil in Frage kommen. In der Summe steckt in dem Mittel, das eigentlich in der Form keine Konkurrenz hat und bei dem sehr hoher Bedarf besteht, also eine Menge Potential. Vor allen Dingen wird die Vermarktung von NeuroSearch alleine bewältigt werden können... und NeuroSearch ist dagegen noch ein relativ kleines Unternehmen.
Zwar soll die Zulassung erst Ende des Jahres beantragt werden, doch soll vorher schon Huntexil einem begrenzten Patientenkreis zugänglich gemacht werden.
mfg ipollit
Antwort auf Beitrag Nr.: 38.851.197 von ipollit am 31.01.10 16:56:25nimm zu deinen medigene, deren börsenwert (178 mio $) kontinuierlich fällt, doch mologene (136 mio $), deren wert kontinuierlich steigt, dazu!
Antwort auf Beitrag Nr.: 38.875.174 von pokemon am 03.02.10 20:47:10@pokemon: sorry, auch wenn ich jetzt riskiere, von dir geschlagen zu werden, aber mologen!? ich gönne dir ein paar hundert prozent, aber befürchte gegenteiliges
@ipollit: gratulation zum jüngsten trade! das zahlt sich im wahrsten sinne aus
@ipollit: gratulation zum jüngsten trade! das zahlt sich im wahrsten sinne aus
Antwort auf Beitrag Nr.: 38.875.591 von PathFinder2 am 03.02.10 21:47:08danke... gehört aber einiges an Glück dazu, hätte auch schief gehen können.
mfg ipollit
mfg ipollit
Genmab... doch leider ein ziemlicher Problemfall geworden. Rückblickend ist es eine klare Fehlinvestition gewesen.
Heute kamen die ersten Zahlen zu Arzerra. Außerdem wurde das wenig aussichtsreiche Zanolimumab noch für ein paar Mios verscherbelt.
4 Feb, 2010 13:09 CET
Genmab A/S Company Announcement Arzerra 2009 Net Sales Figures
Summary: Net sales of Arzerra™ for the fourth quarter of 2009 wereapproximately DKK 29 million, with an expected royalty payment to Genmab of DKK6 million. Copenhagen, Denmark; February 4, 2010 - Genmab A/S (OMX: GEN) announced todaythat the U.S. net sales for Arzerra (ofatumumab) during the fourth quarter of2009 were approximately DKK 29 million (approximately USD 5.5 million). Underthe terms of the collaboration with GlaxoSmithKline (GSK), Genmab expects to receive a royalty payment of approximately DKK 6 million (approximately USD 1.1million). “We are very pleased to see the first sales figures for Arzerra and we believethat they reflect the need for new treatment options for patients withrefractory CLL,” said Lisa N. Drakeman, Ph.D., Chief Executive Officer ofGenmab. Arzerra was approved by the FDA in October, 2009, for the treatment of patientsin the U.S. with chronic lymphocytic leukemia (CLL) that is refractory tofludarabine and alemtuzumab, and was launched by GSK in mid-November, 2009. Arzerra is a monoclonal antibody that causes the body's immune response tofight against normal and cancerous B-cells. Arzerra attaches to the small andlarge loop epitopes - on a molecule called CD20, which is found on the surfaceof B-cells, the type of cell that becomes cancerous in CLL.
************
4 Feb, 2010 17:48 CET
Genmab A/S Investor News Genmab Outlicenses Zanolimumab to TenX Biopharma
Summary: Genmab has licensed zanolimumab to TenX Biopharma, Inc. Copenhagen, Denmark; February 4, 2010 - Genmab A/S (OMX: GEN) announced today that it has closed a license agreement under which Genmab has granted exclusive worldwide rights to develop and commercialize zanolimumab (HuMax-CD4(R)) to TenXBiopharma, Inc. Under the terms of the agreement, Genmab receives a payment of USD 4.5 million and will be entitled to milestones and royalties on sales of zanolimumab. TenX Biopharma will be responsible for all future costs of developing, manufacturing and commercializing zanolimumab. “Genmab is pleased to find a partner who wishes to take zanolimumab forward in development,” said Lisa N. Drakeman, Ph.D., Chief Executive Officer. “This is good news for patients who may one day benefit from treatment with zanolimumab.”“Zanolimumab has promise for treatment of patients with T cell cancers, and potential in other cancer types for which existing therapies are inadequate”, said Gardiner F.H. Smith, Chief Executive Officer of TenX Biopharma. “We are building a pipeline of new medicines for high unmet need, with a focus on the patient through business efficiency.” Asher Nathan, Managing Partner of Zoticon Bioventures, and lead investor in TenX, said “TenX and zanolimumab represent the fourth late stage drug funding which we've led in the past two years. I'm delighted that this important product will be available in the future, and look forward to the management teamat TenX continuing to build value with novel cancer therapeutics.”
mfg ipollit
Heute kamen die ersten Zahlen zu Arzerra. Außerdem wurde das wenig aussichtsreiche Zanolimumab noch für ein paar Mios verscherbelt.
4 Feb, 2010 13:09 CET
Genmab A/S Company Announcement Arzerra 2009 Net Sales Figures
Summary: Net sales of Arzerra™ for the fourth quarter of 2009 wereapproximately DKK 29 million, with an expected royalty payment to Genmab of DKK6 million. Copenhagen, Denmark; February 4, 2010 - Genmab A/S (OMX: GEN) announced todaythat the U.S. net sales for Arzerra (ofatumumab) during the fourth quarter of2009 were approximately DKK 29 million (approximately USD 5.5 million). Underthe terms of the collaboration with GlaxoSmithKline (GSK), Genmab expects to receive a royalty payment of approximately DKK 6 million (approximately USD 1.1million). “We are very pleased to see the first sales figures for Arzerra and we believethat they reflect the need for new treatment options for patients withrefractory CLL,” said Lisa N. Drakeman, Ph.D., Chief Executive Officer ofGenmab. Arzerra was approved by the FDA in October, 2009, for the treatment of patientsin the U.S. with chronic lymphocytic leukemia (CLL) that is refractory tofludarabine and alemtuzumab, and was launched by GSK in mid-November, 2009. Arzerra is a monoclonal antibody that causes the body's immune response tofight against normal and cancerous B-cells. Arzerra attaches to the small andlarge loop epitopes - on a molecule called CD20, which is found on the surfaceof B-cells, the type of cell that becomes cancerous in CLL.
************
4 Feb, 2010 17:48 CET
Genmab A/S Investor News Genmab Outlicenses Zanolimumab to TenX Biopharma
Summary: Genmab has licensed zanolimumab to TenX Biopharma, Inc. Copenhagen, Denmark; February 4, 2010 - Genmab A/S (OMX: GEN) announced today that it has closed a license agreement under which Genmab has granted exclusive worldwide rights to develop and commercialize zanolimumab (HuMax-CD4(R)) to TenXBiopharma, Inc. Under the terms of the agreement, Genmab receives a payment of USD 4.5 million and will be entitled to milestones and royalties on sales of zanolimumab. TenX Biopharma will be responsible for all future costs of developing, manufacturing and commercializing zanolimumab. “Genmab is pleased to find a partner who wishes to take zanolimumab forward in development,” said Lisa N. Drakeman, Ph.D., Chief Executive Officer. “This is good news for patients who may one day benefit from treatment with zanolimumab.”“Zanolimumab has promise for treatment of patients with T cell cancers, and potential in other cancer types for which existing therapies are inadequate”, said Gardiner F.H. Smith, Chief Executive Officer of TenX Biopharma. “We are building a pipeline of new medicines for high unmet need, with a focus on the patient through business efficiency.” Asher Nathan, Managing Partner of Zoticon Bioventures, and lead investor in TenX, said “TenX and zanolimumab represent the fourth late stage drug funding which we've led in the past two years. I'm delighted that this important product will be available in the future, and look forward to the management teamat TenX continuing to build value with novel cancer therapeutics.”
mfg ipollit
Antwort auf Beitrag Nr.: 38.883.581 von ipollit am 04.02.10 20:06:20hier noch ein Artikel, der den Problemfall Genmab ganz gut wiedergibt. Seitdem ist es eher noch schlechter geworden durch das Ende der Entwicklung von Roche's RG1507.
http://www.europharmatoday.com/2009/11/genmabs-star-falls-am…
November 23, 2009
Genmab's Star Falls Amid Setbacks, Cuts And Manufacturing Fire-Sale
What a difference a year makes. In October 2008, Genmab's leaders were being applauded for their prudent strategic review as they cut 101 staff and axed the development of Phase III HuMax-CD4 (zanulimumab) plus a handful of earlier-stage programs, to concentrate instead on the potentially far larger Arzerra (ofatumumab) and zalutumumab, a late-stage EGFR-inhibitor. But now management credibility is being questioned, following the Nov. 5 announcement of a further 300 headcount reduction, setbacks to both remaining key programs, plus the fire-sale of its U.S. manufacturing unit acquired only 18 months earlier.
Genmab was becoming that rare beast in European biotech - a company with home-grown, innovative, late-stage antibody drugs, and significant cash ($333 million at the end of 2008).
As well as securing significant Big Pharma partnerships with GlaxoSmithKline (which paid over $100 million upfront for global rights to ofatumumab in 2006) and Roche (which signed an antibody discovery partnership back in 2001), Genmab had manufacturing capability and, back then, the real possibility of co-promoting its own products in some markets (1 See 'GSK/Genmab: An Impressive Display of Biotech's Increased Leverage,' IN VIVO, Jan. 2007).
On Oct. 27, 2009, the company earned yet another star when FDA approved CD-20 targeting antibody Arzerra for chronic lymphocytic leukaemia (CLL) under an accelerated process (2 'The Pink Sheet' DAILY, Oct. 27, 2009).
A Series of Setbacks
But the full significance of that landmark event was lost in discussions about what has gone wrong at Genmab. Some analysts are even questioning the sustainability of this one-time biotech star.
Granted, Arzerra's approval was anticipated following a positive FDA advisory committee meeting in June and the fact that few treatments are available in this setting. As such, the share price had little movement. And although Arzerra's development and approval path hasn't been smooth, that's hardly unique to Genmab. It's as likely a sign of tougher regulatory hurdles across the board.
Still, Genmab appears - whether through mismanagement, or just bad luck - to have driven itself into a tight corner at a time when investors have minimal appetite for risk.
First, FDA now wants more data from an ongoing CLL study to show that the drug does indeed slow disease progression. And the compound has already stumbled in other indications such as non-Hodgkin's Lymphoma and autoimmune diseases (such as rheumatoid arthritis), both key to the drug's achieving its full potential, yet both highly competitive fields. Some analysts were forecasting peak sales for Arzerra (across all indications) at over $1 billion, although most have cut these forecasts significantly since.
Phase III data in NHL patients refractory to Roche/Genentech's Rituxan (rituximab), released over the summer, showed a disappointingly low response rate - 10 percent versus company expectations of around 20 percent. Thus Genmab was denied the milestone payment from partner GSK and the share price plummeted to a three-year low (3 'The Pink Sheet,' Aug. 24, 2009).
In RA, a market where Arzerra aims to compete against the anti-TNFa drugs (such as Abbott's Humira (adalimumab), neither Genmab nor GSK are clear about which formulation will move forward in trials -intravenous or subcutaneous.
Strategic Errors?
Analysts had been expecting Genmab to react to the Arzerra NHL setback, possibly with a fundraising, but not with a fire-sale. The acquisition of the Brooklyn Park manufacturing site in March 2008 for $240 million now looks to some like a strategic error, which, coupled with the additional headcount reduction, is making many question why Genmab management didn't adequately foresee, during the October 2008 review, the need for these further cuts so soon afterwards. Many point to poor contingency planning and over-optimism.
At the time of the Brooklyn Park purchase, Genmab stated the need to secure "significant manufacturing capacity." Yet Arzerra manufacturing is GSK's responsibility, according to the 2006 deal terms, so Brooklyn Park was in any case used only for producing zalutumumab and the now-shelved HuMax-CD4. To make things worse, antibody manufacturing has since very rapidly shifted from a state of under-capacity to overcapacity, reducing the need for a proprietary facility and, more crucially, making any manufacturing asset hard to sell. Genmab says it hopes to get $145 million for the unit (after $5 million in selling costs) and will take an $83 million impairment charge this quarter.
Chairman and CEO Lisa Drakeman argues that the drastic changes affecting both the financial markets and biologics manufacturing capacity could not have been foreseen.
"We made the best decision we could with all the information we had at the time," she said. "Things changed in a way no one could have predicted and I think that none of us are to be blamed for the world economic climate that has resulted in the kind of actions companies such as Genmab are taking. What we have to do is manage for the world that we live in."
She also acknowledges, however, that most of the company's resources and attention over the past 12 months have been - quite reasonably - focused on Arzerra. "We had a significant role to play in the filing and approval of Arzerra. We had the heaviest workload of any period, especially in terms of the importance of the work. Now that workload has changed dramatically and we are now looking forward, hence the reorganization. This is a major shift, a transformation."
Development Costs Still Significant
Whether or not that's the case, Genmab's about-face on the manufacturing site will at least help it pay what will continue to be hefty development costs, including for its 50 percent share of an Arzerra-Rituxan head-to-head trial in diffuse large B-cell lymphoma (DLBCL), the most common form of NHL, announced Nov. 9. Piper Jaffray analysts estimate that the sale will buy Genmab another two years of operations.
The randomized NHL trial, whose primary endpoint will be progression-free survival, is highly risky. But Arzerra needs to show superiority to Rituxan in order to get any kind of foothold in this indication. The study will also take several years to complete: recruitment alone of 380 patients who are refractory to or have relapsed following first-line treatment with Rituxan plus chemotherapy could take up to two years.
Meanwhile, Genmab's other key drug, zalutumumab, has been hit by a delay in pivotal data on head and neck cancer. Expected before year-end, this will now be released in Q1 2010 - pushing out further partnering revenues. And securing the right partner will be crucial to expand the range of indications (which Genmab narrowed down during its 2008 strategic review) and establish zalutumumab in the already crowded anti-EGFr market, where it will be up against the likes of AstraZeneca's Iressa (gefitinib), Amgen's Vectibix (panitumumab), Roche's Tarceva (erlotinib) and Bristol's Erbitux (cetuximab). Given the relatively small refractory head and neck cancer market and lack of data in any of the larger indications, Piper Jaffray analysts recently slashed their peak sales forecast for this candidate to $150 million from $500 million.
Drakeman, who has been at the company's helm since inception, is putting on a brave face. Perhaps she has some justification: after all, GSK has not returned Arzerra, as no Big Pharma would hesitate to do these days, but is instead pushing ahead with the lengthy and costly Rituxan head-to-head trial.
Yet Genmab's ambition to share in both the costs, and thus more fully in the rewards, of its drug assets worries some investors. "I would rather see GSK take the product in-house, [and] assume all costs in a return for a reduced royalty rate to Genmab. That would make me happier," declares one analyst.
And that's the rub: in this economic environment, risk-taking is being punished, not rewarded. Indeed, Europe's investors have a reputation for being risk-averse at the best of times. Most investors would now much rather Genmab had settled for less upside from its drugs, and faced less financial exposure along the way, whatever their views may have been at the time the GSK deal was signed.
Genmab may yet come through the latest round of setbacks; it has further assets in its pipeline, including three Roche-partnered development-stage programs and a handful of antibodies available for licensing. And Drakeman's right: it's impossible for any biotech CEO to predict the future economic context and thus tailor its deal-terms accordingly.
But deal-terms can be changed, and biotech leaders need big contingencies - they can go for the stars, but need to acknowledge that failures and delays are more likely, and thus plan for those eventualities.
mfg ipollit
http://www.europharmatoday.com/2009/11/genmabs-star-falls-am…
November 23, 2009
Genmab's Star Falls Amid Setbacks, Cuts And Manufacturing Fire-Sale
What a difference a year makes. In October 2008, Genmab's leaders were being applauded for their prudent strategic review as they cut 101 staff and axed the development of Phase III HuMax-CD4 (zanulimumab) plus a handful of earlier-stage programs, to concentrate instead on the potentially far larger Arzerra (ofatumumab) and zalutumumab, a late-stage EGFR-inhibitor. But now management credibility is being questioned, following the Nov. 5 announcement of a further 300 headcount reduction, setbacks to both remaining key programs, plus the fire-sale of its U.S. manufacturing unit acquired only 18 months earlier.
Genmab was becoming that rare beast in European biotech - a company with home-grown, innovative, late-stage antibody drugs, and significant cash ($333 million at the end of 2008).
As well as securing significant Big Pharma partnerships with GlaxoSmithKline (which paid over $100 million upfront for global rights to ofatumumab in 2006) and Roche (which signed an antibody discovery partnership back in 2001), Genmab had manufacturing capability and, back then, the real possibility of co-promoting its own products in some markets (1 See 'GSK/Genmab: An Impressive Display of Biotech's Increased Leverage,' IN VIVO, Jan. 2007).
On Oct. 27, 2009, the company earned yet another star when FDA approved CD-20 targeting antibody Arzerra for chronic lymphocytic leukaemia (CLL) under an accelerated process (2 'The Pink Sheet' DAILY, Oct. 27, 2009).
A Series of Setbacks
But the full significance of that landmark event was lost in discussions about what has gone wrong at Genmab. Some analysts are even questioning the sustainability of this one-time biotech star.
Granted, Arzerra's approval was anticipated following a positive FDA advisory committee meeting in June and the fact that few treatments are available in this setting. As such, the share price had little movement. And although Arzerra's development and approval path hasn't been smooth, that's hardly unique to Genmab. It's as likely a sign of tougher regulatory hurdles across the board.
Still, Genmab appears - whether through mismanagement, or just bad luck - to have driven itself into a tight corner at a time when investors have minimal appetite for risk.
First, FDA now wants more data from an ongoing CLL study to show that the drug does indeed slow disease progression. And the compound has already stumbled in other indications such as non-Hodgkin's Lymphoma and autoimmune diseases (such as rheumatoid arthritis), both key to the drug's achieving its full potential, yet both highly competitive fields. Some analysts were forecasting peak sales for Arzerra (across all indications) at over $1 billion, although most have cut these forecasts significantly since.
Phase III data in NHL patients refractory to Roche/Genentech's Rituxan (rituximab), released over the summer, showed a disappointingly low response rate - 10 percent versus company expectations of around 20 percent. Thus Genmab was denied the milestone payment from partner GSK and the share price plummeted to a three-year low (3 'The Pink Sheet,' Aug. 24, 2009).
In RA, a market where Arzerra aims to compete against the anti-TNFa drugs (such as Abbott's Humira (adalimumab), neither Genmab nor GSK are clear about which formulation will move forward in trials -intravenous or subcutaneous.
Strategic Errors?
Analysts had been expecting Genmab to react to the Arzerra NHL setback, possibly with a fundraising, but not with a fire-sale. The acquisition of the Brooklyn Park manufacturing site in March 2008 for $240 million now looks to some like a strategic error, which, coupled with the additional headcount reduction, is making many question why Genmab management didn't adequately foresee, during the October 2008 review, the need for these further cuts so soon afterwards. Many point to poor contingency planning and over-optimism.
At the time of the Brooklyn Park purchase, Genmab stated the need to secure "significant manufacturing capacity." Yet Arzerra manufacturing is GSK's responsibility, according to the 2006 deal terms, so Brooklyn Park was in any case used only for producing zalutumumab and the now-shelved HuMax-CD4. To make things worse, antibody manufacturing has since very rapidly shifted from a state of under-capacity to overcapacity, reducing the need for a proprietary facility and, more crucially, making any manufacturing asset hard to sell. Genmab says it hopes to get $145 million for the unit (after $5 million in selling costs) and will take an $83 million impairment charge this quarter.
Chairman and CEO Lisa Drakeman argues that the drastic changes affecting both the financial markets and biologics manufacturing capacity could not have been foreseen.
"We made the best decision we could with all the information we had at the time," she said. "Things changed in a way no one could have predicted and I think that none of us are to be blamed for the world economic climate that has resulted in the kind of actions companies such as Genmab are taking. What we have to do is manage for the world that we live in."
She also acknowledges, however, that most of the company's resources and attention over the past 12 months have been - quite reasonably - focused on Arzerra. "We had a significant role to play in the filing and approval of Arzerra. We had the heaviest workload of any period, especially in terms of the importance of the work. Now that workload has changed dramatically and we are now looking forward, hence the reorganization. This is a major shift, a transformation."
Development Costs Still Significant
Whether or not that's the case, Genmab's about-face on the manufacturing site will at least help it pay what will continue to be hefty development costs, including for its 50 percent share of an Arzerra-Rituxan head-to-head trial in diffuse large B-cell lymphoma (DLBCL), the most common form of NHL, announced Nov. 9. Piper Jaffray analysts estimate that the sale will buy Genmab another two years of operations.
The randomized NHL trial, whose primary endpoint will be progression-free survival, is highly risky. But Arzerra needs to show superiority to Rituxan in order to get any kind of foothold in this indication. The study will also take several years to complete: recruitment alone of 380 patients who are refractory to or have relapsed following first-line treatment with Rituxan plus chemotherapy could take up to two years.
Meanwhile, Genmab's other key drug, zalutumumab, has been hit by a delay in pivotal data on head and neck cancer. Expected before year-end, this will now be released in Q1 2010 - pushing out further partnering revenues. And securing the right partner will be crucial to expand the range of indications (which Genmab narrowed down during its 2008 strategic review) and establish zalutumumab in the already crowded anti-EGFr market, where it will be up against the likes of AstraZeneca's Iressa (gefitinib), Amgen's Vectibix (panitumumab), Roche's Tarceva (erlotinib) and Bristol's Erbitux (cetuximab). Given the relatively small refractory head and neck cancer market and lack of data in any of the larger indications, Piper Jaffray analysts recently slashed their peak sales forecast for this candidate to $150 million from $500 million.
Drakeman, who has been at the company's helm since inception, is putting on a brave face. Perhaps she has some justification: after all, GSK has not returned Arzerra, as no Big Pharma would hesitate to do these days, but is instead pushing ahead with the lengthy and costly Rituxan head-to-head trial.
Yet Genmab's ambition to share in both the costs, and thus more fully in the rewards, of its drug assets worries some investors. "I would rather see GSK take the product in-house, [and] assume all costs in a return for a reduced royalty rate to Genmab. That would make me happier," declares one analyst.
And that's the rub: in this economic environment, risk-taking is being punished, not rewarded. Indeed, Europe's investors have a reputation for being risk-averse at the best of times. Most investors would now much rather Genmab had settled for less upside from its drugs, and faced less financial exposure along the way, whatever their views may have been at the time the GSK deal was signed.
Genmab may yet come through the latest round of setbacks; it has further assets in its pipeline, including three Roche-partnered development-stage programs and a handful of antibodies available for licensing. And Drakeman's right: it's impossible for any biotech CEO to predict the future economic context and thus tailor its deal-terms accordingly.
But deal-terms can be changed, and biotech leaders need big contingencies - they can go for the stars, but need to acknowledge that failures and delays are more likely, and thus plan for those eventualities.
mfg ipollit
Antwort auf Beitrag Nr.: 38.883.581 von ipollit am 04.02.10 20:06:20hi ipollit,
ja, glück gehört einfach dazu, gerade im biotech-bereich.
was hältst du eigentlich vom daratumumab-programm von Genmab?
ja, glück gehört einfach dazu, gerade im biotech-bereich.
was hältst du eigentlich vom daratumumab-programm von Genmab?
ARRY...
Array BioPharma Reports Financial Results for the Second Quarter of Fiscal 2010
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (Nasdaq: ARRY - News) today reported financial results for the second quarter of fiscal 2010.
Array reported revenue of $9.6 million for the second quarter of fiscal 2010, compared to revenue of $7.7 million for the same period in fiscal 2009. Array spent $19.1 million for proprietary research and development during the quarter to advance its wholly-owned drugs in clinical development and select discovery programs. This compares to $23.7 million spent for research and development during the second quarter of fiscal 2009. Array reported a net loss of $21.8 million, or ($0.44) per share, for the second quarter, compared to a net loss of $37.8 million, or ($0.79) per share, for the second quarter in fiscal 2009. Array ended the second quarter of fiscal 2010 with $115 million in cash, cash equivalents and marketable securities.
Array reported revenue of $17.5 million for the six-month period ended December 31, 2009, compared to revenue of $13.4 million for the same period in fiscal 2009. Net loss for the six months ended December 31, 2009, was $46.6 million, or ($0.96) per share, compared to a net loss of $71.5 million, or ($1.50) per share, reported in the same six-month period in fiscal 2009.
“We are delighted to partner with Amgen on our type 2 diabetes program, including AMG 151 / ARRY-403, which provided a $60 million up-front payment with additional potential milestones of $666 million and a double-digit royalty,” said Robert E. Conway, Chief Executive Officer. “We continued to implement our partnering strategy with our Amgen deal, which significantly added to our cash balance and reduced our burn. Our partnerships, which include seven Array-invented drugs in clinical trials, provide Array with significant upside of $1.9 billion in potential milestones and royalties that range up to 15 percent. As a result of our partnering efforts, we are revising our guidance for the second half of the fiscal year, increasing our revenue and reducing our loss per share.”
SUMMARY OF RECENT PROGRESS
Array Partners with Amgen in Type 2 Diabetes:
AMG 151 / ARRY-403 – GK activator for type 2 diabetes: Array entered into an agreement with Amgen Inc. for Array’s small-molecule glucokinase activator program, including AMG 151 / ARRY-403, which is currently being tested in a Phase 1 multiple ascending dose clinical trial in patients with type 2 diabetes.
Under the terms of the agreement, Array received an upfront payment of $60 million and additional contingent payments up to $666 million if clinical and commercial milestones are achieved for AMG 151 and at least one back-up compound. Amgen is responsible for future clinical development and commercialization for AMG 151 and any resulting back-up compounds, with Array having an option to co-promote in the United States. Array will receive double digit royalties on sales of AMG 151. In addition, Amgen is providing research funding over the next two-years to identify and advance second-generation glucokinase activators.
Five clinical programs advanced for the treatment of cancer:
ARRY-162 – MEK inhibitor for cancer: Array completed enrolling four cohorts and reached the maximum tolerated dose in a Phase 1 clinical trial in cancer patients with its most advanced wholly-owned MEK inhibitor, ARRY-162. ARRY-162 is now advancing into an expansion phase of the trial in biliary tract cancer patients at ten clinical sites in North America. The Phase 1 dose escalation study is designed to evaluate safety, pharmacokinetics and pharmacodynamics of ARRY-162 in patients with advanced solid tumors.
ARRY-520 – KSP inhibitor for AML and MM: Array continued a Phase 1 trial of ARRY-520, a novel KSP inhibitor, in patients with solid tumors and two Phase 1/2 trials in patients with acute myelogenous leukemia and multiple myeloma.
ARRY-614 - p38/Tie-2 Inhibitor for MDS: Array continued dosing patients with myelodysplastic syndromes (MDS) in a Phase 1 trial with ARRY-614 to determine the safety, maximum tolerated dose, pharmacokinetics and preliminary estimates of efficacy of the compound in this patient population.
ARRY-543 - ErbB family inhibitor for solid tumors: Array completed a Phase 1 dose-escalation study with tablet formulation in patients with solid tumors and continued three Phase 1b trials in combination with Xeloda® (capecitabine), Taxotere® (docetaxel) and Gemzar® (gemcitabine), respectively.
ARRY-380 – HER2 oral, selective inhibitor for cancer: Array continues dose escalation in a Phase 1 trial with ARRY-380. The trial is designed to evaluate the safety, maximum tolerated dose and pharmacokinetics of ARRY-380 in patients with advanced cancer.
Array announced positive interim results of this trial at the 2009 San Antonio Breast Cancer Symposium. These results showed that ARRY-380 at doses greater than or equal to 200 mg BID demonstrated evidence of tumor regression in eight out of ten heavily pre-treated patients with HER2 expressing cancers. Of these eight patients, four had prolonged stable disease for 16 weeks or longer. ARRY-380 has been well-tolerated; the predominant adverse events have been Grade 1 and included nausea, rash and fatigue. In completed cohorts, no Grade 3 or 4 treatment-related adverse events and no cardiac adverse events have been observed.
mfg ipollit
Array BioPharma Reports Financial Results for the Second Quarter of Fiscal 2010
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (Nasdaq: ARRY - News) today reported financial results for the second quarter of fiscal 2010.
Array reported revenue of $9.6 million for the second quarter of fiscal 2010, compared to revenue of $7.7 million for the same period in fiscal 2009. Array spent $19.1 million for proprietary research and development during the quarter to advance its wholly-owned drugs in clinical development and select discovery programs. This compares to $23.7 million spent for research and development during the second quarter of fiscal 2009. Array reported a net loss of $21.8 million, or ($0.44) per share, for the second quarter, compared to a net loss of $37.8 million, or ($0.79) per share, for the second quarter in fiscal 2009. Array ended the second quarter of fiscal 2010 with $115 million in cash, cash equivalents and marketable securities.
Array reported revenue of $17.5 million for the six-month period ended December 31, 2009, compared to revenue of $13.4 million for the same period in fiscal 2009. Net loss for the six months ended December 31, 2009, was $46.6 million, or ($0.96) per share, compared to a net loss of $71.5 million, or ($1.50) per share, reported in the same six-month period in fiscal 2009.
“We are delighted to partner with Amgen on our type 2 diabetes program, including AMG 151 / ARRY-403, which provided a $60 million up-front payment with additional potential milestones of $666 million and a double-digit royalty,” said Robert E. Conway, Chief Executive Officer. “We continued to implement our partnering strategy with our Amgen deal, which significantly added to our cash balance and reduced our burn. Our partnerships, which include seven Array-invented drugs in clinical trials, provide Array with significant upside of $1.9 billion in potential milestones and royalties that range up to 15 percent. As a result of our partnering efforts, we are revising our guidance for the second half of the fiscal year, increasing our revenue and reducing our loss per share.”
SUMMARY OF RECENT PROGRESS
Array Partners with Amgen in Type 2 Diabetes:
AMG 151 / ARRY-403 – GK activator for type 2 diabetes: Array entered into an agreement with Amgen Inc. for Array’s small-molecule glucokinase activator program, including AMG 151 / ARRY-403, which is currently being tested in a Phase 1 multiple ascending dose clinical trial in patients with type 2 diabetes.
Under the terms of the agreement, Array received an upfront payment of $60 million and additional contingent payments up to $666 million if clinical and commercial milestones are achieved for AMG 151 and at least one back-up compound. Amgen is responsible for future clinical development and commercialization for AMG 151 and any resulting back-up compounds, with Array having an option to co-promote in the United States. Array will receive double digit royalties on sales of AMG 151. In addition, Amgen is providing research funding over the next two-years to identify and advance second-generation glucokinase activators.
Five clinical programs advanced for the treatment of cancer:
ARRY-162 – MEK inhibitor for cancer: Array completed enrolling four cohorts and reached the maximum tolerated dose in a Phase 1 clinical trial in cancer patients with its most advanced wholly-owned MEK inhibitor, ARRY-162. ARRY-162 is now advancing into an expansion phase of the trial in biliary tract cancer patients at ten clinical sites in North America. The Phase 1 dose escalation study is designed to evaluate safety, pharmacokinetics and pharmacodynamics of ARRY-162 in patients with advanced solid tumors.
ARRY-520 – KSP inhibitor for AML and MM: Array continued a Phase 1 trial of ARRY-520, a novel KSP inhibitor, in patients with solid tumors and two Phase 1/2 trials in patients with acute myelogenous leukemia and multiple myeloma.
ARRY-614 - p38/Tie-2 Inhibitor for MDS: Array continued dosing patients with myelodysplastic syndromes (MDS) in a Phase 1 trial with ARRY-614 to determine the safety, maximum tolerated dose, pharmacokinetics and preliminary estimates of efficacy of the compound in this patient population.
ARRY-543 - ErbB family inhibitor for solid tumors: Array completed a Phase 1 dose-escalation study with tablet formulation in patients with solid tumors and continued three Phase 1b trials in combination with Xeloda® (capecitabine), Taxotere® (docetaxel) and Gemzar® (gemcitabine), respectively.
ARRY-380 – HER2 oral, selective inhibitor for cancer: Array continues dose escalation in a Phase 1 trial with ARRY-380. The trial is designed to evaluate the safety, maximum tolerated dose and pharmacokinetics of ARRY-380 in patients with advanced cancer.
Array announced positive interim results of this trial at the 2009 San Antonio Breast Cancer Symposium. These results showed that ARRY-380 at doses greater than or equal to 200 mg BID demonstrated evidence of tumor regression in eight out of ten heavily pre-treated patients with HER2 expressing cancers. Of these eight patients, four had prolonged stable disease for 16 weeks or longer. ARRY-380 has been well-tolerated; the predominant adverse events have been Grade 1 and included nausea, rash and fatigue. In completed cohorts, no Grade 3 or 4 treatment-related adverse events and no cardiac adverse events have been observed.
mfg ipollit
Antwort auf Beitrag Nr.: 38.883.680 von ipollit am 04.02.10 20:15:57und noch ein paar Anmerkungen zur ARRY CC... von iHub...
http://investorshub.advfn.com/boards/read_msg.aspx?message_i…
ARRY - Notes on 2Q10 Call
1. ARRY anticipates milestones from existing collaborations to total about $10M for this year and to double to about $20M in 2011.
2. ARRY anticipates 1 or more significant partnering deals in calendar 2010. There is significant partnering interest around ARRY-162 and ARRY-380, but discussions are also ongoing with respect to ARRY-543 and ARRY-520, in addition to discovery assets. ARRY may prefer to get additional clinical data on 380 before partnering the compound to increase the economics of the deal.
3. The 2010 calendar milestones remain largely unchanged from what was posted in #msg-45521735 .
4. ARRY-162 has moved into biliary cancer patients in Phase 1 trials, because biliary cancers have high IL-6, TNF, angiogenesis, and BRAF mutations. AZD6244 showed success in this indication before with 3 PRs, prolonged PFS, and a quality of life benefit (weight gain in a tumor type that is cachexic) in a prior trial, so ARRY is hopeful that 162 will be successful. Japan has over 78,000 biliary cancer patients.
5. Trial design for 162 in biliary tract cancer will look at biomarkers (BRAF and others) to assess activity of the drug. ARRY wants to see the relationship between mutation status and response and see if 162 spurs weight gain and adds to patient pain relief.
****
ARRY also expects to present data on 162, 543 and 380 by the 1st half of the year.
http://seekingalpha.com/article/186072-array-biopharma-inc-f…
mfg ipollit
http://investorshub.advfn.com/boards/read_msg.aspx?message_i…
ARRY - Notes on 2Q10 Call
1. ARRY anticipates milestones from existing collaborations to total about $10M for this year and to double to about $20M in 2011.
2. ARRY anticipates 1 or more significant partnering deals in calendar 2010. There is significant partnering interest around ARRY-162 and ARRY-380, but discussions are also ongoing with respect to ARRY-543 and ARRY-520, in addition to discovery assets. ARRY may prefer to get additional clinical data on 380 before partnering the compound to increase the economics of the deal.
3. The 2010 calendar milestones remain largely unchanged from what was posted in #msg-45521735 .
4. ARRY-162 has moved into biliary cancer patients in Phase 1 trials, because biliary cancers have high IL-6, TNF, angiogenesis, and BRAF mutations. AZD6244 showed success in this indication before with 3 PRs, prolonged PFS, and a quality of life benefit (weight gain in a tumor type that is cachexic) in a prior trial, so ARRY is hopeful that 162 will be successful. Japan has over 78,000 biliary cancer patients.
5. Trial design for 162 in biliary tract cancer will look at biomarkers (BRAF and others) to assess activity of the drug. ARRY wants to see the relationship between mutation status and response and see if 162 spurs weight gain and adds to patient pain relief.
****
ARRY also expects to present data on 162, 543 and 380 by the 1st half of the year.
http://seekingalpha.com/article/186072-array-biopharma-inc-f…
mfg ipollit
Antwort auf Beitrag Nr.: 38.883.657 von PathFinder2 am 04.02.10 20:12:52daratumumab... hab ich bisher noch nicht viel zu gefunden. Zieht sich ja auch schon länger hin... sind aber damit immerhin weiter als MOR
mfg ipollit
mfg ipollit
Durch den Anstieg ist Neurosearch nun doch zu einer größeren Position geworden. Huntexil bietet meiner Meinung nach weiter Potential. Einen kleinen Rückschlag wird es wohl noch geben, da Glaxo angekündigt hat, generell aus der Indikation Depressionen auszusteigen, zu der sie auch einige Projekte mit NeuroSearch haben.
aktuelle Positionen im Depot...
10,5% Incyte http://finance.yahoo.com/q?s=INCY
10,1% Regeneron http://finance.yahoo.com/q?s=REGN
8,3% Genmab http://finance.yahoo.com/q?s=GEN.CO
8,1% NeuroSearch http://finance.yahoo.com/q?s=NEUR.CO
7,1% Cubist http://finance.yahoo.com/q?s=CBST
5,8% Micromet http://finance.yahoo.com/q?s=MITI
5,8% Allos http://finance.yahoo.com/q?s=ALTH
5,5% Onyx http://finance.yahoo.com/q?s=ONXX
5,1% Rigel http://finance.yahoo.com/q?s=RIGL
4,9% Isis http://finance.yahoo.com/q?s=ISIS
4,9% Exelixis http://finance.yahoo.com/q?s=EXEL
3,5% NeurogesX http://finance.yahoo.com/q?s=NGSX
3,3% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,1% Medigene http://finance.yahoo.com/q?s=MDG.DE
2,9% ArQule http://finance.yahoo.com/q?s=ARQL
2,7% Array http://finance.yahoo.com/q?s=ARRY
2,3% ViroPharma http://finance.yahoo.com/q?s=VPHM
1,9% Arena http://finance.yahoo.com/q?s=ARNA
1,5% NicOx http://finance.yahoo.com/q?s=COX.PA
1,1% Progenics http://finance.yahoo.com/q?s=PGNX
0,9% Addex http://finance.yahoo.com/q?s=ADXN.SW
0,8% Sucampo http://finance.yahoo.com/q?s=SCMP
das Depot hält sich Dank NeuroSearch und der EUR-Schwäche bisher noch ganz gut... YTD Jahres-Entwicklung auf EUR-Basis:
+12,5% Depot
+5,5% NBI
-8,8% Dax
+5,2% USD
+190,8% NeuroSearch ( 8,1% )
+30,8% Micromet ( 5,8% )
+21,2% ViroPharma ( 2,3% )
+18,9% Genmab ( 8,3% )
+17,7% Incyte ( 10,5% )
+12,9% Allos ( 5,8% )
+12,4% Cubist ( 7,1% )
+12,0% Regeneron ( 10,1% )
+5,2% Progenics ( 1,1% )
+4,6% Isis ( 4,9% )
+4,4% Onyx ( 5,5% )
+1,1% NeurogesX ( 3,5% )
-0,3% Medigene ( 3,1% )
-2,7% Seattle Genetics ( 3,3% )
-3,7% Rigel ( 5,1% )
-5,0% NicOx ( 1,5% )
-6,0% Arena ( 1,9% )
-7,5% Array ( 2,7% )
-10,7% ArQule ( 2,9% )
-11,2% Sucampo ( 0,8% )
-11,2% Exelixis ( 4,9% )
-15,0% Addex ( 0,9% )
mfg ipollit
aktuelle Positionen im Depot...
10,5% Incyte http://finance.yahoo.com/q?s=INCY
10,1% Regeneron http://finance.yahoo.com/q?s=REGN
8,3% Genmab http://finance.yahoo.com/q?s=GEN.CO
8,1% NeuroSearch http://finance.yahoo.com/q?s=NEUR.CO
7,1% Cubist http://finance.yahoo.com/q?s=CBST
5,8% Micromet http://finance.yahoo.com/q?s=MITI
5,8% Allos http://finance.yahoo.com/q?s=ALTH
5,5% Onyx http://finance.yahoo.com/q?s=ONXX
5,1% Rigel http://finance.yahoo.com/q?s=RIGL
4,9% Isis http://finance.yahoo.com/q?s=ISIS
4,9% Exelixis http://finance.yahoo.com/q?s=EXEL
3,5% NeurogesX http://finance.yahoo.com/q?s=NGSX
3,3% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,1% Medigene http://finance.yahoo.com/q?s=MDG.DE
2,9% ArQule http://finance.yahoo.com/q?s=ARQL
2,7% Array http://finance.yahoo.com/q?s=ARRY
2,3% ViroPharma http://finance.yahoo.com/q?s=VPHM
1,9% Arena http://finance.yahoo.com/q?s=ARNA
1,5% NicOx http://finance.yahoo.com/q?s=COX.PA
1,1% Progenics http://finance.yahoo.com/q?s=PGNX
0,9% Addex http://finance.yahoo.com/q?s=ADXN.SW
0,8% Sucampo http://finance.yahoo.com/q?s=SCMP
das Depot hält sich Dank NeuroSearch und der EUR-Schwäche bisher noch ganz gut... YTD Jahres-Entwicklung auf EUR-Basis:
+12,5% Depot
+5,5% NBI
-8,8% Dax
+5,2% USD
+190,8% NeuroSearch ( 8,1% )
+30,8% Micromet ( 5,8% )
+21,2% ViroPharma ( 2,3% )
+18,9% Genmab ( 8,3% )
+17,7% Incyte ( 10,5% )
+12,9% Allos ( 5,8% )
+12,4% Cubist ( 7,1% )
+12,0% Regeneron ( 10,1% )
+5,2% Progenics ( 1,1% )
+4,6% Isis ( 4,9% )
+4,4% Onyx ( 5,5% )
+1,1% NeurogesX ( 3,5% )
-0,3% Medigene ( 3,1% )
-2,7% Seattle Genetics ( 3,3% )
-3,7% Rigel ( 5,1% )
-5,0% NicOx ( 1,5% )
-6,0% Arena ( 1,9% )
-7,5% Array ( 2,7% )
-10,7% ArQule ( 2,9% )
-11,2% Sucampo ( 0,8% )
-11,2% Exelixis ( 4,9% )
-15,0% Addex ( 0,9% )
mfg ipollit
auch eine meiner Meinung interessante Long-Position gewesen... idealerweise gestern: AMAG. Leider habe ich dafür im Moment nichts frei, zumal es ja gerade auch generell abwärts geht.
Feraheme ist gerade am Markt und lag bisher über den Erwartungen... die Sicherheitsbedenken scheinen unbegründet zu sein und vielleicht nur gestreut, um den Kurs zu drücken

http://www.reuters.com/article/idCAN0516236520100205?rpc=44
UPDATE 2-AMAG safety update on anemia drug lifts shares
* AMAG says adverse event rate in line with drug label
* AMAG does not believe single death related to Feraheme
* Shares rise 4.5 pct
By Bill Berkrot and Ransdell Pierson
NEW YORK, Feb 5 (Reuters) - AMAG Pharmaceuticals Inc (AMAG.O), whose shares fell 16 percent on Thursday on concerns about the safety of its Feraheme anemia treatment, said the number of serious adverse events seen with the product was in line with what would be expected from the intravenous drug.
AMAG, whose shares rose 4.5 percent after the company issued its safety update late Friday, said 40 serious adverse events had been reported since the iron replacement drug was introduced in the United States in July 2009. That represents a rate of 0.1 percent of the 35,000 "patient exposures" to date, AMAG said.
"A single reported death occurred in a patient two days post-Feraheme treatment, which the company does not believe was the result of Feraheme," the company said in a statement.
"Since the infusion was well tolerated we don't think it (the death) was related to the drug," AMAG's Chief Medical Officer Lee Allen later said on a conference call.
Feraheme is approved for the treatment of iron deficiency anemia in adult patients with chronic kidney disease.
Analysts said they believe the AMAG share sell-off earlier in the week, which was triggered by a research note suggesting there had been a higher-than-expected rate of serious adverse events from the drug, was unfounded.
"Every drug has beneficial effects and unwanted side effects," Needham & Co analyst Mark Monane said, adding that he believes Feraheme has a desirable benefit/risk ratio that favors use of the drug.
Monane said he still expects Feraheme will eventually garner $500 million to $1 billion in annual sales once it gains broader approval for use in a wider variety of patients.
The company said the rate of serious side effects, such as a potentially dangerous drop in blood pressure or allergic reaction "was consistent with that contained in the U.S. package insert."
A drug's label, or package insert, typically includes results from clinical trials as well as adverse events seen with the medicine's use in studies.
Robert W Baird analyst Christopher Raymond said in a note that Friday's safety update "provides a big sigh of relief."
"With safety rumors completely dispelled, we continue to recommend purchase of AMAG shares into the low $50 range," Raymond said.
AMAG shares rose to $39.50 after hours from their Nasdaq close on Friday at $37.77.
Summer Street Research analyst Carol Werther also believes the recent sell-off was "overdone," but she was not quite ready to write off safety concerns as completely dispelled.
"The clinicians I spoke to are going to use the drug more cautiously until they have more information. Whether the information that came out today is enough, I have no idea," she said.
mfg ipollit
Feraheme ist gerade am Markt und lag bisher über den Erwartungen... die Sicherheitsbedenken scheinen unbegründet zu sein und vielleicht nur gestreut, um den Kurs zu drücken
http://www.reuters.com/article/idCAN0516236520100205?rpc=44
UPDATE 2-AMAG safety update on anemia drug lifts shares
* AMAG says adverse event rate in line with drug label
* AMAG does not believe single death related to Feraheme
* Shares rise 4.5 pct
By Bill Berkrot and Ransdell Pierson
NEW YORK, Feb 5 (Reuters) - AMAG Pharmaceuticals Inc (AMAG.O), whose shares fell 16 percent on Thursday on concerns about the safety of its Feraheme anemia treatment, said the number of serious adverse events seen with the product was in line with what would be expected from the intravenous drug.
AMAG, whose shares rose 4.5 percent after the company issued its safety update late Friday, said 40 serious adverse events had been reported since the iron replacement drug was introduced in the United States in July 2009. That represents a rate of 0.1 percent of the 35,000 "patient exposures" to date, AMAG said.
"A single reported death occurred in a patient two days post-Feraheme treatment, which the company does not believe was the result of Feraheme," the company said in a statement.
"Since the infusion was well tolerated we don't think it (the death) was related to the drug," AMAG's Chief Medical Officer Lee Allen later said on a conference call.
Feraheme is approved for the treatment of iron deficiency anemia in adult patients with chronic kidney disease.
Analysts said they believe the AMAG share sell-off earlier in the week, which was triggered by a research note suggesting there had been a higher-than-expected rate of serious adverse events from the drug, was unfounded.
"Every drug has beneficial effects and unwanted side effects," Needham & Co analyst Mark Monane said, adding that he believes Feraheme has a desirable benefit/risk ratio that favors use of the drug.
Monane said he still expects Feraheme will eventually garner $500 million to $1 billion in annual sales once it gains broader approval for use in a wider variety of patients.
The company said the rate of serious side effects, such as a potentially dangerous drop in blood pressure or allergic reaction "was consistent with that contained in the U.S. package insert."
A drug's label, or package insert, typically includes results from clinical trials as well as adverse events seen with the medicine's use in studies.
Robert W Baird analyst Christopher Raymond said in a note that Friday's safety update "provides a big sigh of relief."
"With safety rumors completely dispelled, we continue to recommend purchase of AMAG shares into the low $50 range," Raymond said.
AMAG shares rose to $39.50 after hours from their Nasdaq close on Friday at $37.77.
Summer Street Research analyst Carol Werther also believes the recent sell-off was "overdone," but she was not quite ready to write off safety concerns as completely dispelled.
"The clinicians I spoke to are going to use the drug more cautiously until they have more information. Whether the information that came out today is enough, I have no idea," she said.
mfg ipollit
Antwort auf Beitrag Nr.: 38.869.634 von ipollit am 03.02.10 10:29:48hier auch nochmal zu dem Erfolg von NeuroSearch... genau genommen ein großer Erfolg der Arbeit von Arvid Carlsson.
http://www.thelocal.se/24818/20100205/
Swedes develop Huntington's medicine
Published: 5 Feb 10 13:48 CET
A Swedish biotechnology firm has developed a new medicine which provides hope for sufferers of Huntington's disease, according to a report in Göteborgs-Posten.
The new medicine, based on the findings of Nobel prize winning scientist Arvid Carlsson, is set to replace the only other treatment for the disease.
Huntexil is the name given to the new medicine which has been found to relieve the disease's tension and tremors without the secondary side-effects of existing drugs.
The next stage in the development of Huntexil is to have it approved as a medicine with its prospects to date reported to be bright.
The company responsible for the research is called Neurosearch, a Denmark-based firm with a subsidiary in Gothenburg. The firm emerged from the group of research scientists working with Arvid Carlsson.
Arvid Carlsson is a Swedish scientist who is best known for his work to develop a method for measuring the amount of dopamine in brain tissues and it effects on Parkinson's disease.
Carlsson was awarded the Nobel prize in Physiology or Medicine in 2000.
mfg ipollit
http://www.thelocal.se/24818/20100205/
Swedes develop Huntington's medicine
Published: 5 Feb 10 13:48 CET
A Swedish biotechnology firm has developed a new medicine which provides hope for sufferers of Huntington's disease, according to a report in Göteborgs-Posten.
The new medicine, based on the findings of Nobel prize winning scientist Arvid Carlsson, is set to replace the only other treatment for the disease.
Huntexil is the name given to the new medicine which has been found to relieve the disease's tension and tremors without the secondary side-effects of existing drugs.
The next stage in the development of Huntexil is to have it approved as a medicine with its prospects to date reported to be bright.
The company responsible for the research is called Neurosearch, a Denmark-based firm with a subsidiary in Gothenburg. The firm emerged from the group of research scientists working with Arvid Carlsson.
Arvid Carlsson is a Swedish scientist who is best known for his work to develop a method for measuring the amount of dopamine in brain tissues and it effects on Parkinson's disease.
Carlsson was awarded the Nobel prize in Physiology or Medicine in 2000.
mfg ipollit
zu ISIS:
Genzyme Corporation and Isis Pharmaceuticals Say Cholesterol Drug Met Trial Goal-Reuters
9:07am EST
Reuters reported that Genzyme Corporation and Isis Pharmaceuticals said that their drug to treat a subset of patients with high cholesterol met the main goal of a late stage clinical trial. The drug is being tested in patients with severe, inherited high cholesterol, known as familial hypercholesterolemia, in which patients are unable to properly metabolize LDL due to dysfunctional LDL receptors, which are responsible for clearing LDL from plasma. The companies said elevated liver enzymes were seen in some patients, at about the same rate as in previous trials. The Company, which is handling the regulatory filings for the drug, will first seek approval for patients with hoFH. A late stage study of the drug previously met its main goal of a 25% average reduction in LDL cholesterol. The Company expects to file for U.S. and European approval for the drug in the first half of 2011. - - - - - (repeated)
----------
Kurs ist nach der Meldung 15% im Minus... Liegts am gefetteten Satz?
Genzyme Corporation and Isis Pharmaceuticals Say Cholesterol Drug Met Trial Goal-Reuters
9:07am EST
Reuters reported that Genzyme Corporation and Isis Pharmaceuticals said that their drug to treat a subset of patients with high cholesterol met the main goal of a late stage clinical trial. The drug is being tested in patients with severe, inherited high cholesterol, known as familial hypercholesterolemia, in which patients are unable to properly metabolize LDL due to dysfunctional LDL receptors, which are responsible for clearing LDL from plasma. The companies said elevated liver enzymes were seen in some patients, at about the same rate as in previous trials. The Company, which is handling the regulatory filings for the drug, will first seek approval for patients with hoFH. A late stage study of the drug previously met its main goal of a 25% average reduction in LDL cholesterol. The Company expects to file for U.S. and European approval for the drug in the first half of 2011. - - - - - (repeated)
----------
Kurs ist nach der Meldung 15% im Minus... Liegts am gefetteten Satz?
Antwort auf Beitrag Nr.: 38.920.147 von SLGramann am 10.02.10 17:37:07Genau ...
The companies spooked Wall Street by noting that elevated levels of liver enzymes called transaminases were observed in the study that were similar in magnitude and duration to those seen in earlier studies.
Such elevated enzymes are considered to be a marker of potential liver toxicity and are seen with standard "statin" pills used to treat cholesterol and with many other medicines.
But elevated enzymes have torpedoed approvals of some promising drugs .
The companies spooked Wall Street by noting that elevated levels of liver enzymes called transaminases were observed in the study that were similar in magnitude and duration to those seen in earlier studies.
Such elevated enzymes are considered to be a marker of potential liver toxicity and are seen with standard "statin" pills used to treat cholesterol and with many other medicines.
But elevated enzymes have torpedoed approvals of some promising drugs .
Antwort auf Beitrag Nr.: 38.920.175 von BrauchGeld am 10.02.10 17:40:21http://www.thestreet.com/story/10678655/1/isis-genzyme-fall-…
(...) On a conference call Wednesday morning, Isis CEO Stanley Crooke downplayed the mipomersen safety worries, stating that "no new concerns" were raised in this study that were not detected in previous studies. Furthermore, Crooke said none of the patients treated with mipomersen experienced liver dysfunction.
Yet, at the same time, Crooke, in reply to a question, conceded that MRI scans of mipomersen patients in the study detected fat deposits on their livers -- a safety signal that wasn't found in prior mipomersen studies.
"We're still analyzing the data," said Crooke of the liver-fat findings, adding that it's consistent with what's been shown when mipomersen was tested in animals.
Isis and Genzyme previously conducted a phase III study of mipomersen in patients with homozygous familial hypercholesterolemia (HoFH) -- a rarer and much more severe form of the same disease in which patients inherit two defective genes that can cause fatal levels of cholesterol elevation. In that study, mipomersen lowered cholesterol levels by 25% but patients also experienced elevated liver enzymes.
HeFH patients are less sick than HoFH patients, so expectations going into Wednesday results were that mipomersen would cause less liver toxicity in HeFH patients. But in its announcement Wednesday and on the conference call, Isis described the liver safety data as being "similar" between the two patient groups. (...)
(...) On a conference call Wednesday morning, Isis CEO Stanley Crooke downplayed the mipomersen safety worries, stating that "no new concerns" were raised in this study that were not detected in previous studies. Furthermore, Crooke said none of the patients treated with mipomersen experienced liver dysfunction.
Yet, at the same time, Crooke, in reply to a question, conceded that MRI scans of mipomersen patients in the study detected fat deposits on their livers -- a safety signal that wasn't found in prior mipomersen studies.
"We're still analyzing the data," said Crooke of the liver-fat findings, adding that it's consistent with what's been shown when mipomersen was tested in animals.
Isis and Genzyme previously conducted a phase III study of mipomersen in patients with homozygous familial hypercholesterolemia (HoFH) -- a rarer and much more severe form of the same disease in which patients inherit two defective genes that can cause fatal levels of cholesterol elevation. In that study, mipomersen lowered cholesterol levels by 25% but patients also experienced elevated liver enzymes.
HeFH patients are less sick than HoFH patients, so expectations going into Wednesday results were that mipomersen would cause less liver toxicity in HeFH patients. But in its announcement Wednesday and on the conference call, Isis described the liver safety data as being "similar" between the two patient groups. (...)
Hallo - bin neu hier.
Frage an Euch Bio-Freaks.
Schon was von Sino European Biotechnology AG, ISIN CH0042354727
gehört??
Frage an Euch Bio-Freaks.
Schon was von Sino European Biotechnology AG, ISIN CH0042354727
gehört??
Antwort auf Beitrag Nr.: 38.929.203 von raphel am 11.02.10 19:59:51hallo,
nein, aber der name klingt echt unseriös
nein, aber der name klingt echt unseriös
Noch mal zu ISIS und mipomersen:
A Wonder Drug With Not-So-Wonderful Side Effects
Brian Orelli, Ph.D.
February 11, 2010
Side effects may not kill Isis Pharmaceuticals (Nasdaq: ISIS) and Genzyme's (Nasdaq: GENZ) cholesterol drug mipomersen, but they could severely diminish the potential patient population that would be willing to take it.
News of the side effects dampened Isis' shares yesterday, which fell more than 18%. As marketing partner Genzyme already has several drugs on the market, its shares were hardly touched by the news.
Let's start with the good news about the drug's great efficacy data. In patients who have a genetic disorder that results in high cholesterol levels even while taking cholesterol-lowering drugs -- like Pfizer's (NYSE: PFE) Lipitor, AstraZeneca's (NYSE: AZN) Crestor, Merck's (NYSE: MRK) Zetia, and Abbott Labs' (NYSE: ABT) Niaspan -- mipomersen was able to reduce cholesterol by 28%. Patients taking placebo saw their cholesterol levels go up 5%.
The bad news is that increased liver enzymes continue to plague mipomersen -- 14% of patients taking mipomersen saw liver enzyme levels at least three-times the normal level. Elevated liver enzymes are a sign of liver toxicity and a reason that many drugs die in clinical trials. Most of the patients only had one measurement that was high and there weren't other signs of liver damage, which puts the companies in an awkward position. The side effects may not be severe enough to keep mipomersen from getting approved, but only the most severe patients may be willing to roll the dice given the side effect signal.
We should know a little more when the full data set is presented at a medical meeting. Here are the two most important things to look out for:
* The number of patients that rolled over to the open label study. After completion of the 26-week trial, both patients that got mipomersen and those on placebo are given the option of taking mipomersen, so the company can get long-term safety data from the patients. For investors, a high rollover rate indicates that doctors and patients think the benefits outweigh the risk.
* The results of MRIs looking at fat build up in the liver. Management said that there was an initial build up of fat, but they're hoping images from later in the study show that the fat decreases over time like it does with other cholesterol drugs.
After the first trial, I said that investors might be overreacting. Now that we've had two trials with increased liver enzymes, I'd say a little paranoia is justified. The data to date is likely good enough to get past the FDA, but if mipomersen can only be used on the most severe patients, sales will be limited.
-----------------
Tja... ISIS wird jetzt noch mit 850 Mio. bewertet. Zieht man die 550 Mio. Cash ab (was auch nicht ganz sauber ist), dann kostet das Unternehmen jetzt noch ganze 300 Mio. Dollar.
Was sagt uns das jetzt? Keine Ahnung. Leider.
A Wonder Drug With Not-So-Wonderful Side Effects
Brian Orelli, Ph.D.
February 11, 2010
Side effects may not kill Isis Pharmaceuticals (Nasdaq: ISIS) and Genzyme's (Nasdaq: GENZ) cholesterol drug mipomersen, but they could severely diminish the potential patient population that would be willing to take it.
News of the side effects dampened Isis' shares yesterday, which fell more than 18%. As marketing partner Genzyme already has several drugs on the market, its shares were hardly touched by the news.
Let's start with the good news about the drug's great efficacy data. In patients who have a genetic disorder that results in high cholesterol levels even while taking cholesterol-lowering drugs -- like Pfizer's (NYSE: PFE) Lipitor, AstraZeneca's (NYSE: AZN) Crestor, Merck's (NYSE: MRK) Zetia, and Abbott Labs' (NYSE: ABT) Niaspan -- mipomersen was able to reduce cholesterol by 28%. Patients taking placebo saw their cholesterol levels go up 5%.
The bad news is that increased liver enzymes continue to plague mipomersen -- 14% of patients taking mipomersen saw liver enzyme levels at least three-times the normal level. Elevated liver enzymes are a sign of liver toxicity and a reason that many drugs die in clinical trials. Most of the patients only had one measurement that was high and there weren't other signs of liver damage, which puts the companies in an awkward position. The side effects may not be severe enough to keep mipomersen from getting approved, but only the most severe patients may be willing to roll the dice given the side effect signal.
We should know a little more when the full data set is presented at a medical meeting. Here are the two most important things to look out for:
* The number of patients that rolled over to the open label study. After completion of the 26-week trial, both patients that got mipomersen and those on placebo are given the option of taking mipomersen, so the company can get long-term safety data from the patients. For investors, a high rollover rate indicates that doctors and patients think the benefits outweigh the risk.
* The results of MRIs looking at fat build up in the liver. Management said that there was an initial build up of fat, but they're hoping images from later in the study show that the fat decreases over time like it does with other cholesterol drugs.
After the first trial, I said that investors might be overreacting. Now that we've had two trials with increased liver enzymes, I'd say a little paranoia is justified. The data to date is likely good enough to get past the FDA, but if mipomersen can only be used on the most severe patients, sales will be limited.
-----------------
Tja... ISIS wird jetzt noch mit 850 Mio. bewertet. Zieht man die 550 Mio. Cash ab (was auch nicht ganz sauber ist), dann kostet das Unternehmen jetzt noch ganze 300 Mio. Dollar.
Was sagt uns das jetzt? Keine Ahnung. Leider.
Antwort auf Beitrag Nr.: 38.929.203 von raphel am 11.02.10 19:59:51Diese Sino Scheiße müllt mir seit Wochen meinen Briefkasten zu.
Wieder eine Biotech Hülle ohne inneren Wert, aber dafür mit ausgezeichneter PR. Haben für alles ein Mittel und können alles heilen.
Finger weg! Unseriös! Totalverlust vorprogrammiert! Der Sitz wird wieder in Zug / CH sein und die Personen dahinter werden auch dieses mal keine Unbekannten sein.
Wieder eine Biotech Hülle ohne inneren Wert, aber dafür mit ausgezeichneter PR. Haben für alles ein Mittel und können alles heilen.
Finger weg! Unseriös! Totalverlust vorprogrammiert! Der Sitz wird wieder in Zug / CH sein und die Personen dahinter werden auch dieses mal keine Unbekannten sein.
Antwort auf Beitrag Nr.: 38.929.203 von raphel am 11.02.10 19:59:51auf deren homepage konnte ich keine bllanz noch geschäftsbericht finden!!
wohl neustart
wohl neustart
RIGL... eine Sache, die sehr unsicher war, scheint trotz allem wohl erfolgreich gewesen zu sein
Rigel schafft den erhofften Deal... 100 Mio USD Upfront, zweistellige Royalties und über 1 Mrd USD Meilensteine. AstraZeneca übernimmt die komplette Entwicklung und Vermarktung von R788, was bei dieser umfangreichen Indikation RA sehr wichtig ist.
Der Haken an der Sache war, dass es offensichtlich wissenschaftlich Bedenken gegen den chronischen Einsatz von Syk-Hemmern gibt. Z.B. gab es zuletzt Studien, in denen Syk Brustkrebs verhindert und die Hemmung entsprechend zur Förderung von Brustkrebs führte. Mit diesem Deal liegt das aber jetzt in der Hand von AZ.
http://www.reuters.com/article/idCNSGE61F05420100216?rpc=44
UPDATE 2-Astra pays up to $1.2 bln for Rigel arthritis drug
Tue Feb 16, 2010 3:28am
* Rigel to receive $100 mln upfront payment from AstraZeneca
* Up to $1.145 bln in milestones plus double-digit royalties
* AstraZeneca aims to file new drug for approval in 2013
* R788 a potential rival to Pfizer oral arthritis medicine
By Ben Hirschler
LONDON, Feb 16 (Reuters) - AstraZeneca Plc (AZN.L) waded into the competitive rheumatoid arthritis market on Tuesday by signing a deal worth up to $1.245 billion for rights to Rigel Pharmaceuticals Inc's (RIGL.O) next-generation drug R788.
The deal is a big vote of confidence in the Rigel product, known chemically as fostamatinib disodium, which failed in a mid-stage Phase II study last July.
Despite that setback, AstraZeneca and Rigel believe it can still win through in final-stage Phase III tests, as an oral alternative to injections of anti-TNF medicines.
An AstraZeneca spokesman said there was evidence that results in the failed trial were due to technical issues with the study design and interpretation.
R788 is the furthest developed in a new class of drugs called oral spleen tyrosine kinase inhibitors being evaluated for rheumatoid arthritis.
Pfizer Inc (PFE.N) is developing a rival oral rheumatoid arthritis pill that works through a different mechanism of action.
The big licensing deal is another example of Chief Executive David Brennan's drive to "externalise" research at the Anglo-Swedish group, which is cutting back in-house staffing as it deals with a coming wave of patent losses on top medicines.
Under the agreement, AstraZeneca will make an upfront payment of $100 million to Rigel, with up to $345 million more payable after achieving regulatory and first sale milestones, the two companies said in a statement.
Rigel will also be eligible to receive up to an additional $800 million and double-digit royalties on specific sales-related payments.
AstraZeneca will design a Phase III programme, anticipated to begin in the second half of 2010, with the goal of filing a new drug application in 2013.
The London-based firm is responsible for all development, manufacturing and global commercialisation activities under the contract.
Rigel believes its drug has multi-billion dollar sales potential as an alternative to injections of blockbuster anti-TNF drugs like Amgen Inc's (AMGN.O) Enbrel, Johnson & Johnson's (JNJ.N) Remicade and Abbott Laboratories' (ABT.N) Humira. These drugs work by blocking an inflammatory protein known as a tumour necrosis factor, or TNF.
R788 is designed to help patients who do not respond to the older drug methotrexate, which is the current standard of care for rheumatoid arthritis patients.
For AstraZeneca, buying rights to R788 marks a significant step up in its investment in arthritis, although it is not its first foray in the field.
Its biotech division, MedImmune, has a monoclonal antibody in Phase I trials and AstraZeneca is also seeking approval for a new painkiller, Vimovo, to treat the symptoms of both rheumatoid and osteoarthritis.
mfg ipollit
Rigel schafft den erhofften Deal... 100 Mio USD Upfront, zweistellige Royalties und über 1 Mrd USD Meilensteine. AstraZeneca übernimmt die komplette Entwicklung und Vermarktung von R788, was bei dieser umfangreichen Indikation RA sehr wichtig ist.
Der Haken an der Sache war, dass es offensichtlich wissenschaftlich Bedenken gegen den chronischen Einsatz von Syk-Hemmern gibt. Z.B. gab es zuletzt Studien, in denen Syk Brustkrebs verhindert und die Hemmung entsprechend zur Förderung von Brustkrebs führte. Mit diesem Deal liegt das aber jetzt in der Hand von AZ.
http://www.reuters.com/article/idCNSGE61F05420100216?rpc=44
UPDATE 2-Astra pays up to $1.2 bln for Rigel arthritis drug
Tue Feb 16, 2010 3:28am
* Rigel to receive $100 mln upfront payment from AstraZeneca
* Up to $1.145 bln in milestones plus double-digit royalties
* AstraZeneca aims to file new drug for approval in 2013
* R788 a potential rival to Pfizer oral arthritis medicine
By Ben Hirschler
LONDON, Feb 16 (Reuters) - AstraZeneca Plc (AZN.L) waded into the competitive rheumatoid arthritis market on Tuesday by signing a deal worth up to $1.245 billion for rights to Rigel Pharmaceuticals Inc's (RIGL.O) next-generation drug R788.
The deal is a big vote of confidence in the Rigel product, known chemically as fostamatinib disodium, which failed in a mid-stage Phase II study last July.
Despite that setback, AstraZeneca and Rigel believe it can still win through in final-stage Phase III tests, as an oral alternative to injections of anti-TNF medicines.
An AstraZeneca spokesman said there was evidence that results in the failed trial were due to technical issues with the study design and interpretation.
R788 is the furthest developed in a new class of drugs called oral spleen tyrosine kinase inhibitors being evaluated for rheumatoid arthritis.
Pfizer Inc (PFE.N) is developing a rival oral rheumatoid arthritis pill that works through a different mechanism of action.
The big licensing deal is another example of Chief Executive David Brennan's drive to "externalise" research at the Anglo-Swedish group, which is cutting back in-house staffing as it deals with a coming wave of patent losses on top medicines.
Under the agreement, AstraZeneca will make an upfront payment of $100 million to Rigel, with up to $345 million more payable after achieving regulatory and first sale milestones, the two companies said in a statement.
Rigel will also be eligible to receive up to an additional $800 million and double-digit royalties on specific sales-related payments.
AstraZeneca will design a Phase III programme, anticipated to begin in the second half of 2010, with the goal of filing a new drug application in 2013.
The London-based firm is responsible for all development, manufacturing and global commercialisation activities under the contract.
Rigel believes its drug has multi-billion dollar sales potential as an alternative to injections of blockbuster anti-TNF drugs like Amgen Inc's (AMGN.O) Enbrel, Johnson & Johnson's (JNJ.N) Remicade and Abbott Laboratories' (ABT.N) Humira. These drugs work by blocking an inflammatory protein known as a tumour necrosis factor, or TNF.
R788 is designed to help patients who do not respond to the older drug methotrexate, which is the current standard of care for rheumatoid arthritis patients.
For AstraZeneca, buying rights to R788 marks a significant step up in its investment in arthritis, although it is not its first foray in the field.
Its biotech division, MedImmune, has a monoclonal antibody in Phase I trials and AstraZeneca is also seeking approval for a new painkiller, Vimovo, to treat the symptoms of both rheumatoid and osteoarthritis.
mfg ipollit
Antwort auf Beitrag Nr.: 38.949.184 von ipollit am 16.02.10 10:41:35
Hi ipollit,
RIGL sind derzeit beim 10fachen Handelsvolumen (bis jetzt!) 7% im Minus. NIcht unbedingt das, was man erwartet hätte würde ich sagen...
Hast Du dafür eine Erklärung? Hat der Markt einfach einen noch besseren Deal erwartet? Das ist nach den jüngsten Problemen doch wohl kaum anzunehmen, oder doch?
Ich denke gerade über eine kleine Position bei RIGL nach. Eigentlich war mein Depot für 2010 ja fertig, aber...
RIGL scheint mir derzeit nicht so wahnsinnig teuer zu sein. Die Marktkapitalisierung dürfte nach Abzug des vorhandenen und des neu reinkommenden Cash bei ca. 250 Mio. Dollar liegen.
Du hattest RIGL ja eh relativ hoch gewichtet...
Gruß
Hi ipollit,
RIGL sind derzeit beim 10fachen Handelsvolumen (bis jetzt!) 7% im Minus. NIcht unbedingt das, was man erwartet hätte würde ich sagen...
Hast Du dafür eine Erklärung? Hat der Markt einfach einen noch besseren Deal erwartet? Das ist nach den jüngsten Problemen doch wohl kaum anzunehmen, oder doch?
Ich denke gerade über eine kleine Position bei RIGL nach. Eigentlich war mein Depot für 2010 ja fertig, aber...
RIGL scheint mir derzeit nicht so wahnsinnig teuer zu sein. Die Marktkapitalisierung dürfte nach Abzug des vorhandenen und des neu reinkommenden Cash bei ca. 250 Mio. Dollar liegen.
Du hattest RIGL ja eh relativ hoch gewichtet...
Gruß
BANGALORE, Feb 16 (Reuters) - Biotechnology company Rigel Pharmaceuticals Inc (RIGL.O) said it was close to signing a partner to pursue development of its experimental psoriasis treatment.
"We are continuing to work on R348 as a topical agent for psoriasis. It is likely that in the next three months we will announce a deal on R348," Chief Operating Officer Raul Rodriguez told Reuters. Rodriguez said the company would start mid-stage studies on the compound only with a partner and was looking for a global collaboration with a dermatological and/or ophthalmic focus. He said the company, which separately announced a collaboration on its rheumatoid arthritis drug R788 with AstraZeneca PLC (AZN.L) on Tuesday [ID:SGE61F054], was still in the process of determining the terms of the deal.
Shares of the South San Francisco, California-based company were down 8 percent at $8.71 Tuesday afternoon on Nasdaq.
------------------
Ist das eigentlich üblich, während laufender Gespräche solche Andeutungen zu machen? Finde ich nicht so toll...
Wie auch immer - ich habe mir gerade ein paar wenige Stücke ins Depot gelegt.
"We are continuing to work on R348 as a topical agent for psoriasis. It is likely that in the next three months we will announce a deal on R348," Chief Operating Officer Raul Rodriguez told Reuters. Rodriguez said the company would start mid-stage studies on the compound only with a partner and was looking for a global collaboration with a dermatological and/or ophthalmic focus. He said the company, which separately announced a collaboration on its rheumatoid arthritis drug R788 with AstraZeneca PLC (AZN.L) on Tuesday [ID:SGE61F054], was still in the process of determining the terms of the deal.
Shares of the South San Francisco, California-based company were down 8 percent at $8.71 Tuesday afternoon on Nasdaq.
------------------
Ist das eigentlich üblich, während laufender Gespräche solche Andeutungen zu machen? Finde ich nicht so toll...
Wie auch immer - ich habe mir gerade ein paar wenige Stücke ins Depot gelegt.
Antwort auf Beitrag Nr.: 38.953.801 von SLGramann am 16.02.10 18:52:01ich finde die Reaktion des Marktes auch ein wenig unlogisch. Den Kursrutsch kann ich mir eigentlich nur dadurch erklären, dass wohl schon seit längerem viele in der Aktie sind, die auf einen Deal spekuliert haben (der ja schon für Anfang letzten Jahres angepeilt war) und man jetzt erstmal keine wesentlichen Nachrichten in der nächsten Zeit sieht, so dass die Leute dann diese Meldung zum Verkauf genutzt haben.
Der Deal liegt eigentlich über meinen Erwartungen... keine Ahnung, was der Markt erwartet hat. Ich kann mir aber nicht vorstellen, dass das der Grund für das Minus sein soll. Es geht hier ja nicht nur um die Vorauszahlung, die z.B. höher als bei INCY ausgefallen ist, sondern darum, dass AZ die komplette Entwicklung mit allen Kosten übernimmt. Dieser Betrag steht ja nirgendwo. RIGL kann ab jetzt also nur gewinnen und das vorhandene Geld in andere Projekte stecken. Gut finde ich auch, dass AZ offensichtlich davon ausgeht, dass der Fehler in TASKi3 nicht bedeutet, dass R788 nicht bei Patienten wirkt, bei denen z.B. die TNF-Hemmer versagen. Dass der Deal schon eingepreist gewesen sein soll, finde ich unlogisch, da R788 schon einige Haken hat... da zeigt der Deal eigentlich eher die Zuversicht, dass R788 ein Erfolg wird.
*********
CS: Rigel Pharmaceuticals Inc. (RIGL) OUTPERFORM [V] M. Aberman
CP: US$ 9.43 TP: US$ 16 CAP: US$ 344.9m
Rigel Delivers A Great Deal For R788 - Raising Estimates
* Action/Event: Rigel announced an exclusive worldwide license agreement with AstraZeneca for R788 in rheumatoid arthritis and other indications. AstraZeneca will make an upfront payment of $100M with up to an additional $345 million in
development, regulatory and first commercial sale milestones. Rigel will also be eligible to receive up to additional $800
million of specified sales related milestone payments if the product achieves considerable levels of commercial success.
AstraZeneca is responsible for all development, regulatory filings, manufacturing and global commercialization.
* Investment Case: The deal met (if not exceeded) our expectations. The upfront of $100M is in-line with similar large deals but the fact that Astra Zeneca will pay for the future development is an equally, if not more, important term given the high
cost of a Phase II program. Astra Zeneca is one of the largest major pharma companies in the world and brings the
development capability as well as a far-reaching commercial infrastructure that is required for a market like rheumatoid
arthritis. Its patent expiries in the next several years also suggest that AstraZeneca will be fully committed to R788
development and will push forward as quickly as possible. Ultimately, we think the deal consummation validates the
potential of R788 as an oral treatment for rheumatoid arthritis and thus our investment thesis in Rigel. While several
companies have sold off following deal announcement, we believe the skepticism around R788 compound will lead the
stock to trade up on this piece of eagerly anticipated news.
* Catalysts: The next catalyst is initiation of R788 P III clinical program in 2H10.
* Valuation: We have updated our model to incorporate the partnership and conservatively do not include commercial
milestones. Our $16 target price for Rigel is based on a probability-adjusted, sum-of-the-parts discounted cash flow (DCF)
and net present value (NPV) analysis.
********
j. birchenough/r.martins, barclays
Equity Research
February 16, 2010
Rigel Pharmaceuticals (RIGL - US$ 9.43) 1-Overweight
Company Update
Large Pharma Deal Better Than Expected
Investment Conclusion
We are reiterating our 1-Overweight rating on
RIGL following today’s partnership with AZN for
R788 in RA and other rheumatologic disorders.
We believe the deal is one of the best for a biotech
company, with full funding, clear timelines to
commercialization and attractive financial terms.
We believe the deal is better than expected and
would buy RIGL here.
Summary
RIGL announced today a global development and
commercialization agreement with Astra Zeneca
for syk kinase inhibitor R788 as well as back up
compounds for rheumatoid arthritis and other
rheumatologic disorders. Terms of the deal
include $100M upfront, $345M in development
milestones, $800M in sales milestones and tiered
double digit royalties that we expect are at the
high end for late stage deals of this type.
With AZN funding all development and
commercialization of R788 and back up
compounds, we see significant upside potential for
RIGL here. Expert feedback suggests a
competitive profile for R788 in the $13B RA
market and with phase III initiation in 2H10 and
NDA/EMEA filing expected in 2013 outcomes
study requirements appear unlikely.
*********
http://www.minyanville.com/businessmarkets/articles/astrazen…
Rigel Investors Balk at AstraZeneca Deal
By Lisa LaMotta Feb 16, 2010 12:45 pm
They're unable to focus on the overall payout, and seem stuck on lower-than-expected upfront payment.
Sometimes it’s hard for investors to see the big picture, which seems to be the case with Rigel Pharmaceuticals (RIGL).
The stock fell like snow on Tuesday, dropping an initial 8% only to recover slightly to stick around $9. The precipitous drop in the stock price came after the drug-development company announced that it's inked a deal with the Anglo-Swedish drugmaker AstraZeneca (AZN) for its late-stage rheumatoid arthritis drug.
According to the terms of the deal, AstraZeneca will pay $100 million upfront for fostamatinib disodium, or R788, with an additional $345 million once certain developmental milestones are reached, as well as the possibility of $800 million once certain sales milestones are reached, and royalty payments -- totaling an upwards of $1.24 billion for Rigel when all is said and done.
The deal with AstraZeneca seems to fulfill all of a small drugmaker’s wildest dreams -- it allows the small pharmaceutical developer to partner with a large-cap pharma that's backed by a significant sales force and will assume all of the development and regulatory costs of the drug.
Yet, Rigel investors had a tough time focusing on the overall payout of the deal and seem to be stuck on lower-than-expected upfront payment, as well as AstraZeneca’s lack of expertise in the RA field. “Our initial take is that the terms are solid; while the upfront payment of $100M may be slightly below Street expectations,” writes Oppenheimer analyst Brian Abrahams in a note to investors on Tuesday, "the fact that AZN will fund 100% of the substantial future development costs is a positive surprise and should significantly reduce RIGL's go-forward cash burn.”
Beyond the smaller-than-expected upfront payout for the drug, investors seem to have qualms with the way the royalty payments are structured. "Rigel had hoped for a deal with profit sharing, 50/50, economics,” wrote RBC Capital Markets analyst Jason Kantor in a note to investors. “This would equate to mid-20% royalties. Although the exact rate was not disclosed, the release stated 'significant stepped double-digit royalties' which we interpret as at least very high teens and likely above 20% on average”
While AstraZeneca is one of the world’s leading pharmaceutical manufacturers with a market cap of $63 billion, the it's far from a leader in the rheumatology field.
Rheumatoid arthritis is an autoimmune inflammatory disease that causes damage to the joints, as well as other organs. The deal stands to be potentially lucrative for both companies; the rheumatoid arthritis market was estimated to be about $13 billion globally in 2009, up from $1.3 billion a decade earlier. The drug has currently competed three phase 2 clinical trials and AstraZeneca will develop a phase 3 program to be initiated this year. The company expects to file for approval with the FDA in 2013.
The collaboration with Rigel comes at the perfect time for AstraZeneca, which is expected to see a significant drop in sales once its blockbuster statin Crestor loses patent protection. Crestor is already one of the best-selling drugs in the world with revenues of $4.5 billion in 2009, up 25% from $3.6 billion in 2008, but the blockbuster will lose patent protection in 2016. The company is already fighting plenty of court battles trying to defend that 2016 date as generic companies like Israel’s Teva Pharmaceuticals (TEVA) try to get the patent expiration pushed up.
mfg ipollit
Der Deal liegt eigentlich über meinen Erwartungen... keine Ahnung, was der Markt erwartet hat. Ich kann mir aber nicht vorstellen, dass das der Grund für das Minus sein soll. Es geht hier ja nicht nur um die Vorauszahlung, die z.B. höher als bei INCY ausgefallen ist, sondern darum, dass AZ die komplette Entwicklung mit allen Kosten übernimmt. Dieser Betrag steht ja nirgendwo. RIGL kann ab jetzt also nur gewinnen und das vorhandene Geld in andere Projekte stecken. Gut finde ich auch, dass AZ offensichtlich davon ausgeht, dass der Fehler in TASKi3 nicht bedeutet, dass R788 nicht bei Patienten wirkt, bei denen z.B. die TNF-Hemmer versagen. Dass der Deal schon eingepreist gewesen sein soll, finde ich unlogisch, da R788 schon einige Haken hat... da zeigt der Deal eigentlich eher die Zuversicht, dass R788 ein Erfolg wird.
*********
CS: Rigel Pharmaceuticals Inc. (RIGL) OUTPERFORM [V] M. Aberman
CP: US$ 9.43 TP: US$ 16 CAP: US$ 344.9m
Rigel Delivers A Great Deal For R788 - Raising Estimates
* Action/Event: Rigel announced an exclusive worldwide license agreement with AstraZeneca for R788 in rheumatoid arthritis and other indications. AstraZeneca will make an upfront payment of $100M with up to an additional $345 million in
development, regulatory and first commercial sale milestones. Rigel will also be eligible to receive up to additional $800
million of specified sales related milestone payments if the product achieves considerable levels of commercial success.
AstraZeneca is responsible for all development, regulatory filings, manufacturing and global commercialization.
* Investment Case: The deal met (if not exceeded) our expectations. The upfront of $100M is in-line with similar large deals but the fact that Astra Zeneca will pay for the future development is an equally, if not more, important term given the high
cost of a Phase II program. Astra Zeneca is one of the largest major pharma companies in the world and brings the
development capability as well as a far-reaching commercial infrastructure that is required for a market like rheumatoid
arthritis. Its patent expiries in the next several years also suggest that AstraZeneca will be fully committed to R788
development and will push forward as quickly as possible. Ultimately, we think the deal consummation validates the
potential of R788 as an oral treatment for rheumatoid arthritis and thus our investment thesis in Rigel. While several
companies have sold off following deal announcement, we believe the skepticism around R788 compound will lead the
stock to trade up on this piece of eagerly anticipated news.
* Catalysts: The next catalyst is initiation of R788 P III clinical program in 2H10.
* Valuation: We have updated our model to incorporate the partnership and conservatively do not include commercial
milestones. Our $16 target price for Rigel is based on a probability-adjusted, sum-of-the-parts discounted cash flow (DCF)
and net present value (NPV) analysis.
********
j. birchenough/r.martins, barclays
Equity Research
February 16, 2010
Rigel Pharmaceuticals (RIGL - US$ 9.43) 1-Overweight
Company Update
Large Pharma Deal Better Than Expected
Investment Conclusion
We are reiterating our 1-Overweight rating on
RIGL following today’s partnership with AZN for
R788 in RA and other rheumatologic disorders.
We believe the deal is one of the best for a biotech
company, with full funding, clear timelines to
commercialization and attractive financial terms.
We believe the deal is better than expected and
would buy RIGL here.
Summary
RIGL announced today a global development and
commercialization agreement with Astra Zeneca
for syk kinase inhibitor R788 as well as back up
compounds for rheumatoid arthritis and other
rheumatologic disorders. Terms of the deal
include $100M upfront, $345M in development
milestones, $800M in sales milestones and tiered
double digit royalties that we expect are at the
high end for late stage deals of this type.
With AZN funding all development and
commercialization of R788 and back up
compounds, we see significant upside potential for
RIGL here. Expert feedback suggests a
competitive profile for R788 in the $13B RA
market and with phase III initiation in 2H10 and
NDA/EMEA filing expected in 2013 outcomes
study requirements appear unlikely.
*********
http://www.minyanville.com/businessmarkets/articles/astrazen…
Rigel Investors Balk at AstraZeneca Deal
By Lisa LaMotta Feb 16, 2010 12:45 pm
They're unable to focus on the overall payout, and seem stuck on lower-than-expected upfront payment.
Sometimes it’s hard for investors to see the big picture, which seems to be the case with Rigel Pharmaceuticals (RIGL).
The stock fell like snow on Tuesday, dropping an initial 8% only to recover slightly to stick around $9. The precipitous drop in the stock price came after the drug-development company announced that it's inked a deal with the Anglo-Swedish drugmaker AstraZeneca (AZN) for its late-stage rheumatoid arthritis drug.
According to the terms of the deal, AstraZeneca will pay $100 million upfront for fostamatinib disodium, or R788, with an additional $345 million once certain developmental milestones are reached, as well as the possibility of $800 million once certain sales milestones are reached, and royalty payments -- totaling an upwards of $1.24 billion for Rigel when all is said and done.
The deal with AstraZeneca seems to fulfill all of a small drugmaker’s wildest dreams -- it allows the small pharmaceutical developer to partner with a large-cap pharma that's backed by a significant sales force and will assume all of the development and regulatory costs of the drug.
Yet, Rigel investors had a tough time focusing on the overall payout of the deal and seem to be stuck on lower-than-expected upfront payment, as well as AstraZeneca’s lack of expertise in the RA field. “Our initial take is that the terms are solid; while the upfront payment of $100M may be slightly below Street expectations,” writes Oppenheimer analyst Brian Abrahams in a note to investors on Tuesday, "the fact that AZN will fund 100% of the substantial future development costs is a positive surprise and should significantly reduce RIGL's go-forward cash burn.”
Beyond the smaller-than-expected upfront payout for the drug, investors seem to have qualms with the way the royalty payments are structured. "Rigel had hoped for a deal with profit sharing, 50/50, economics,” wrote RBC Capital Markets analyst Jason Kantor in a note to investors. “This would equate to mid-20% royalties. Although the exact rate was not disclosed, the release stated 'significant stepped double-digit royalties' which we interpret as at least very high teens and likely above 20% on average”
While AstraZeneca is one of the world’s leading pharmaceutical manufacturers with a market cap of $63 billion, the it's far from a leader in the rheumatology field.
Rheumatoid arthritis is an autoimmune inflammatory disease that causes damage to the joints, as well as other organs. The deal stands to be potentially lucrative for both companies; the rheumatoid arthritis market was estimated to be about $13 billion globally in 2009, up from $1.3 billion a decade earlier. The drug has currently competed three phase 2 clinical trials and AstraZeneca will develop a phase 3 program to be initiated this year. The company expects to file for approval with the FDA in 2013.
The collaboration with Rigel comes at the perfect time for AstraZeneca, which is expected to see a significant drop in sales once its blockbuster statin Crestor loses patent protection. Crestor is already one of the best-selling drugs in the world with revenues of $4.5 billion in 2009, up 25% from $3.6 billion in 2008, but the blockbuster will lose patent protection in 2016. The company is already fighting plenty of court battles trying to defend that 2016 date as generic companies like Israel’s Teva Pharmaceuticals (TEVA) try to get the patent expiration pushed up.
mfg ipollit
Antwort auf Beitrag Nr.: 38.962.988 von ipollit am 18.02.10 00:18:43
Hi ipollit, Du triffst mit Deiner Erklärung "sell on good news" wahrscheinlich ins Schwarze. Denn die Diskussion über die Höhe des upfront ist müßig. Wie Du ja auch schreibst, ist es für RIGL viel wertvoller, dass Astra den "Rest" der Entwicklung allein stemmt. Wenn RIGL am Ende zwischen 15 und 20% Royalties bekommt, ist es ein fantastischer Deal!
Meine Überlegung geht so: Astra hat sich die R788-Daten sehr, sehr genau angesehen und entschieden, dass ihnen das Projekt 100 Mio. upfront wert ist und dass sie eine P III finanzieren. Ich glaube, es trifft schon einigermaßen die Größenordnung, wenn ich denke, dass Astra damit insgesamt irgendwas zwischen 300 und 500 Millionen Dollar in das Projekt steckt. Bei RA stelle ich mir die P III jedenfalls sehr(!) teuer vor.
Unter diesen Voraussetzungen kann ich als "Schütze Arsch" ja meine 3 Cents auch noch riskieren.
Gehen wir mal von einem Erfolg aus - welche Perspektiven ergeben sich dann daraus?
R788 hat prinzipiell Blockbuster-Potential. Bei wirklich guten Daten sind m.E. sogar mehrere Milliarden Dollar Umsatz binnen weniger Jahre realistisch. Das wären Royaltie-Einnahmen (also eigentlich direkt ins EBT) von mehreren 100 Mio. für RIGL - ohne die Meilensteine.
Die Marktkapitalisierung würde dann zwingend im Milliardenbereich liegen (eher 2 Milliarden, als eine). Heute liegt sie bei cash-bereinigten 250 Mio. oder so. Eine Übernahme wäre auch wahrscheinlich.
So viel zum best case...
Gruß
Hi ipollit, Du triffst mit Deiner Erklärung "sell on good news" wahrscheinlich ins Schwarze. Denn die Diskussion über die Höhe des upfront ist müßig. Wie Du ja auch schreibst, ist es für RIGL viel wertvoller, dass Astra den "Rest" der Entwicklung allein stemmt. Wenn RIGL am Ende zwischen 15 und 20% Royalties bekommt, ist es ein fantastischer Deal!
Meine Überlegung geht so: Astra hat sich die R788-Daten sehr, sehr genau angesehen und entschieden, dass ihnen das Projekt 100 Mio. upfront wert ist und dass sie eine P III finanzieren. Ich glaube, es trifft schon einigermaßen die Größenordnung, wenn ich denke, dass Astra damit insgesamt irgendwas zwischen 300 und 500 Millionen Dollar in das Projekt steckt. Bei RA stelle ich mir die P III jedenfalls sehr(!) teuer vor.
Unter diesen Voraussetzungen kann ich als "Schütze Arsch" ja meine 3 Cents auch noch riskieren.
Gehen wir mal von einem Erfolg aus - welche Perspektiven ergeben sich dann daraus?
R788 hat prinzipiell Blockbuster-Potential. Bei wirklich guten Daten sind m.E. sogar mehrere Milliarden Dollar Umsatz binnen weniger Jahre realistisch. Das wären Royaltie-Einnahmen (also eigentlich direkt ins EBT) von mehreren 100 Mio. für RIGL - ohne die Meilensteine.
Die Marktkapitalisierung würde dann zwingend im Milliardenbereich liegen (eher 2 Milliarden, als eine). Heute liegt sie bei cash-bereinigten 250 Mio. oder so. Eine Übernahme wäre auch wahrscheinlich.
So viel zum best case...
Gruß
zwei Meldungen von REGN:
VEGF-Trap-Eye Ergebnisse aus einer P II:
TARRYTOWN, N.Y. and LEVERKUSEN, Germany, Feb 18, 2010 /PRNewswire via COMTEX News Network/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) and Bayer HealthCare AG today announced that VEGF Trap-Eye showed positive results in a Phase 2 study in patients with diabetic macular edema (DME). The primary endpoint of the study, a statistically significant improvement in visual acuity over 24 weeks compared to the standard of care in DME, macular laser therapy, was met. Visual acuity improvement was measured by the mean number of letters gained over the initial 24 weeks of the study.
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=44…
Jahresergebnis 2009:
TARRYTOWN, N.Y., Feb 18, 2010 /PRNewswire via COMTEX News Network/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) today announced financial and operating results for the full year and fourth quarter of 2009. The Company reported a net loss of $67.8 million, or $0.85 per share (basic and diluted), for the year ended December 31, 2009 compared with a net loss of $79.1 million, or $1.00 per share (basic and diluted), for the year ended December 31, 2008. The Company reported a net loss of $36.5 million, or $0.46 per share (basic and diluted), for the fourth quarter of 2009 compared with a net loss of $29.5 million, or $0.37 per share (basic and diluted), for the fourth quarter of 2008.
At December 31, 2009, cash, restricted cash, and marketable securities totaled $390.0 million compared with $527.5 million at December 31, 2008.
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=44…
------------
Kurs ist 11% im Minus. Ich hoffe, es liegt wirklich nur an der Meldung Nr. 2...
VEGF-Trap-Eye Ergebnisse aus einer P II:
TARRYTOWN, N.Y. and LEVERKUSEN, Germany, Feb 18, 2010 /PRNewswire via COMTEX News Network/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) and Bayer HealthCare AG today announced that VEGF Trap-Eye showed positive results in a Phase 2 study in patients with diabetic macular edema (DME). The primary endpoint of the study, a statistically significant improvement in visual acuity over 24 weeks compared to the standard of care in DME, macular laser therapy, was met. Visual acuity improvement was measured by the mean number of letters gained over the initial 24 weeks of the study.
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=44…
Jahresergebnis 2009:
TARRYTOWN, N.Y., Feb 18, 2010 /PRNewswire via COMTEX News Network/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) today announced financial and operating results for the full year and fourth quarter of 2009. The Company reported a net loss of $67.8 million, or $0.85 per share (basic and diluted), for the year ended December 31, 2009 compared with a net loss of $79.1 million, or $1.00 per share (basic and diluted), for the year ended December 31, 2008. The Company reported a net loss of $36.5 million, or $0.46 per share (basic and diluted), for the fourth quarter of 2009 compared with a net loss of $29.5 million, or $0.37 per share (basic and diluted), for the fourth quarter of 2008.
At December 31, 2009, cash, restricted cash, and marketable securities totaled $390.0 million compared with $527.5 million at December 31, 2008.
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=44…
------------
Kurs ist 11% im Minus. Ich hoffe, es liegt wirklich nur an der Meldung Nr. 2...
Antwort auf Beitrag Nr.: 38.968.989 von SLGramann am 18.02.10 18:58:42"Kurs ist 11% im Minus. Ich hoffe, es liegt wirklich nur an der Meldung Nr. 2"
ich glaube, weder noch... die PII sieht meiner Meinung nach gut aus und war bereits vor der Eröffnung bekannt, ohne dass es eine Reaktion gegeben hätte. Ich glaube eher, dass es charttechnische Gründe gibt... der Kurs war zuletzt ja ohne Pause gestiegen und da haben vielleicht viele jetzt Kasse gemacht. Wenn sich das beruhigt hat, wird es meiner Meinung nach auch wieder nach oben gehen.


*****
"Regeneron ended 2009 with many late-stage Phase 3 trials, a diversified pipeline, and a healthy balance sheet," said Leonard S. Schleifer, M.D., Ph.D., President and Chief Executive Officer. "Our eight drug candidates in development for 17 indications and our expanded antibody collaboration with sanofi-aventis position the Company for continued growth. 2010 should be especially eventful for Regeneron as we anticipate, among other clinical results, Phase 3 data from two of our four trials in gout and from our two studies in wet AMD (age-related macular degeneration), as well as potential interim news from our Phase 3 cancer program."
Current Business Highlights
ARCALYST(R) (rilonacept) - CAPS
The Company shipped $20.0 million of ARCALYST(R) (rilonacept) Injection for Subcutaneous Use to its distributors in 2009, including $5.0 million in the fourth quarter, for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) in adults and children 12 and older in the United States. This compares to shipments of $10.7 million in 2008, including $4.0 million in the fourth quarter of 2008. CAPS is a group of rare, inherited, auto-inflammatory conditions characterized by life-long, recurrent symptoms of rash, fever/chills, joint pain, eye redness/pain, and fatigue. In October, rilonacept was approved under exceptional circumstances by the European Medicines Agency (EMEA) for the treatment of CAPS with severe symptoms in adults and children aged 12 years and older. Rilonacept is not currently marketed in the European Union. ARCALYST is a fusion protein that blocks the cytokine interleukin-1 (IL-1).
Rilonacept - Gout
Rilonacept is in a Phase 3 clinical development program for the treatment of gout. The program includes four clinical trials. Two Phase 3 clinical trials (called PRE-SURGE 1 and PRE-SURGE 2) are evaluating rilonacept versus placebo for the prevention of gout flares in patients initiating urate-lowering drug therapy. A third Phase 3 trial in acute gout (SURGE) is evaluating treatment with rilonacept alone versus rilonacept in combination with a nonsteroidal anti-inflammatory drug (NSAID) versus an NSAID alone. The fourth Phase 3 trial is a placebo-controlled safety study (RE-SURGE) of rilonacept in patients receiving urate-lowering therapy. PRE-SURGE 1 and SURGE are fully enrolled. The Company expects to report initial data from SURGE and PRE-SURGE 1 during the first half of 2010 and from PRE-SURGE 2 and RE-SURGE during the first half of 2011. Regeneron owns worldwide rights to rilonacept.
VEGF Trap-Eye - Ophthalmologic Diseases
VEGF Trap-Eye is a specially purified and formulated form of VEGF Trap for use in the intraocular treatment of retinal diseases. VEGF Trap-Eye blocks vascular endothelial growth factor A (VEGF-A), a secreted protein which promotes the growth of blood vessels. It also binds other mediators of angiogenesis, including VEGF-B and Placental Growth Factor (PlGF). VEGF Trap-Eye is being developed by Regeneron in collaboration with Bayer HealthCare. Bayer HealthCare has rights to market VEGF Trap-Eye outside the United States, where the companies will share equally in profits from any future sales of VEGF Trap-Eye. Regeneron maintains exclusive rights to VEGF Trap-Eye in the United States.
Two Phase 3 studies (VIEW 1 and VIEW 2) evaluating VEGF Trap-Eye in patients with the neovascular form of age-related macular degeneration (wet AMD) are fully enrolled, and initial data from these studies are expected in late 2010. In addition, two Phase 3 studies (COPERNICUS and GALILEO) in central retinal vein occlusion (CRVO) are enrolling patients, and initial data are anticipated in early 2011.
In a separate press release issued today, Regeneron and Bayer HealthCare announced that in a Phase 2 study (called DA VINCI) in patients with clinically significant diabetic macular edema (DME), VEGF Trap-Eye achieved the primary study endpoint, a statistically significant improvement in visual acuity over 24 weeks compared to focal laser therapy, the standard of care in DME. VEGF Trap-Eye was generally well-tolerated, and no ocular or non-ocular drug-related serious adverse events were reported in the study.
Aflibercept (VEGF Trap) - Oncology
Aflibercept (VEGF Trap) is being developed worldwide by Regeneron and its collaborator, sanofi-aventis, for the potential treatment of solid tumors. Three Phase 3 trials are evaluating combinations of aflibercept with standard chemotherapy regimens for the treatment of cancer. One trial (called VELOUR) is evaluating aflibercept as a 2nd line treatment for metastatic colorectal cancer in combination with FOLFIRI (folinic acid (leucovorin), 5-fluorouracil, and irinotecan). A second trial (VITAL) is evaluating aflibercept as a 2nd line treatment for metastatic non-small cell lung cancer in combination with docetaxel. The third trial (VENICE) is evaluating aflibercept as a 1st line treatment for metastatic androgen-independent prostate cancer in combination with docetaxel/prednisone. All three trials are studying the current standard of chemotherapy care for the cancer being studied with and without aflibercept. VITAL and VENICE are fully enrolled, and the VELOUR study is approximately 95 percent enrolled. Based on current enrollment and event rates, an interim analysis of VELOUR is expected to be conducted by an independent data monitoring committee (IDMC) in the second half of 2010. Final results from the VITAL study are anticipated in the first half of 2011 and from the VELOUR study in the second half of 2011. Based on projected event rates, an interim analysis of VENICE is expected to be conducted by an IDMC in mid-2011, with final results anticipated in 2012.
In addition, a Phase 2 study (AFFIRM) is evaluating aflibercept as a 1st line treatment for metastatic colorectal cancer in combination with FOLFOX (folinic acid (leucovorin), 5-fluorouracil, and oxaliplatin). The AFFIRM study is approximately 75 percent enrolled.
Monoclonal Antibodies
Since 2007, Regeneron and sanofi-aventis have collaborated on the discovery, development, and commercialization of fully human monoclonal antibodies generated by Regeneron using its VelocImmune(R) technology. During the fourth quarter of 2009, Regeneron and sanofi-aventis expanded and extended their collaboration with the objective to advance an average of four to five antibodies into clinical development each year between 2010 and 2017. There are five antibody candidates currently in development under the collaboration:
REGN475, an antibody to nerve growth factor (NGF), is being evaluated in Phase 2 studies in osteoarthritis of the knee, sciatic pain, vertebral fracture pain, chronic pancreatitis pain, and thermal injury pain.
REGN88, an antibody to the interleukin-6 receptor (IL-6R), has completed Phase 1 studies. A Phase 2/3 study of REGN88 in rheumatoid arthritis and a Phase 2 study in ankylosing spondylitis, a form of arthritis that primarily affects the spine, are open for enrollment.
REGN421, an antibody to Delta-like ligand-4 (Dll4), a novel anti-angiogenesis target, is in a Phase 1 study in patients with advanced malignancies.
REGN727, an antibody to PCSK9, a novel target for LDL cholesterol reduction, is in a Phase 1 study.
REGN668, an antibody to the interleukin-4 receptor (IL-4R), a target for allergic and immune conditions, is in a Phase 1 study.
********
und die ersten VEGF Trap-Eye PII-Ergebnisse bei DME:
In this double-masked, prospective, randomized, multi-center Phase 2 trial, entitled DA VINCI (DME And VEGF Trap-Eye: INvestigation of Clinical Impact), 219 patients with clinically significant DME with central macular involvement were randomized to five groups. The control group received macular laser therapy at week one, and patients were eligible for repeat laser treatments, but no more frequently than at 16 week intervals. Two groups received monthly doses of 0.5 or 2.0 milligrams (mg) of VEGF Trap-Eye throughout the 6-month dosing period. Two groups received three initial monthly doses of 2.0 mg of VEGF Trap-Eye (at baseline and weeks 4 and 8), followed through week 24 by either every 8-week dosing or as-needed (PRN) dosing with specific repeat dosing criteria. The following summarizes the mean gain in visual acuity at week 24 by dosing arm and the mean number of treatments received by patients over the first six monthly visits:
* Standard-of-care macular laser therapy (n=44; 1.7 treatments): +2.5 letters gained
* VEGF Trap Eye 0.5 mg monthly (n=44; 5.6 injections): +8.6 letters gained
* VEGF Trap-Eye 2 mg monthly (n=44; 5.5 injections): +11.4 letters gained
* VEGF Trap-Eye 2 mg every other month, following 3 monthly injections (n=42: 3.8 injections): +8.5 letters gained
* VEGF Trap-Eye 2 mg as-needed, following 3 monthly injections (n=45; 4.4 injections): +10.3 letters gained
mfg ipollit
ich glaube, weder noch... die PII sieht meiner Meinung nach gut aus und war bereits vor der Eröffnung bekannt, ohne dass es eine Reaktion gegeben hätte. Ich glaube eher, dass es charttechnische Gründe gibt... der Kurs war zuletzt ja ohne Pause gestiegen und da haben vielleicht viele jetzt Kasse gemacht. Wenn sich das beruhigt hat, wird es meiner Meinung nach auch wieder nach oben gehen.
*****
"Regeneron ended 2009 with many late-stage Phase 3 trials, a diversified pipeline, and a healthy balance sheet," said Leonard S. Schleifer, M.D., Ph.D., President and Chief Executive Officer. "Our eight drug candidates in development for 17 indications and our expanded antibody collaboration with sanofi-aventis position the Company for continued growth. 2010 should be especially eventful for Regeneron as we anticipate, among other clinical results, Phase 3 data from two of our four trials in gout and from our two studies in wet AMD (age-related macular degeneration), as well as potential interim news from our Phase 3 cancer program."
Current Business Highlights
ARCALYST(R) (rilonacept) - CAPS
The Company shipped $20.0 million of ARCALYST(R) (rilonacept) Injection for Subcutaneous Use to its distributors in 2009, including $5.0 million in the fourth quarter, for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) in adults and children 12 and older in the United States. This compares to shipments of $10.7 million in 2008, including $4.0 million in the fourth quarter of 2008. CAPS is a group of rare, inherited, auto-inflammatory conditions characterized by life-long, recurrent symptoms of rash, fever/chills, joint pain, eye redness/pain, and fatigue. In October, rilonacept was approved under exceptional circumstances by the European Medicines Agency (EMEA) for the treatment of CAPS with severe symptoms in adults and children aged 12 years and older. Rilonacept is not currently marketed in the European Union. ARCALYST is a fusion protein that blocks the cytokine interleukin-1 (IL-1).
Rilonacept - Gout
Rilonacept is in a Phase 3 clinical development program for the treatment of gout. The program includes four clinical trials. Two Phase 3 clinical trials (called PRE-SURGE 1 and PRE-SURGE 2) are evaluating rilonacept versus placebo for the prevention of gout flares in patients initiating urate-lowering drug therapy. A third Phase 3 trial in acute gout (SURGE) is evaluating treatment with rilonacept alone versus rilonacept in combination with a nonsteroidal anti-inflammatory drug (NSAID) versus an NSAID alone. The fourth Phase 3 trial is a placebo-controlled safety study (RE-SURGE) of rilonacept in patients receiving urate-lowering therapy. PRE-SURGE 1 and SURGE are fully enrolled. The Company expects to report initial data from SURGE and PRE-SURGE 1 during the first half of 2010 and from PRE-SURGE 2 and RE-SURGE during the first half of 2011. Regeneron owns worldwide rights to rilonacept.
VEGF Trap-Eye - Ophthalmologic Diseases
VEGF Trap-Eye is a specially purified and formulated form of VEGF Trap for use in the intraocular treatment of retinal diseases. VEGF Trap-Eye blocks vascular endothelial growth factor A (VEGF-A), a secreted protein which promotes the growth of blood vessels. It also binds other mediators of angiogenesis, including VEGF-B and Placental Growth Factor (PlGF). VEGF Trap-Eye is being developed by Regeneron in collaboration with Bayer HealthCare. Bayer HealthCare has rights to market VEGF Trap-Eye outside the United States, where the companies will share equally in profits from any future sales of VEGF Trap-Eye. Regeneron maintains exclusive rights to VEGF Trap-Eye in the United States.
Two Phase 3 studies (VIEW 1 and VIEW 2) evaluating VEGF Trap-Eye in patients with the neovascular form of age-related macular degeneration (wet AMD) are fully enrolled, and initial data from these studies are expected in late 2010. In addition, two Phase 3 studies (COPERNICUS and GALILEO) in central retinal vein occlusion (CRVO) are enrolling patients, and initial data are anticipated in early 2011.
In a separate press release issued today, Regeneron and Bayer HealthCare announced that in a Phase 2 study (called DA VINCI) in patients with clinically significant diabetic macular edema (DME), VEGF Trap-Eye achieved the primary study endpoint, a statistically significant improvement in visual acuity over 24 weeks compared to focal laser therapy, the standard of care in DME. VEGF Trap-Eye was generally well-tolerated, and no ocular or non-ocular drug-related serious adverse events were reported in the study.
Aflibercept (VEGF Trap) - Oncology
Aflibercept (VEGF Trap) is being developed worldwide by Regeneron and its collaborator, sanofi-aventis, for the potential treatment of solid tumors. Three Phase 3 trials are evaluating combinations of aflibercept with standard chemotherapy regimens for the treatment of cancer. One trial (called VELOUR) is evaluating aflibercept as a 2nd line treatment for metastatic colorectal cancer in combination with FOLFIRI (folinic acid (leucovorin), 5-fluorouracil, and irinotecan). A second trial (VITAL) is evaluating aflibercept as a 2nd line treatment for metastatic non-small cell lung cancer in combination with docetaxel. The third trial (VENICE) is evaluating aflibercept as a 1st line treatment for metastatic androgen-independent prostate cancer in combination with docetaxel/prednisone. All three trials are studying the current standard of chemotherapy care for the cancer being studied with and without aflibercept. VITAL and VENICE are fully enrolled, and the VELOUR study is approximately 95 percent enrolled. Based on current enrollment and event rates, an interim analysis of VELOUR is expected to be conducted by an independent data monitoring committee (IDMC) in the second half of 2010. Final results from the VITAL study are anticipated in the first half of 2011 and from the VELOUR study in the second half of 2011. Based on projected event rates, an interim analysis of VENICE is expected to be conducted by an IDMC in mid-2011, with final results anticipated in 2012.
In addition, a Phase 2 study (AFFIRM) is evaluating aflibercept as a 1st line treatment for metastatic colorectal cancer in combination with FOLFOX (folinic acid (leucovorin), 5-fluorouracil, and oxaliplatin). The AFFIRM study is approximately 75 percent enrolled.
Monoclonal Antibodies
Since 2007, Regeneron and sanofi-aventis have collaborated on the discovery, development, and commercialization of fully human monoclonal antibodies generated by Regeneron using its VelocImmune(R) technology. During the fourth quarter of 2009, Regeneron and sanofi-aventis expanded and extended their collaboration with the objective to advance an average of four to five antibodies into clinical development each year between 2010 and 2017. There are five antibody candidates currently in development under the collaboration:
REGN475, an antibody to nerve growth factor (NGF), is being evaluated in Phase 2 studies in osteoarthritis of the knee, sciatic pain, vertebral fracture pain, chronic pancreatitis pain, and thermal injury pain.
REGN88, an antibody to the interleukin-6 receptor (IL-6R), has completed Phase 1 studies. A Phase 2/3 study of REGN88 in rheumatoid arthritis and a Phase 2 study in ankylosing spondylitis, a form of arthritis that primarily affects the spine, are open for enrollment.
REGN421, an antibody to Delta-like ligand-4 (Dll4), a novel anti-angiogenesis target, is in a Phase 1 study in patients with advanced malignancies.
REGN727, an antibody to PCSK9, a novel target for LDL cholesterol reduction, is in a Phase 1 study.
REGN668, an antibody to the interleukin-4 receptor (IL-4R), a target for allergic and immune conditions, is in a Phase 1 study.
********
und die ersten VEGF Trap-Eye PII-Ergebnisse bei DME:
In this double-masked, prospective, randomized, multi-center Phase 2 trial, entitled DA VINCI (DME And VEGF Trap-Eye: INvestigation of Clinical Impact), 219 patients with clinically significant DME with central macular involvement were randomized to five groups. The control group received macular laser therapy at week one, and patients were eligible for repeat laser treatments, but no more frequently than at 16 week intervals. Two groups received monthly doses of 0.5 or 2.0 milligrams (mg) of VEGF Trap-Eye throughout the 6-month dosing period. Two groups received three initial monthly doses of 2.0 mg of VEGF Trap-Eye (at baseline and weeks 4 and 8), followed through week 24 by either every 8-week dosing or as-needed (PRN) dosing with specific repeat dosing criteria. The following summarizes the mean gain in visual acuity at week 24 by dosing arm and the mean number of treatments received by patients over the first six monthly visits:
* Standard-of-care macular laser therapy (n=44; 1.7 treatments): +2.5 letters gained
* VEGF Trap Eye 0.5 mg monthly (n=44; 5.6 injections): +8.6 letters gained
* VEGF Trap-Eye 2 mg monthly (n=44; 5.5 injections): +11.4 letters gained
* VEGF Trap-Eye 2 mg every other month, following 3 monthly injections (n=42: 3.8 injections): +8.5 letters gained
* VEGF Trap-Eye 2 mg as-needed, following 3 monthly injections (n=45; 4.4 injections): +10.3 letters gained
mfg ipollit
INCY...
http://finance.yahoo.com/news/Incyte-Reports-2009-Financial-…
2010 Clinical Program Goals
Oncology Programs
JAK1/JAK2 Inhibitor: INCB18424 (oral formulation)
Complete and present results from the Comfort-I Phase III US trial and begin preparation of the New Drug Application for MF to insure earliest possible filing in 2011
Confirm regulatory requirements with the FDA for approval in two other myeloproliferative neoplasms, first in PV followed by ET
In conjunction with the Children’s Oncology Group at the National Cancer Institute, support initiation of a Phase I/II trial in children with relapsed or refractory solid tumors, hematological malignancies and myeloproliferative neoplasms
Sheddase Inhibitor: INCB7839
Meet with the FDA to discuss registration requirements for use in patients with p95 HER2 positive breast cancer provided results from the ongoing clinical development program in this subset of patients continues to demonstrate positive results
Early Stage Oncology Programs
cMET Inhibitor, INCB28060: Complete an initial Phase I/II trial in patients with solid tumors and transfer the program to Novartis
IDO Inhibitor, INCB24360: Initiate a Phase I/II trial in patients with solid tumors
Discovery: File an Investigational New Drug Application for another oncology compound that addresses a new target
Inflammation Programs
JAK1/JAK2 Inhibitor: INCB28050
Complete the Phase II trial and present top-line results for the three-month portion of the study in the first half of this year; present the full six month results at the American College of Rheumatology Annual Meeting
Based on these results, decide whether to exercise our co-development option and participate in the Phase IIb program
JAK1/JAK2 Inhibitor: INCB18424 (topical formulation)
Present full results from the three-month Phase IIb trial at a scientific meeting
Decide on the appropriate next clinical trials
Evaluate potential strategic alliance opportunities
mfg ipollit
http://finance.yahoo.com/news/Incyte-Reports-2009-Financial-…
2010 Clinical Program Goals
Oncology Programs
JAK1/JAK2 Inhibitor: INCB18424 (oral formulation)
Complete and present results from the Comfort-I Phase III US trial and begin preparation of the New Drug Application for MF to insure earliest possible filing in 2011
Confirm regulatory requirements with the FDA for approval in two other myeloproliferative neoplasms, first in PV followed by ET
In conjunction with the Children’s Oncology Group at the National Cancer Institute, support initiation of a Phase I/II trial in children with relapsed or refractory solid tumors, hematological malignancies and myeloproliferative neoplasms
Sheddase Inhibitor: INCB7839
Meet with the FDA to discuss registration requirements for use in patients with p95 HER2 positive breast cancer provided results from the ongoing clinical development program in this subset of patients continues to demonstrate positive results
Early Stage Oncology Programs
cMET Inhibitor, INCB28060: Complete an initial Phase I/II trial in patients with solid tumors and transfer the program to Novartis
IDO Inhibitor, INCB24360: Initiate a Phase I/II trial in patients with solid tumors
Discovery: File an Investigational New Drug Application for another oncology compound that addresses a new target
Inflammation Programs
JAK1/JAK2 Inhibitor: INCB28050
Complete the Phase II trial and present top-line results for the three-month portion of the study in the first half of this year; present the full six month results at the American College of Rheumatology Annual Meeting
Based on these results, decide whether to exercise our co-development option and participate in the Phase IIb program
JAK1/JAK2 Inhibitor: INCB18424 (topical formulation)
Present full results from the three-month Phase IIb trial at a scientific meeting
Decide on the appropriate next clinical trials
Evaluate potential strategic alliance opportunities
mfg ipollit
Exelixis mit Restrukturierung und umfassendem Personalabbau:
SOUTH SAN FRANCISCO, Calif.--(BUSINESS WIRE)--Exelixis, Inc. (Nasdaq:EXEL) today announced a restructuring as a consequence of its continued strategy to focus resources on the development of its key late-stage compounds. As its first priority, the company will aggressively advance XL184, XL147 and XL765, each of which is the subject of a large clinical development program. Additionally, Exelixis retains a fully integrated R&D organization, and will continue to advance new compounds into development, although the number will be reduced for the foreseeable future. Exelixis has retained the ability to meet all of its obligations to existing partners. Further, as a result of its retained R&D capabilities and its numerous unpartnered clinical and preclinical compounds, the company expects that its ongoing and future business development discussions will be unaffected.
The restructuring announced today results in a reduction of Exelixis’ workforce by approximately 40%, or 270 employees. This restructuring increases the company’s financial strength and positions the company for longer-term sustainable growth. As a result of the restructuring, the company estimates that its cumulative cash expenditures through 2011 will by reduced by approximately $90 million after payment of restructuring costs. The savings are primarily related to reductions in compensation and benefits, laboratory supplies and clinical trial costs. The company expects to record a restructuring charge of approximately $15 million in the first quarter of 2010, which may increase later in the year, depending on potential facility-related charges and other write downs that have not yet been finalized.
“The restructuring reflects our commitment to the strategic goals that we presented at our R&D Day in December 2009. We are focused on the aggressive development of our most advanced clinical compounds XL184, XL147 and XL765. Along with our investigators and partners, we remain enthusiastic about their potential as new anti-cancer agents and have retained the development expertise and capabilities to advance these compounds expeditiously. Additionally, we have scaled our fully integrated drug discovery and preclinical development organization to deliver one new IND per year. Our priority is to see ourselves through to the anticipated filing of our first NDA for XL184 in the second half of 2011,” said George A. Scangos, president and CEO of Exelixis, Inc. “We are convinced that the restructuring is the right step for the company and positions us well to move into the future. However, it is extremely difficult to release many people who have contributed substantially to the company over the years and who are our friends and colleagues.”
In addition to XL184, XL147 and XL765, Exelixis will continue development of XL888, an orally available small molecule inhibitor of HSP90 currently in phase 1, XL139 and XL413, compounds co-developed with BMS, as well as its preclinical program focused on PI3K delta.
SOUTH SAN FRANCISCO, Calif.--(BUSINESS WIRE)--Exelixis, Inc. (Nasdaq:EXEL) today announced a restructuring as a consequence of its continued strategy to focus resources on the development of its key late-stage compounds. As its first priority, the company will aggressively advance XL184, XL147 and XL765, each of which is the subject of a large clinical development program. Additionally, Exelixis retains a fully integrated R&D organization, and will continue to advance new compounds into development, although the number will be reduced for the foreseeable future. Exelixis has retained the ability to meet all of its obligations to existing partners. Further, as a result of its retained R&D capabilities and its numerous unpartnered clinical and preclinical compounds, the company expects that its ongoing and future business development discussions will be unaffected.
The restructuring announced today results in a reduction of Exelixis’ workforce by approximately 40%, or 270 employees. This restructuring increases the company’s financial strength and positions the company for longer-term sustainable growth. As a result of the restructuring, the company estimates that its cumulative cash expenditures through 2011 will by reduced by approximately $90 million after payment of restructuring costs. The savings are primarily related to reductions in compensation and benefits, laboratory supplies and clinical trial costs. The company expects to record a restructuring charge of approximately $15 million in the first quarter of 2010, which may increase later in the year, depending on potential facility-related charges and other write downs that have not yet been finalized.
“The restructuring reflects our commitment to the strategic goals that we presented at our R&D Day in December 2009. We are focused on the aggressive development of our most advanced clinical compounds XL184, XL147 and XL765. Along with our investigators and partners, we remain enthusiastic about their potential as new anti-cancer agents and have retained the development expertise and capabilities to advance these compounds expeditiously. Additionally, we have scaled our fully integrated drug discovery and preclinical development organization to deliver one new IND per year. Our priority is to see ourselves through to the anticipated filing of our first NDA for XL184 in the second half of 2011,” said George A. Scangos, president and CEO of Exelixis, Inc. “We are convinced that the restructuring is the right step for the company and positions us well to move into the future. However, it is extremely difficult to release many people who have contributed substantially to the company over the years and who are our friends and colleagues.”
In addition to XL184, XL147 and XL765, Exelixis will continue development of XL888, an orally available small molecule inhibitor of HSP90 currently in phase 1, XL139 and XL413, compounds co-developed with BMS, as well as its preclinical program focused on PI3K delta.
Antwort auf Beitrag Nr.: 39.088.620 von SLGramann am 08.03.10 22:12:44tja, krasse sache. die wahren motivationsgründe sind mir momentan noch komplett unklar.
übernahmevorbereitung?
scheitern von einem oder mehreren großen programmen?
???
wer denkt was?
übernahmevorbereitung?
scheitern von einem oder mehreren großen programmen?
???
wer denkt was?
Antwort auf Beitrag Nr.: 39.088.773 von PathFinder2 am 08.03.10 22:29:34hmmm, alle BMY-programme bleiben. eine roche/DNA-programm und ein eigenprogramm (beide PI) werden nicht weiter verfolgt.
sieht ziemlich nach einer übernahmevorbereitung aus, aber ich mag ganz falsch liegen...
würde auch die dealflaute der letzten monate erklären (zuvor wurde noch groß darüber gesprochen).
vielleicht aber auch nur wishful thinking...
sieht ziemlich nach einer übernahmevorbereitung aus, aber ich mag ganz falsch liegen...
würde auch die dealflaute der letzten monate erklären (zuvor wurde noch groß darüber gesprochen).
vielleicht aber auch nur wishful thinking...
Antwort auf Beitrag Nr.: 39.088.883 von PathFinder2 am 08.03.10 22:44:38
Ich rätsele auch herum. Mein erster Gedanke war, dass ihnen ein wichtiger Partner abhanden kommt - bspw. BMS. Aber so liest es sich eigentlich nicht.
Vielleicht ist es einfach so gemeint, wie es geschrieben ist. Obwohl, darf man so naiv denken?
Vielleicht klärt sich heute schon einiges auf?
Ich rätsele auch herum. Mein erster Gedanke war, dass ihnen ein wichtiger Partner abhanden kommt - bspw. BMS. Aber so liest es sich eigentlich nicht.
Vielleicht ist es einfach so gemeint, wie es geschrieben ist. Obwohl, darf man so naiv denken?
Vielleicht klärt sich heute schon einiges auf?
EXEL - lasst uns mal indizien zusammen tragen:
1. aussage von CEO Scangos auf R&D-day 2.12.2009:
"So, what are our goals? Well, look, we live in the real world. We have an attractive pipeline. We know that lots of things can happen. We know that this goal of building a profitable company we may never get to see because other things might happen, if they do, thats fine, right? But we are operating the company in a way that we can have a sustainable business. We have a balanced pipeline now. We have late stage compounds, we have earlier clinical programs. We have compounds moving thru lead optimization. We have potential revenue from 4 of these codevelopment projects. We have milestone and royalty revenue potential coming down the pike. Eventually we may take a compound forward on our own and we have the ability to keep replenishing that pipeline."
2. optionszuteilungen:
Von 8k-filing Dez 2009 (siehe http://biz.yahoo.com/e/091211/exel8-k.html )
"Due principally to the expiration of the 2000 Plan in January 2010 together with the Company’s expectation that it will not seek approval for a new equity compensation plan at the Company’s 2010 Annual Meeting of Stockholders (the “2010 Annual Meeting”), as well as certain other factors, the Compensation Committee, following discussion with the Board, determined to grant to the Company’s employees, including the named executive officers, equity awards that in the aggregate represent two years of historical year-end equity awards (i.e., for both fiscal 2009 and 2010). (....) Each of the named executive officers is a party to the Company’s Change in Control and Severance Benefit Plan, which provides for acceleration of vesting of the Awards in the event of certain specified change in control events involving the Company"
und eben viele optionszuteilungen:
http://ir.exelixis.com/phoenix.zhtml?c=120923&p=irol-sec&con…
3. Verzögerungen bei angekündigten Deals
4. krasse Restrukturierung:
40% der Belegschaft (!) werden gekündigt. Schwerpunkt auf drug discovery group (siehe press release "the workforce reduction was weighted towards the Company's drug discovery group")
===============
wer hat noch was?
und, was bedeutet das? meinungen?
1. aussage von CEO Scangos auf R&D-day 2.12.2009:
"So, what are our goals? Well, look, we live in the real world. We have an attractive pipeline. We know that lots of things can happen. We know that this goal of building a profitable company we may never get to see because other things might happen, if they do, thats fine, right? But we are operating the company in a way that we can have a sustainable business. We have a balanced pipeline now. We have late stage compounds, we have earlier clinical programs. We have compounds moving thru lead optimization. We have potential revenue from 4 of these codevelopment projects. We have milestone and royalty revenue potential coming down the pike. Eventually we may take a compound forward on our own and we have the ability to keep replenishing that pipeline."
2. optionszuteilungen:
Von 8k-filing Dez 2009 (siehe http://biz.yahoo.com/e/091211/exel8-k.html )
"Due principally to the expiration of the 2000 Plan in January 2010 together with the Company’s expectation that it will not seek approval for a new equity compensation plan at the Company’s 2010 Annual Meeting of Stockholders (the “2010 Annual Meeting”), as well as certain other factors, the Compensation Committee, following discussion with the Board, determined to grant to the Company’s employees, including the named executive officers, equity awards that in the aggregate represent two years of historical year-end equity awards (i.e., for both fiscal 2009 and 2010). (....) Each of the named executive officers is a party to the Company’s Change in Control and Severance Benefit Plan, which provides for acceleration of vesting of the Awards in the event of certain specified change in control events involving the Company"
und eben viele optionszuteilungen:
http://ir.exelixis.com/phoenix.zhtml?c=120923&p=irol-sec&con…
3. Verzögerungen bei angekündigten Deals
4. krasse Restrukturierung:
40% der Belegschaft (!) werden gekündigt. Schwerpunkt auf drug discovery group (siehe press release "the workforce reduction was weighted towards the Company's drug discovery group")
===============
wer hat noch was?
und, was bedeutet das? meinungen?
Antwort auf Beitrag Nr.: 39.098.512 von PathFinder2 am 09.03.10 21:10:56
Ich hab mal das CC-Transcript bei Seeking Alpha überflogen. Gibt leider nicht viel her imho.
Mein Bauchgefühl ist, dass sie wirklich einfach nur finanziellen Spielraum schaffen, um XL 184 mit voller Kraft weiterentwickeln zu können. Im CC haben sie gesagt, dass BMS 65% der Entwicklungskosten schultert. Das heißt aber eben auch, dass EXEL immerhin 35% der Kosten tragen muss. Nehmen wir mal, der Kandidat taugt wirklich so viel, wie erhofft, dann laufen da demnächst noch mehr P III-Studien in mehreren Indikationen. Das wird natürlich teuer. Wenn EXEL dem Kandidaten wirklich vertraut, macht es Sinn, sich jetzt darauf zu konzentrieren.
Aber vielleicht bin ich auch einfach nur zu naiv.
Ich hab mal das CC-Transcript bei Seeking Alpha überflogen. Gibt leider nicht viel her imho.
Mein Bauchgefühl ist, dass sie wirklich einfach nur finanziellen Spielraum schaffen, um XL 184 mit voller Kraft weiterentwickeln zu können. Im CC haben sie gesagt, dass BMS 65% der Entwicklungskosten schultert. Das heißt aber eben auch, dass EXEL immerhin 35% der Kosten tragen muss. Nehmen wir mal, der Kandidat taugt wirklich so viel, wie erhofft, dann laufen da demnächst noch mehr P III-Studien in mehreren Indikationen. Das wird natürlich teuer. Wenn EXEL dem Kandidaten wirklich vertraut, macht es Sinn, sich jetzt darauf zu konzentrieren.
Aber vielleicht bin ich auch einfach nur zu naiv.
INCY hat wirklich einen nachhaltig starken Lauf. Der Markt preist hier meines Erachtens jetzt bereits ein, dass das Unternehmen in zwei Jahren oder so nicht mehr development-stage sein wird, sondern richtig Geld verdienen wird.
Na ja... hoffentlich...

Na ja... hoffentlich...
wow! 

SGEN
wer denkst es wäre ein guter zeitpunkt zum verkaufen? gap bei USD 13... market cap mit 1,2 mrd. auch nicht mehr ganz günstig.
meinungen?
wer denkst es wäre ein guter zeitpunkt zum verkaufen? gap bei USD 13... market cap mit 1,2 mrd. auch nicht mehr ganz günstig.
meinungen?
Antwort auf Beitrag Nr.: 39.179.612 von PathFinder2 am 19.03.10 17:28:06
Tja, sag mir, wie die Lintuzumab-Daten sein werden und ich sag Dir, ob es ein guter Zeitpunkt ist, um jetzt zu verkaufen.
Ganz im ernst: Der SGEN-Kurs wird dieses Jahr aus fundamentalen Gründen erheblich bewegt werden imho. Binnen der nächsten drei Monate durch Lintuzumab und bis Jahresende nochmals durch Brentuximab.
Aus meiner Sicht sind beide Ereignisse dazu geeignet, den Kurs entweder deutlich steigen oder fallen zu lassen - je nach dem. Es ist also die Frage, ob man jetzt aussteigt und den Spatz in der Hand mitnimmt oder "den langen Weg geht".
Ich bleibe dabei - mit allen Risiken.
Tja, sag mir, wie die Lintuzumab-Daten sein werden und ich sag Dir, ob es ein guter Zeitpunkt ist, um jetzt zu verkaufen.
Ganz im ernst: Der SGEN-Kurs wird dieses Jahr aus fundamentalen Gründen erheblich bewegt werden imho. Binnen der nächsten drei Monate durch Lintuzumab und bis Jahresende nochmals durch Brentuximab.
Aus meiner Sicht sind beide Ereignisse dazu geeignet, den Kurs entweder deutlich steigen oder fallen zu lassen - je nach dem. Es ist also die Frage, ob man jetzt aussteigt und den Spatz in der Hand mitnimmt oder "den langen Weg geht".
Ich bleibe dabei - mit allen Risiken.
Antwort auf Beitrag Nr.: 39.118.129 von SLGramann am 11.03.10 20:26:18
Während ich bei INCY immer noch 18424-fixiert bin, schaut der Markt (angeblich) derzeit vor allem auf INCB 28050.
Dazu folgende Meinung:
Biotech investors should have Incyte (INCY) on their radar. Here’s why:
The drug discovery company will likely be announcing the results of a mid-stage study for an oral rheumatoid arthritis drug in April and analysts are pretty bullish on its prospects.
The drug program, currently referred to as INCB28050, or just '050, is similar to the later-stage drug '424 that Incyte is developing, which has already shown plenty of promise. While '424 is currently being tested in myelofibrosis, a bone marrow disease, and psoriasis, a condition that is closely related to rheumatoid arthritis, both drugs are Janus kinase (JAK) inhibitors. The JAK inhibitors target one or several of four enzymes that play a role in inflammatory diseases, myeloproliferative disorders, and malignancies.
The data that Incyte will be releasing soon is for a phase 2b study of '050 -- earlier data has been promising, but many investors are still a bit tentative about putting money on the program because of its infancy. They shouldn’t be though, because data from a '424 study in RA showed clear positive results for patients over a placebo. Cowen analyst Ziad Bakri told Minyanville that '050 is clearly efficacious.
Eli Lilly (LLY) certainly thought so -- the company paid $90 million upfront in December for the worldwide rights to ‘050, and the deal is potentially worth $665 million.
“Many investors are waiting for the results of this study before assigning value to this program. We are confident that '050 will show excellent activity in this indication,” wrote Leerink Swann analyst Joshua Schimmer in a recent note.
Yet, correlations with ‘424 and early data aren’t the only reasons to be checking out this program. RA is currently treated by injection with drugs like Abbott’s Humira (ABT) and Amgen’s Enbrel (AMGN). An oral treatment for the two million plus Americans who are plagued by constant inflammation of the joints and organs would be a huge convenience.
Incyte isn’t the only company that realizes the potential of an oral RA treatment; Pfizer (PFE), Celgene (CELG), and Rigel Pharmaceuticals (RIGL) are all developing drugs that would compete with ‘050. Pfizer’s JAK3 inhibitor is the furthest along of the group and has shown to be effective in treating RA; it is currently in late-stage studies.
“Pfizer's compound inhibits JAK3, but also inhibits JAK1 and JAK2. We see this as a close cousin to Incyte's JAK 1/2 inhibitor, and the success to date with [Pfizer’s compound] validates JAK inhibition for treating RA,” adds Schimmer.
While the new class of drugs is showing plenty of promise, no drug is a sure thing. Bakri points to safety questions that have popped up in the testing of the other oral RA drugs. “These have all been very potent drugs, but now the questions will be about safety,” he says. “Physicians generally want to see longer-term safety data and how they stack up against the current treatments. The oral drugs may be safe, but we really need to follow them up for longer to see how they do.”
Incyte is currently trading near $13.00. It has fluctuated between a low of $1.96 and a high of $13.25 over the last 52 weeks. The rheumatoid arthritis market is expected to reach $27 billion by 2015. Incyte is currently set to receive 11% to 20% in royalties from Lilly on ‘050 should it be approved, but the company has the option to bump that percentage up to 20% to 29% should it fund a bit more of the program costs.
Nochmals die Pipeline:

Während ich bei INCY immer noch 18424-fixiert bin, schaut der Markt (angeblich) derzeit vor allem auf INCB 28050.
Dazu folgende Meinung:
Biotech investors should have Incyte (INCY) on their radar. Here’s why:
The drug discovery company will likely be announcing the results of a mid-stage study for an oral rheumatoid arthritis drug in April and analysts are pretty bullish on its prospects.
The drug program, currently referred to as INCB28050, or just '050, is similar to the later-stage drug '424 that Incyte is developing, which has already shown plenty of promise. While '424 is currently being tested in myelofibrosis, a bone marrow disease, and psoriasis, a condition that is closely related to rheumatoid arthritis, both drugs are Janus kinase (JAK) inhibitors. The JAK inhibitors target one or several of four enzymes that play a role in inflammatory diseases, myeloproliferative disorders, and malignancies.
The data that Incyte will be releasing soon is for a phase 2b study of '050 -- earlier data has been promising, but many investors are still a bit tentative about putting money on the program because of its infancy. They shouldn’t be though, because data from a '424 study in RA showed clear positive results for patients over a placebo. Cowen analyst Ziad Bakri told Minyanville that '050 is clearly efficacious.
Eli Lilly (LLY) certainly thought so -- the company paid $90 million upfront in December for the worldwide rights to ‘050, and the deal is potentially worth $665 million.
“Many investors are waiting for the results of this study before assigning value to this program. We are confident that '050 will show excellent activity in this indication,” wrote Leerink Swann analyst Joshua Schimmer in a recent note.
Yet, correlations with ‘424 and early data aren’t the only reasons to be checking out this program. RA is currently treated by injection with drugs like Abbott’s Humira (ABT) and Amgen’s Enbrel (AMGN). An oral treatment for the two million plus Americans who are plagued by constant inflammation of the joints and organs would be a huge convenience.
Incyte isn’t the only company that realizes the potential of an oral RA treatment; Pfizer (PFE), Celgene (CELG), and Rigel Pharmaceuticals (RIGL) are all developing drugs that would compete with ‘050. Pfizer’s JAK3 inhibitor is the furthest along of the group and has shown to be effective in treating RA; it is currently in late-stage studies.
“Pfizer's compound inhibits JAK3, but also inhibits JAK1 and JAK2. We see this as a close cousin to Incyte's JAK 1/2 inhibitor, and the success to date with [Pfizer’s compound] validates JAK inhibition for treating RA,” adds Schimmer.
While the new class of drugs is showing plenty of promise, no drug is a sure thing. Bakri points to safety questions that have popped up in the testing of the other oral RA drugs. “These have all been very potent drugs, but now the questions will be about safety,” he says. “Physicians generally want to see longer-term safety data and how they stack up against the current treatments. The oral drugs may be safe, but we really need to follow them up for longer to see how they do.”
Incyte is currently trading near $13.00. It has fluctuated between a low of $1.96 and a high of $13.25 over the last 52 weeks. The rheumatoid arthritis market is expected to reach $27 billion by 2015. Incyte is currently set to receive 11% to 20% in royalties from Lilly on ‘050 should it be approved, but the company has the option to bump that percentage up to 20% to 29% should it fund a bit more of the program costs.
Nochmals die Pipeline:
In den letzten Wochen oder Monaten ist mir aufgefallen, dass zwei ziemlich kleine Unternehmen eine Stabilisierung ihrer Kurse erlebt haben. Nämlich Immunogen und Celldex.
Mir geht es nicht um Chart-Deutung. In beiden Fällen gibt es fundamentale Gründe zu großen Hoffnungen (ob am Ende berechtigt, kann kein Mensch wissen, das ist wie immer klar).
Zum einen Immunogen, die "kleine Schwester" unserer SGEN:

IMGN ist immerhin Roches Partner bei der Entwicklung einer ADC-Version von Herceptin, nämlich Trastuzumab-DM1 (T-DM1). Da sind sie mittlerweile in der P III angekommen. Man hatte gehofft, dass für das 3rd-line Treatment bereits P II-Daten für die Zulassung reichen würden, aber Roche scheint zu zögern, damit auf die FDA zuzugehen. Jedenfalls ist der Status da nicht ganz klar, was den Kurs seit Dezember belastet hatte.
Dennoch spricht einiges dafür, dass T-DM1 sehr werthaltig ist.
Feuerstein dazu:
I like ImmunoGen, but I feel like the stock is stuck in limbo as Roche/Genentech decides on a regulatory filing strategy for the breast cancer drug T-DM1.
I will be surprised if Roche decides against seeking U.S. approval for T-DM1 based on the very strong results from phase II study in third-line metastatic breast cancer patients that were presented last December. The uncertainty remains around timing of the filing. I expect T-DM1 to be submitted to the FDA in the first half of this year, but until Roche comes out and says something definitive, ImmunoGen's stock price will be weighed down.
When T-DM1 is filed, I'd think it's entirely possible that ImmunoGen's stock price will return to the $8-9 level.
Outside of third-line breast cancer, Roche is still very much engaged in advancing T-DM1 into larger breast cancer treatment opportunities. Essentially, T-DM1 could one day replace Herceptin as the best-in-class drug for Her-2 positive breast cancer treatment. That's a very good thing for ImmunoGen, long term.
Dann gibt es noch Celldex:

Da geht es vor allem um dieses Programm:
CDX-011: Clinical Program
TREATMENT FOR METASTATIC BREAST CANCER AND ADVANCED MELANOMA
Our first antibody-drug conjugate (ADC) product candidate, CDX-011, formerly CR011-vcMMAE, is in development for the treatment of locally advanced or metastatic breast cancer and stage III and IV melanoma. CDX-011 is a fully-human monoclonal ADC that targets glycoprotein NMB, also known as osteoactivin, a protein overexpressed on the surface of cancer cells, including melanoma, breast cancer and gliomas. The antibody, CDX-011, is linked to a potent chemotherapeutic, monomethyl auristatin E (MMAE), using Seattle Genetics’ proprietary technology. The ADC is designed to be stable in the bloodstream, but to release MMAE upon internalization into GPNMB-expressing tumor cells, resulting in a targeted cell-killing effect.
Es gibt erst ein paar P Ib-Daten, die aber - sagen andere - sehr interessant aussehen. Die Indikationen sind auf jeden Fall stark. Einen Wirkstoff auf das Melanom loszulassen, ist in jedem Fall mutig. Das sollte man im Auge behalten - ist ja auch aus SGEN-Sicht spannend.
Mir geht es nicht um Chart-Deutung. In beiden Fällen gibt es fundamentale Gründe zu großen Hoffnungen (ob am Ende berechtigt, kann kein Mensch wissen, das ist wie immer klar).
Zum einen Immunogen, die "kleine Schwester" unserer SGEN:
IMGN ist immerhin Roches Partner bei der Entwicklung einer ADC-Version von Herceptin, nämlich Trastuzumab-DM1 (T-DM1). Da sind sie mittlerweile in der P III angekommen. Man hatte gehofft, dass für das 3rd-line Treatment bereits P II-Daten für die Zulassung reichen würden, aber Roche scheint zu zögern, damit auf die FDA zuzugehen. Jedenfalls ist der Status da nicht ganz klar, was den Kurs seit Dezember belastet hatte.
Dennoch spricht einiges dafür, dass T-DM1 sehr werthaltig ist.
Feuerstein dazu:
I like ImmunoGen, but I feel like the stock is stuck in limbo as Roche/Genentech decides on a regulatory filing strategy for the breast cancer drug T-DM1.
I will be surprised if Roche decides against seeking U.S. approval for T-DM1 based on the very strong results from phase II study in third-line metastatic breast cancer patients that were presented last December. The uncertainty remains around timing of the filing. I expect T-DM1 to be submitted to the FDA in the first half of this year, but until Roche comes out and says something definitive, ImmunoGen's stock price will be weighed down.
When T-DM1 is filed, I'd think it's entirely possible that ImmunoGen's stock price will return to the $8-9 level.
Outside of third-line breast cancer, Roche is still very much engaged in advancing T-DM1 into larger breast cancer treatment opportunities. Essentially, T-DM1 could one day replace Herceptin as the best-in-class drug for Her-2 positive breast cancer treatment. That's a very good thing for ImmunoGen, long term.
Dann gibt es noch Celldex:
Da geht es vor allem um dieses Programm:
CDX-011: Clinical Program
TREATMENT FOR METASTATIC BREAST CANCER AND ADVANCED MELANOMA
Our first antibody-drug conjugate (ADC) product candidate, CDX-011, formerly CR011-vcMMAE, is in development for the treatment of locally advanced or metastatic breast cancer and stage III and IV melanoma. CDX-011 is a fully-human monoclonal ADC that targets glycoprotein NMB, also known as osteoactivin, a protein overexpressed on the surface of cancer cells, including melanoma, breast cancer and gliomas. The antibody, CDX-011, is linked to a potent chemotherapeutic, monomethyl auristatin E (MMAE), using Seattle Genetics’ proprietary technology. The ADC is designed to be stable in the bloodstream, but to release MMAE upon internalization into GPNMB-expressing tumor cells, resulting in a targeted cell-killing effect.
Es gibt erst ein paar P Ib-Daten, die aber - sagen andere - sehr interessant aussehen. Die Indikationen sind auf jeden Fall stark. Einen Wirkstoff auf das Melanom loszulassen, ist in jedem Fall mutig. Das sollte man im Auge behalten - ist ja auch aus SGEN-Sicht spannend.
REGN
Ein lauter Mensch... zu recht?
PDATE 3-Regeneron shares jump after CEO touts drugs
Tue Mar 23, 2010 6:45pm EDT
* Expects aflibercept colon, lung cancer data next year
* Says aflibercept has best chance against colon cancer
* Says drug may have less blood-clot risk than Avastin
* Shares close up 5.6 pct (Adds CEO quotes from interview, details on drugs)
By Ransdell Pierson
NEW YORK, March 23 (Reuters) - The CEO of Regeneron Inc (REGN.O) said on Tuesday his company expects late-stage data by the first half of 2011 on its experimental drug for colorectal and lung cancer, and that it could show a safety advantage over Roche Holding AG's (ROG.VX) blockbuster Avastin.
Shares of Tarrytown, New York-based Regeneron closed up 5.6 percent at $25.72 Tuesday on the Nasdaq, after Chief Executive Leonard Schleifer put the spotlight on aflibercept and other company drugs now winding their way through late-stage trials.
Partner Sanofi-Aventis (SASY.PA) is paying all costs of developing the oncology product, aflibercept, which is also being tested against prostate cancer. Interim data from late-stage trials for that cancer are expected in mid-2011.
But Schleifer said aflibercept probably has the best chance of succeeding against colorectal cancer. He noted it works through a similar mechanism as Avastin, which is one of the leading approved treatments for that cancer.
Aflibercept and Avastin both block a protein called VEGF that tumors use to build blood vessels capable of supplying themselves nutrients.
But Avastin and other anti-VEGF drugs cause blood clots, presumably by causing sticky blood cells called platelets to clump together, Schleifer said in a presentation at the annual Barclays Capital Global Healthcare Conference in Miami.
"There is reason to expect our drug doesn't activate platelets," providing a possible safety advantage over rival anti-VEGF drugs, Schleifer said.
Regeneron is also conducting late-stage trials of drugs meant to treat gout and macular degeneration, a leading cause of blindness.
Schleifer said the company expects late-stage data in the the second quarter on use of its drug, Arcalyst (rilonacept), to prevent gout flares and to treat acute gout -- an intensely painful form of arthritis.
He said Regeneron expects late-state data in the second half of this year on its medicine for macular degeneration, called VEGF-TRAP, whose active ingredient is aflibercept.
By developing an eye drug with its VEGF-inhibitor, Regeneron is following a trail blazed by Roche. The Swiss drugmaker also sells a treatment for macular degeneration, Lucentis, whose active ingredient is very similar to Avastin.
Regeneron also hopes to receive "proof-of-concept" data in the second quarter from an early-stage human trial of an experimental drug that cut "bad" LDL cholesterol 70 percent in primate studies. It works by blocking a protein called PCSK9.
"It's one of the most exciting new targets in biotechnology," Schleifer told Reuters in an interview, noting the drug is among a handful of monoclonal antibodies being tested in human trials under a 3-year-old deal with Sanofi.
Regeneron aims to begin trials of 30 to 40 other antibodies in the next eight years, under the Sanofi partnership.
"This is a year chock full of data for us and the depth and breadth of our pipeline, with eight biologics in development, is really just beginning to be appreciated," Schleifer said.
"I would aspire over time to achieve what some of the larger and more successful biotechnology companies have done, such as Genentech and Amgen," he said, adding that success will be measured by how many significant drugs Regeneron launches.
Sanofi provides Regeneron $160 million a year that it uses to discover new antibodies, and pays 100 percent of later costs to test the experimental drugs in clinical trials. In return, Schleifer said Regeneron will be entitled to very hefty royalties on drugs that succeed in trials and are approved.
Sanofi's funding represents a "substantial" percentage of the $750 million Regeneron will spend this year on research and development, Schleifer said, a strong endorsement of the company by far-larger Sanofi.
"It's also a big vote of confidence of us in Sanofi because we hooked up with them," said Schleifer. "And I feel strongly about that." (Reporting by Ransdell Pierson; editing by Matthew Lewis, Bernard Orr)
(die Cholesterin-Sache habe ich bisher überhaupt noch nicht registriert gehabt...)
Ein lauter Mensch... zu recht?
PDATE 3-Regeneron shares jump after CEO touts drugs
Tue Mar 23, 2010 6:45pm EDT
* Expects aflibercept colon, lung cancer data next year
* Says aflibercept has best chance against colon cancer
* Says drug may have less blood-clot risk than Avastin
* Shares close up 5.6 pct (Adds CEO quotes from interview, details on drugs)
By Ransdell Pierson
NEW YORK, March 23 (Reuters) - The CEO of Regeneron Inc (REGN.O) said on Tuesday his company expects late-stage data by the first half of 2011 on its experimental drug for colorectal and lung cancer, and that it could show a safety advantage over Roche Holding AG's (ROG.VX) blockbuster Avastin.
Shares of Tarrytown, New York-based Regeneron closed up 5.6 percent at $25.72 Tuesday on the Nasdaq, after Chief Executive Leonard Schleifer put the spotlight on aflibercept and other company drugs now winding their way through late-stage trials.
Partner Sanofi-Aventis (SASY.PA) is paying all costs of developing the oncology product, aflibercept, which is also being tested against prostate cancer. Interim data from late-stage trials for that cancer are expected in mid-2011.
But Schleifer said aflibercept probably has the best chance of succeeding against colorectal cancer. He noted it works through a similar mechanism as Avastin, which is one of the leading approved treatments for that cancer.
Aflibercept and Avastin both block a protein called VEGF that tumors use to build blood vessels capable of supplying themselves nutrients.
But Avastin and other anti-VEGF drugs cause blood clots, presumably by causing sticky blood cells called platelets to clump together, Schleifer said in a presentation at the annual Barclays Capital Global Healthcare Conference in Miami.
"There is reason to expect our drug doesn't activate platelets," providing a possible safety advantage over rival anti-VEGF drugs, Schleifer said.
Regeneron is also conducting late-stage trials of drugs meant to treat gout and macular degeneration, a leading cause of blindness.
Schleifer said the company expects late-stage data in the the second quarter on use of its drug, Arcalyst (rilonacept), to prevent gout flares and to treat acute gout -- an intensely painful form of arthritis.
He said Regeneron expects late-state data in the second half of this year on its medicine for macular degeneration, called VEGF-TRAP, whose active ingredient is aflibercept.
By developing an eye drug with its VEGF-inhibitor, Regeneron is following a trail blazed by Roche. The Swiss drugmaker also sells a treatment for macular degeneration, Lucentis, whose active ingredient is very similar to Avastin.
Regeneron also hopes to receive "proof-of-concept" data in the second quarter from an early-stage human trial of an experimental drug that cut "bad" LDL cholesterol 70 percent in primate studies. It works by blocking a protein called PCSK9.
"It's one of the most exciting new targets in biotechnology," Schleifer told Reuters in an interview, noting the drug is among a handful of monoclonal antibodies being tested in human trials under a 3-year-old deal with Sanofi.
Regeneron aims to begin trials of 30 to 40 other antibodies in the next eight years, under the Sanofi partnership.
"This is a year chock full of data for us and the depth and breadth of our pipeline, with eight biologics in development, is really just beginning to be appreciated," Schleifer said.
"I would aspire over time to achieve what some of the larger and more successful biotechnology companies have done, such as Genentech and Amgen," he said, adding that success will be measured by how many significant drugs Regeneron launches.
Sanofi provides Regeneron $160 million a year that it uses to discover new antibodies, and pays 100 percent of later costs to test the experimental drugs in clinical trials. In return, Schleifer said Regeneron will be entitled to very hefty royalties on drugs that succeed in trials and are approved.
Sanofi's funding represents a "substantial" percentage of the $750 million Regeneron will spend this year on research and development, Schleifer said, a strong endorsement of the company by far-larger Sanofi.
"It's also a big vote of confidence of us in Sanofi because we hooked up with them," said Schleifer. "And I feel strongly about that." (Reporting by Ransdell Pierson; editing by Matthew Lewis, Bernard Orr)
(die Cholesterin-Sache habe ich bisher überhaupt noch nicht registriert gehabt...)
habe mal eine kleine Position AMAG hinzugenommen und INCY leicht reduziert...

12,1% Incyte http://finance.yahoo.com/q?s=INCY
9,6% Regeneron http://finance.yahoo.com/q?s=REGN
7,5% Cubist http://finance.yahoo.com/q?s=CBST
5,7% NeuroSearch http://finance.yahoo.com/q?s=NEUR.CO
5,7% Allos http://finance.yahoo.com/q?s=ALTH
5,6% Micromet http://finance.yahoo.com/q?s=MITI
5,6% Genmab http://finance.yahoo.com/q?s=GEN.CO
5,4% Onyx http://finance.yahoo.com/q?s=ONXX
4,4% Exelixis http://finance.yahoo.com/q?s=EXEL
4,3% Rigel http://finance.yahoo.com/q?s=RIGL
4,3% Isis http://finance.yahoo.com/q?s=ISIS
4,1% NeurogesX http://finance.yahoo.com/q?s=NGSX
3,9% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,4% AMAG http://finance.yahoo.com/q?s=AMAG
3,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,0% ArQule http://finance.yahoo.com/q?s=ARQL
2,9% Array http://finance.yahoo.com/q?s=ARRY
2,8% Medigene http://finance.yahoo.com/q?s=MDG.DE
1,9% Arena http://finance.yahoo.com/q?s=ARNA
1,6% NicOx http://finance.yahoo.com/q?s=COX.PA
1,3% Progenics http://finance.yahoo.com/q?s=PGNX
1,0% Addex http://finance.yahoo.com/q?s=ADXN.SW
0,8% Sucampo http://finance.yahoo.com/q?s=SCMP
Entwicklung seit Jahresanfang auf EUR-Basis...
22,1% Depot
20,1% NBI
2,7% Dax
7,4% USD
124,0% NeuroSearch ( 5,7% )
80,6% ViroPharma ( 3,1% )
65,9% Incyte ( 12,1% )
36,9% Micromet ( 5,6% )
28,8% NeurogesX ( 4,1% )
27,8% Cubist ( 7,5% )
27,2% Progenics ( 1,3% )
23,2% Seattle Genetics ( 3,9% )
20,3% Allos ( 5,7% )
16,5% Regeneron ( 9,6% )
12,7% Onyx ( 5,4% )
10,1% NicOx ( 1,6% )
7,0% Array ( 2,9% )
3,2% Addex ( 1,0% )
2,6% Arena ( 1,9% )
0,1% Isis ( 4,3% )
-0,8% ArQule ( 3,0% )
-5,0% Medigene ( 2,8% )
-5,2% AMAG ( 3,4% )
-5,4% Sucampo ( 0,8% )
-11,0% Rigel ( 4,3% )
-12,4% Exelixis ( 4,4% )
-13,2% Genmab ( 5,6% )
mfg ipollit
12,1% Incyte http://finance.yahoo.com/q?s=INCY
9,6% Regeneron http://finance.yahoo.com/q?s=REGN
7,5% Cubist http://finance.yahoo.com/q?s=CBST
5,7% NeuroSearch http://finance.yahoo.com/q?s=NEUR.CO
5,7% Allos http://finance.yahoo.com/q?s=ALTH
5,6% Micromet http://finance.yahoo.com/q?s=MITI
5,6% Genmab http://finance.yahoo.com/q?s=GEN.CO
5,4% Onyx http://finance.yahoo.com/q?s=ONXX
4,4% Exelixis http://finance.yahoo.com/q?s=EXEL
4,3% Rigel http://finance.yahoo.com/q?s=RIGL
4,3% Isis http://finance.yahoo.com/q?s=ISIS
4,1% NeurogesX http://finance.yahoo.com/q?s=NGSX
3,9% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,4% AMAG http://finance.yahoo.com/q?s=AMAG
3,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,0% ArQule http://finance.yahoo.com/q?s=ARQL
2,9% Array http://finance.yahoo.com/q?s=ARRY
2,8% Medigene http://finance.yahoo.com/q?s=MDG.DE
1,9% Arena http://finance.yahoo.com/q?s=ARNA
1,6% NicOx http://finance.yahoo.com/q?s=COX.PA
1,3% Progenics http://finance.yahoo.com/q?s=PGNX
1,0% Addex http://finance.yahoo.com/q?s=ADXN.SW
0,8% Sucampo http://finance.yahoo.com/q?s=SCMP
Entwicklung seit Jahresanfang auf EUR-Basis...
22,1% Depot
20,1% NBI
2,7% Dax
7,4% USD
124,0% NeuroSearch ( 5,7% )
80,6% ViroPharma ( 3,1% )
65,9% Incyte ( 12,1% )
36,9% Micromet ( 5,6% )
28,8% NeurogesX ( 4,1% )
27,8% Cubist ( 7,5% )
27,2% Progenics ( 1,3% )
23,2% Seattle Genetics ( 3,9% )
20,3% Allos ( 5,7% )
16,5% Regeneron ( 9,6% )
12,7% Onyx ( 5,4% )
10,1% NicOx ( 1,6% )
7,0% Array ( 2,9% )
3,2% Addex ( 1,0% )
2,6% Arena ( 1,9% )
0,1% Isis ( 4,3% )
-0,8% ArQule ( 3,0% )
-5,0% Medigene ( 2,8% )
-5,2% AMAG ( 3,4% )
-5,4% Sucampo ( 0,8% )
-11,0% Rigel ( 4,3% )
-12,4% Exelixis ( 4,4% )
-13,2% Genmab ( 5,6% )
mfg ipollit
ARQL... positiv oder negativ?
wichtige PII-Ergebnisse bei Lungenkrebs in Kombination mit Tarceva... eigentlich steht da "nicht statistisch signifikant", aber adjustiert wieder signifikant???

ArQule Announces Results of Phase 2 Trial with ARQ 197 in Advanced Non-Small Cell Lung Cancer
ARQ 197 in combination with erlotinib prolongs median progression free survival
Press Release Source: ArQule, Inc. On Wednesday March 31, 2010, 7:00 am
WOBURN, Mass.--(BUSINESS WIRE)--ArQule, Inc. (Nasdaq: ARQL - News) today announced that ARQ 197, when used in combination with erlotinib, demonstrated a 66% improvement in median Progression-Free Survival (PFS) in patients with advanced, refractory non-small cell lung cancer (NSCLC). In the intent to treat (ITT) population (n = 167), median PFS was 16.1 weeks in the ARQ 197 plus erlotinib arm, compared with 9.7 weeks in the erlotinib plus placebo arm.
The difference in PFS between the two arms did not achieve statistical significance (hazard ratio = 0.809) by applying a log-rank test. When adjusting for imbalances in the distribution of key prognostic factors, the difference in PFS was statistically significant (hazard ratio = 0.675) by applying a Cox regression analysis specified for secondary efficacy analyses.
Improvement in median PFS was more pronounced in the pre-defined sub-group of patients with non-squamous histology (n = 117); median PFS was 18.9 weeks in the treatment arm versus 9.7 weeks in the control arm, which represents a 94% improvement. Based on an exploratory Cox regression analysis, the endpoint of PFS was met in the sub-group and achieved statistical significance (hazard ratio = 0.613).
There were no clinically relevant differences in adverse event rates between the treatment and control arms. The majority of adverse events were mild in intensity and included rash, diarrhea and fatigue.
“We believe the treatment benefit observed in this trial would represent a meaningful clinical improvement over standard therapy if replicated in Phase 3 trials,” said Dr. Brian Schwartz, chief medical officer of ArQule. “We are especially encouraged by the potential benefit for the large sub-group of non-squamous cell patients. We will thoroughly analyze our extensive database from this trial, including additional patient sub-group characteristics, to optimize ongoing and future trials of ARQ 197.”
Complete data from this trial, which will include biomarker analyses, will be presented at a future medical meeting during 2010.
One hundred sixty-seven patients were evaluated in the Phase 2 trial. Participating patients were EGFR (epidermal growth factor receptor) inhibitor naïve and were randomized one-to-one to receive either the combination of ARQ 197 plus erlotinib or placebo plus erlotinib in second and third line settings. ARQ 197 is an orally available, small molecule inhibitor of the c-Met receptor tyrosine kinase. Erlotinib, marketed as Tarceva™, is an inhibitor of the EGFR tyrosine kinase.
ARQ 197 is also currently being evaluated in clinical trials as a single agent and in combination with other anti-cancer therapies in a number of indications, including c-Met-associated soft-tissue sarcomas, hepatocellular carcinoma, pancreatic adenocarcinoma, germ cell tumors and colorectal cancer.
mfg ipollit
wichtige PII-Ergebnisse bei Lungenkrebs in Kombination mit Tarceva... eigentlich steht da "nicht statistisch signifikant", aber adjustiert wieder signifikant???
ArQule Announces Results of Phase 2 Trial with ARQ 197 in Advanced Non-Small Cell Lung Cancer
ARQ 197 in combination with erlotinib prolongs median progression free survival
Press Release Source: ArQule, Inc. On Wednesday March 31, 2010, 7:00 am
WOBURN, Mass.--(BUSINESS WIRE)--ArQule, Inc. (Nasdaq: ARQL - News) today announced that ARQ 197, when used in combination with erlotinib, demonstrated a 66% improvement in median Progression-Free Survival (PFS) in patients with advanced, refractory non-small cell lung cancer (NSCLC). In the intent to treat (ITT) population (n = 167), median PFS was 16.1 weeks in the ARQ 197 plus erlotinib arm, compared with 9.7 weeks in the erlotinib plus placebo arm.
The difference in PFS between the two arms did not achieve statistical significance (hazard ratio = 0.809) by applying a log-rank test. When adjusting for imbalances in the distribution of key prognostic factors, the difference in PFS was statistically significant (hazard ratio = 0.675) by applying a Cox regression analysis specified for secondary efficacy analyses.
Improvement in median PFS was more pronounced in the pre-defined sub-group of patients with non-squamous histology (n = 117); median PFS was 18.9 weeks in the treatment arm versus 9.7 weeks in the control arm, which represents a 94% improvement. Based on an exploratory Cox regression analysis, the endpoint of PFS was met in the sub-group and achieved statistical significance (hazard ratio = 0.613).
There were no clinically relevant differences in adverse event rates between the treatment and control arms. The majority of adverse events were mild in intensity and included rash, diarrhea and fatigue.
“We believe the treatment benefit observed in this trial would represent a meaningful clinical improvement over standard therapy if replicated in Phase 3 trials,” said Dr. Brian Schwartz, chief medical officer of ArQule. “We are especially encouraged by the potential benefit for the large sub-group of non-squamous cell patients. We will thoroughly analyze our extensive database from this trial, including additional patient sub-group characteristics, to optimize ongoing and future trials of ARQ 197.”
Complete data from this trial, which will include biomarker analyses, will be presented at a future medical meeting during 2010.
One hundred sixty-seven patients were evaluated in the Phase 2 trial. Participating patients were EGFR (epidermal growth factor receptor) inhibitor naïve and were randomized one-to-one to receive either the combination of ARQ 197 plus erlotinib or placebo plus erlotinib in second and third line settings. ARQ 197 is an orally available, small molecule inhibitor of the c-Met receptor tyrosine kinase. Erlotinib, marketed as Tarceva™, is an inhibitor of the EGFR tyrosine kinase.
ARQ 197 is also currently being evaluated in clinical trials as a single agent and in combination with other anti-cancer therapies in a number of indications, including c-Met-associated soft-tissue sarcomas, hepatocellular carcinoma, pancreatic adenocarcinoma, germ cell tumors and colorectal cancer.
mfg ipollit
Antwort auf Beitrag Nr.: 39.254.233 von ipollit am 31.03.10 14:04:54
Scheint positiv angenommen zu werden. Ob das hält? Pre-market ist so ne Sache...
March 31 (Reuters) - Biotechnology company ArQule Inc (ARQL.O) said its experimental lung cancer drug showed positive results in a mid-stage trial, sending its shares up 57 percent in pre-market trade.
The drug, ARQ 197, when used with another lung cancer drug, erlotinib, showed a 66 percent improvement in median progression-free survival (PFS) -- the time without cancer growth or death -- in patients with advanced, refractory non-small cell lung cancer, ArQule said.
The median PFS was 16.1 weeks in the ARQ 197 plus erlotinib arm, compared with 9.7 weeks in the erlotinib plus placebo arm, the company said in a statement.
The company, however, said the difference in PFS between the two arms did not achieve statistical significance by applying a log-rank test.
"There were no clinically relevant differences in adverse event rates between the treatment and control arms," the ArQule said.
Shares of the company were up $2 to $5.50 in pre-market trade. They closed at $3.50 Tuesday on Nasdaq. (Reporting by Anand Basu in Bangalore; Editing by Unnikrishnan Nair)
-----------
ISIS meldet heute eine neue Kooperation mit GSK. Auch schön.
Scheint positiv angenommen zu werden. Ob das hält? Pre-market ist so ne Sache...
March 31 (Reuters) - Biotechnology company ArQule Inc (ARQL.O) said its experimental lung cancer drug showed positive results in a mid-stage trial, sending its shares up 57 percent in pre-market trade.
The drug, ARQ 197, when used with another lung cancer drug, erlotinib, showed a 66 percent improvement in median progression-free survival (PFS) -- the time without cancer growth or death -- in patients with advanced, refractory non-small cell lung cancer, ArQule said.
The median PFS was 16.1 weeks in the ARQ 197 plus erlotinib arm, compared with 9.7 weeks in the erlotinib plus placebo arm, the company said in a statement.
The company, however, said the difference in PFS between the two arms did not achieve statistical significance by applying a log-rank test.
"There were no clinically relevant differences in adverse event rates between the treatment and control arms," the ArQule said.
Shares of the company were up $2 to $5.50 in pre-market trade. They closed at $3.50 Tuesday on Nasdaq. (Reporting by Anand Basu in Bangalore; Editing by Unnikrishnan Nair)
-----------
ISIS meldet heute eine neue Kooperation mit GSK. Auch schön.
Antwort auf Beitrag Nr.: 39.254.233 von ipollit am 31.03.10 14:04:54
Es wurden heute etwas mehr Aktien gehandelt, als insgesamt ausstehen. Da wurde wohl auch wirklich heftigst getradet.
Mal sehen, wo sich ARQL jetzt kurstechnisch mittelfristig einsortiert.
Es wurden heute etwas mehr Aktien gehandelt, als insgesamt ausstehen. Da wurde wohl auch wirklich heftigst getradet.
Mal sehen, wo sich ARQL jetzt kurstechnisch mittelfristig einsortiert.
Antwort auf Beitrag Nr.: 39.254.728 von SLGramann am 31.03.10 14:49:56hier noch der ISIS GSK Deal...
UPDATE 2-Glaxo, Isis in drug development deal of up to $1.5 bln
Wed Mar 31, 2010 1:04pm
* Glaxo to develop drugs using Isis's antisense technology
* Isis to receive $35 mln as upfront payment
* Isis shares up 11 percent
By Shailesh Kuber
BANGALORE, March 31 (Reuters) - Isis Pharmaceuticals Inc (ISIS.O) said it entered into an agreement worth up to $1.5 billion with GlaxoSmithKline (GSK.N) (GSK.L) to develop treatments for infectious diseases, using its antisense drug discovery platform.
Shares of Isis rose as much as 11 percent to $11.43 Wednesday on Nasdaq. They later pared some gains and were trading up 9 percent at $11.20.
"The deal with GlaxoSmithKline shall validate Isis' antisense platform and helps provide visibility to what is a very deep pipeline," Barclays Capital analyst Jim Birchenough said.
"It provides further funding to bring antisense opportunities to fruition," added Birchenough, who maintained his "overweight" rating and price target of $17 on Isis stock.
Under the deal, Isis will receive an upfront payment of $35 million from Glaxo and is eligible to receive on average up to $20 million per program up to mid-stage proof of concept (PoC) as milestone payments.
Also, Isis will be eligible to receive license fees and milestone payments of about $1.5 billion.
Glaxo will have the option to license compounds, and will be responsible for further development and marketing, while Isis will receive up to double-digit royalties on sales from any product that is successfully commercialized, the company said.
RNA-targeted therapeutics, or antisense therapies, aim to interfere at the genetic level to prevent rogue proteins from being formed. (Reporting by Anand Basu and Shailesh Kuber in Bangalore; Editing by Vinu Pilakkott, Anne Pallivathuckal)
mfg ipollit
UPDATE 2-Glaxo, Isis in drug development deal of up to $1.5 bln
Wed Mar 31, 2010 1:04pm
* Glaxo to develop drugs using Isis's antisense technology
* Isis to receive $35 mln as upfront payment
* Isis shares up 11 percent
By Shailesh Kuber
BANGALORE, March 31 (Reuters) - Isis Pharmaceuticals Inc (ISIS.O) said it entered into an agreement worth up to $1.5 billion with GlaxoSmithKline (GSK.N) (GSK.L) to develop treatments for infectious diseases, using its antisense drug discovery platform.
Shares of Isis rose as much as 11 percent to $11.43 Wednesday on Nasdaq. They later pared some gains and were trading up 9 percent at $11.20.
"The deal with GlaxoSmithKline shall validate Isis' antisense platform and helps provide visibility to what is a very deep pipeline," Barclays Capital analyst Jim Birchenough said.
"It provides further funding to bring antisense opportunities to fruition," added Birchenough, who maintained his "overweight" rating and price target of $17 on Isis stock.
Under the deal, Isis will receive an upfront payment of $35 million from Glaxo and is eligible to receive on average up to $20 million per program up to mid-stage proof of concept (PoC) as milestone payments.
Also, Isis will be eligible to receive license fees and milestone payments of about $1.5 billion.
Glaxo will have the option to license compounds, and will be responsible for further development and marketing, while Isis will receive up to double-digit royalties on sales from any product that is successfully commercialized, the company said.
RNA-targeted therapeutics, or antisense therapies, aim to interfere at the genetic level to prevent rogue proteins from being formed. (Reporting by Anand Basu and Shailesh Kuber in Bangalore; Editing by Vinu Pilakkott, Anne Pallivathuckal)
mfg ipollit
CBST... das von Dyax einlizensierte Ecallantide ist in der PII gescheitert (zuvor war die Patientenaufnahme in die PII wegen einer höheren Sterblichkeit bereits gestoppt worden), die Entwicklung und der Deal mit Dyax wird beendet
Cubist Pharmaceuticals to End Development of CB-500,929
Press Release Source: Cubist Pharmaceuticals, Inc. On Wednesday March 31, 2010, 4:00 pm EDT
LEXINGTON, Mass.--(BUSINESS WIRE)--Cubist Pharmaceuticals, Inc. (NASDAQ: CBST - News) today announced that it plans to stop investing in the clinical development of CB-500,929 (ecallantide) as a therapy to reduce blood loss in patients undergoing cardiac surgery using cardiopulmonary bypass. The company made this decision after reviewing top line efficacy and safety data from the CB-500,929 Phase 2 CONSERV™ 1 and CONSERV 2 trials. This decision does not impact the 2010 R & D cost guidance announced by the company in January 2010 as the guidance did not include costs for potential development of CB-500,929 beyond the CONSERV trials.
Top Line results
CONSERV 1: This study treated 249 patients undergoing primary Coronary Artery Bypass Graft (CABG) Surgery—while on cardiopulmonary bypass (CPB). This patient population was considered to be at a relatively low risk of bleeding complications in comparison to the CONSERV 2 population. Three doses of CB-500,929 (approximately 5, 25 or 75 mg depending on a patient’s weight and time on bypass) were evaluated vs. placebo. The results for this trial showed no improvement in blood loss (measured by the need for transfusion of blood and blood products) vs. placebo for any of the three doses studied. CONSERV 1 results showed similar safety results in patients who received study drug and those who received placebo.
CONSERV 2: This study treated 218 patients undergoing cardiac surgery while on CPB for procedures including repeat sternotomy, CABG + valve replacement or more than one valve replacement. This patient population was considered to be at a higher risk of bleeding complications than those undergoing a primary CABG procedure. In CONSERV 2, a dose of approximately 75 mg of CB-500,929 was studied vs. tranexamic acid as a comparator. The results of this trial also showed no treatment benefit for the CB-500,929 group relative to the comparator arm. A statistically significant higher rate of mortality was observed in patients in the study arm relative to those treated with tranexamic acid. This imbalance in mortality was observed by the Data Safety Monitoring Board late last year, and led Cubist to end enrollment in the CONSERV 2 trial early — see December 3, 2009 news release here. At that time, the company had also decided to end enrollment early in CONSERV 1 as a precaution although there was no imbalance in mortality observed in the CONSERV 1 results.
Given the decision by Cubist to end its development of CB-500,929, Cubist intends to terminate the 2008 agreement with Dyax under which Cubist in-licensed development and commercialization rights to CB-500,929 for surgical indications in the U.S. and the EU in accordance with the terms of the agreement.
mfg ipollit
Cubist Pharmaceuticals to End Development of CB-500,929
Press Release Source: Cubist Pharmaceuticals, Inc. On Wednesday March 31, 2010, 4:00 pm EDT
LEXINGTON, Mass.--(BUSINESS WIRE)--Cubist Pharmaceuticals, Inc. (NASDAQ: CBST - News) today announced that it plans to stop investing in the clinical development of CB-500,929 (ecallantide) as a therapy to reduce blood loss in patients undergoing cardiac surgery using cardiopulmonary bypass. The company made this decision after reviewing top line efficacy and safety data from the CB-500,929 Phase 2 CONSERV™ 1 and CONSERV 2 trials. This decision does not impact the 2010 R & D cost guidance announced by the company in January 2010 as the guidance did not include costs for potential development of CB-500,929 beyond the CONSERV trials.
Top Line results
CONSERV 1: This study treated 249 patients undergoing primary Coronary Artery Bypass Graft (CABG) Surgery—while on cardiopulmonary bypass (CPB). This patient population was considered to be at a relatively low risk of bleeding complications in comparison to the CONSERV 2 population. Three doses of CB-500,929 (approximately 5, 25 or 75 mg depending on a patient’s weight and time on bypass) were evaluated vs. placebo. The results for this trial showed no improvement in blood loss (measured by the need for transfusion of blood and blood products) vs. placebo for any of the three doses studied. CONSERV 1 results showed similar safety results in patients who received study drug and those who received placebo.
CONSERV 2: This study treated 218 patients undergoing cardiac surgery while on CPB for procedures including repeat sternotomy, CABG + valve replacement or more than one valve replacement. This patient population was considered to be at a higher risk of bleeding complications than those undergoing a primary CABG procedure. In CONSERV 2, a dose of approximately 75 mg of CB-500,929 was studied vs. tranexamic acid as a comparator. The results of this trial also showed no treatment benefit for the CB-500,929 group relative to the comparator arm. A statistically significant higher rate of mortality was observed in patients in the study arm relative to those treated with tranexamic acid. This imbalance in mortality was observed by the Data Safety Monitoring Board late last year, and led Cubist to end enrollment in the CONSERV 2 trial early — see December 3, 2009 news release here. At that time, the company had also decided to end enrollment early in CONSERV 1 as a precaution although there was no imbalance in mortality observed in the CONSERV 1 results.
Given the decision by Cubist to end its development of CB-500,929, Cubist intends to terminate the 2008 agreement with Dyax under which Cubist in-licensed development and commercialization rights to CB-500,929 for surgical indications in the U.S. and the EU in accordance with the terms of the agreement.
mfg ipollit
Antwort auf Beitrag Nr.: 39.229.332 von ipollit am 27.03.10 16:31:26AMAG lizensiert Feraheme für einen großen Teil des ex-US Markts an Takeda...
AMAG Pharma Inks Licensing Deal With Takeda For Anemia Drug - Update
4/1/2010 6:55 AM ET
(RTTNews) - Biopharmaceutical companies AMAG Pharmaceuticals, Inc. (AMAG: News ,AVM: News ) and Takeda Pharmaceutical Co. Ltd. (TKPHF.PK: News ) on Thursday jointly announced that the companies have entered into a license, development and commercialization deal for anemia drug Feraheme injection in all therapeutic indications.
Under the terms of the agreement, Osaka, Japan-based Takeda will receive an exclusive license to Feraheme for all therapeutic applications in 5 regions, including Europe, Canada, Turkey, the Commonwealth of Independent States and Asia Pacific countries, which excludes Japan, China and Taiwan.
As per the deal, Lexington, Massachusetts-based AMAG will be receiving an upfront payment of $60 million and will be eligible to receive up to $220 million in development and commercial milestones. Further, AMAG will also receive tiered, double-digit royalties based on Feraheme's net sales in the licensed territories.
Additionally, AMAG will execute and fund Feraheme's global clinical development in all potential therapeutic indications, and will also be initially responsible for filing of Feraheme's regulatory applications in Europe and Canada.
Takeda will be responsible for the regulatory filings in all other regions covered by the agreement, and will eventually hold all marketing authorizations in the licensed territories.
The agreement also states that Takeda will be responsible for commercializing Feraheme in all regions included in the licensed territories.
Feraheme was approved by the FDA as a treatment of iron deficiency anemia in chronic kidney disease patients last June. For the fourth-quarter ended December 31, 2009, Feraheme sales totaled $12.8 million, including $1.3 million of the $11.5 million in previously deferred product revenues.
AMAG also intends to submit a marketing authorization application to the European Medicines Agency or EMA for Feraheme for the treatment of IDA (iron deficiency anemia) in adult patients with chronic kidney disease in Europe in mid-2010. AMAG also added that it plans to initiate a broad global registrational program for Feraheme for the treatment of IDA regardless of underlying cause in mid-2010.
Takeda chief executive officer and international operations executive vice president Alan MacKenzie said, "Takeda is poised to maximize Feraheme's entry into the selected countries following approval."
In 2008, AMAG entered into a license and supply agreement with 3SBio Inc. (SSRX) with respect to the development and commercialization of Feraheme as an intravenous iron replacement therapeutic agent in China. 3SBio submitted its application for a registrational trial of Feraheme to the Chinese State Food & Drug Administration early this year.
mfg ipollit
AMAG Pharma Inks Licensing Deal With Takeda For Anemia Drug - Update
4/1/2010 6:55 AM ET
(RTTNews) - Biopharmaceutical companies AMAG Pharmaceuticals, Inc. (AMAG: News ,AVM: News ) and Takeda Pharmaceutical Co. Ltd. (TKPHF.PK: News ) on Thursday jointly announced that the companies have entered into a license, development and commercialization deal for anemia drug Feraheme injection in all therapeutic indications.
Under the terms of the agreement, Osaka, Japan-based Takeda will receive an exclusive license to Feraheme for all therapeutic applications in 5 regions, including Europe, Canada, Turkey, the Commonwealth of Independent States and Asia Pacific countries, which excludes Japan, China and Taiwan.
As per the deal, Lexington, Massachusetts-based AMAG will be receiving an upfront payment of $60 million and will be eligible to receive up to $220 million in development and commercial milestones. Further, AMAG will also receive tiered, double-digit royalties based on Feraheme's net sales in the licensed territories.
Additionally, AMAG will execute and fund Feraheme's global clinical development in all potential therapeutic indications, and will also be initially responsible for filing of Feraheme's regulatory applications in Europe and Canada.
Takeda will be responsible for the regulatory filings in all other regions covered by the agreement, and will eventually hold all marketing authorizations in the licensed territories.
The agreement also states that Takeda will be responsible for commercializing Feraheme in all regions included in the licensed territories.
Feraheme was approved by the FDA as a treatment of iron deficiency anemia in chronic kidney disease patients last June. For the fourth-quarter ended December 31, 2009, Feraheme sales totaled $12.8 million, including $1.3 million of the $11.5 million in previously deferred product revenues.
AMAG also intends to submit a marketing authorization application to the European Medicines Agency or EMA for Feraheme for the treatment of IDA (iron deficiency anemia) in adult patients with chronic kidney disease in Europe in mid-2010. AMAG also added that it plans to initiate a broad global registrational program for Feraheme for the treatment of IDA regardless of underlying cause in mid-2010.
Takeda chief executive officer and international operations executive vice president Alan MacKenzie said, "Takeda is poised to maximize Feraheme's entry into the selected countries following approval."
In 2008, AMAG entered into a license and supply agreement with 3SBio Inc. (SSRX) with respect to the development and commercialization of Feraheme as an intravenous iron replacement therapeutic agent in China. 3SBio submitted its application for a registrational trial of Feraheme to the Chinese State Food & Drug Administration early this year.
mfg ipollit
Antwort auf Beitrag Nr.: 39.262.845 von ipollit am 01.04.10 14:31:15AMAG Finds Ex-U.S. Feraheme Partner in $280M Takeda Deal
By Jennifer Boggs
Assistant Managing Editor
AMAG Pharmaceuticals Inc. is picking up $60 million up front in a potential $280 million deal with Takeda Pharmaceutical Co. Ltd. for iron replacement therapy Feraheme (ferumoxytol), some much-needed good news for the small biotech still dealing with fallout from the drug's safety data reported earlier this year.
The drug had an impressive launch following its July approval, but a single analyst downgrade in February based on reports of adverse events sent the stock dropping. Lexington, Mass.-based AMAG held an emergency conference call and managed to convince analysts that the serious adverse events rate of 0.1 percent was well within expectations, but the damage was already done.
AMAG now faces a class-action lawsuit alleging that it withheld information about adverse reactions in some patients taking Feraheme, though the company responded in an SEC filing that the claims were "without merit," and said it intends to "vigorously" defend against those allegations.
Takeda clearly isn't worried. The Osaka, Japan-based company offered financial terms that Leerink Swann analyst Joseph Schwartz called "better than we expected, given the genericized nature" of the iron replacement markets outside the U.S.," and will add Feraheme to its growing armament of anemia treatments.
The pharma firm already has rights to Hematide, a Phase III-stage peptide-based alternative to existing erythropoiesis-stimulating agents from Affymax Inc., of Palo Alto, Calif. That drug would be a nice complement to Feraheme in the chronic kidney disease space, though AMAG CEO Brian Pereira assured investors that Feraheme would still be able to "stand on its own" if Hematide encounters a development or regulatory hitch.
"The synergies in the nephrology space are great, but they're not a must" for Feraheme, he added. Takeda also has a "broad reach in multiple segments," which will be important when the companies seek to expand the indications beyond the CKD market.
Under the terms, Takeda will be responsible for commercialization in Europe, Canada, Turkey, the Commonwealth of Independent States and the Asia Pacific countries excluding Japan, China and Taiwan. It's possible that Japan could be added later, though, for now, that territory has not been a priority in Feraheme's development plan.
Given its previous clinical success with Feraheme, AMAG will continue being responsible for development, with trials in iron deficiency anemia slated to start sometime this year. The biotech also will handle the regulatory filing for Europe, expected around midyear. A new drug application was submitted to Canadian authorities late in 2009.
Since its launch in the U.S., Feraheme sales have bested expectations. For the fourth quarter, AMAG posted net revenues of $12.8 million.
Part of that is due to the fact Feraheme is priced at a premium, about 40 percent higher than market-leading Venofer (iron sucrose) from Bad Homburg, Germany-based Fresenius Biotech. That's unlikely to happen in Europe, where there already are low-cost iron replacement generics.
But, on the plus side, the iron replacement market in Europe is growing, Pereira estimated the EU iron market currently to be about $150 million. And, he pointed out, physicians overseas are more likely than U.S. doctors to prescribe an iron therapy before increasing ESA doses.
Takeda has final authority over pricing the drug in its territories.
AMAG has not disclosed how it will recognize funding from the transaction, though David Arkowitz, the firm's chief financial officer and chief business officer, said none of the up-front money will be recognized in first-quarter earnings.
Not that AMAG is hurting for cash. The company reported cash, equivalents and short-term investments totaling $129.5 million as of Dec. 31.
Shares of AMAG (NASDAQ:AMAG) closed Thursday at $35.02, up 11 cents.
Published April 2, 2010
By Jennifer Boggs
Assistant Managing Editor
AMAG Pharmaceuticals Inc. is picking up $60 million up front in a potential $280 million deal with Takeda Pharmaceutical Co. Ltd. for iron replacement therapy Feraheme (ferumoxytol), some much-needed good news for the small biotech still dealing with fallout from the drug's safety data reported earlier this year.
The drug had an impressive launch following its July approval, but a single analyst downgrade in February based on reports of adverse events sent the stock dropping. Lexington, Mass.-based AMAG held an emergency conference call and managed to convince analysts that the serious adverse events rate of 0.1 percent was well within expectations, but the damage was already done.
AMAG now faces a class-action lawsuit alleging that it withheld information about adverse reactions in some patients taking Feraheme, though the company responded in an SEC filing that the claims were "without merit," and said it intends to "vigorously" defend against those allegations.
Takeda clearly isn't worried. The Osaka, Japan-based company offered financial terms that Leerink Swann analyst Joseph Schwartz called "better than we expected, given the genericized nature" of the iron replacement markets outside the U.S.," and will add Feraheme to its growing armament of anemia treatments.
The pharma firm already has rights to Hematide, a Phase III-stage peptide-based alternative to existing erythropoiesis-stimulating agents from Affymax Inc., of Palo Alto, Calif. That drug would be a nice complement to Feraheme in the chronic kidney disease space, though AMAG CEO Brian Pereira assured investors that Feraheme would still be able to "stand on its own" if Hematide encounters a development or regulatory hitch.
"The synergies in the nephrology space are great, but they're not a must" for Feraheme, he added. Takeda also has a "broad reach in multiple segments," which will be important when the companies seek to expand the indications beyond the CKD market.
Under the terms, Takeda will be responsible for commercialization in Europe, Canada, Turkey, the Commonwealth of Independent States and the Asia Pacific countries excluding Japan, China and Taiwan. It's possible that Japan could be added later, though, for now, that territory has not been a priority in Feraheme's development plan.
Given its previous clinical success with Feraheme, AMAG will continue being responsible for development, with trials in iron deficiency anemia slated to start sometime this year. The biotech also will handle the regulatory filing for Europe, expected around midyear. A new drug application was submitted to Canadian authorities late in 2009.
Since its launch in the U.S., Feraheme sales have bested expectations. For the fourth quarter, AMAG posted net revenues of $12.8 million.
Part of that is due to the fact Feraheme is priced at a premium, about 40 percent higher than market-leading Venofer (iron sucrose) from Bad Homburg, Germany-based Fresenius Biotech. That's unlikely to happen in Europe, where there already are low-cost iron replacement generics.
But, on the plus side, the iron replacement market in Europe is growing, Pereira estimated the EU iron market currently to be about $150 million. And, he pointed out, physicians overseas are more likely than U.S. doctors to prescribe an iron therapy before increasing ESA doses.
Takeda has final authority over pricing the drug in its territories.
AMAG has not disclosed how it will recognize funding from the transaction, though David Arkowitz, the firm's chief financial officer and chief business officer, said none of the up-front money will be recognized in first-quarter earnings.
Not that AMAG is hurting for cash. The company reported cash, equivalents and short-term investments totaling $129.5 million as of Dec. 31.
Shares of AMAG (NASDAQ:AMAG) closed Thursday at $35.02, up 11 cents.
Published April 2, 2010
Antwort auf Beitrag Nr.: 39.262.845 von ipollit am 01.04.10 14:31:15
Hi ipollit,
wie viel Potential kann man den mittel- und langfristig für Feraheme annehmen?
AMAG ist ja immerhin schon mit über 700 Mio. bewertet. Reden wir hier über mehr als ein Nischenprodukt?
Viele Grüße.
Hi ipollit,
wie viel Potential kann man den mittel- und langfristig für Feraheme annehmen?
AMAG ist ja immerhin schon mit über 700 Mio. bewertet. Reden wir hier über mehr als ein Nischenprodukt?
Viele Grüße.
Antwort auf Beitrag Nr.: 39.268.111 von SLGramann am 02.04.10 10:33:28hi SLGramann!
Peak sales werden wohl so auf ein paar 100 Mio USD geschätzt... also es könnte mehr als ein Nischenprodukt werden. Anfang des Jahres gab es den Anstig von 40 auf 50 USD, weil die Q4 Umsätze über den Erwartungen lagen. Danach ging es aber wieder deutlich runter, weil andersherum die Umsätze in den ersten beiden Monaten eher schwach gewesen sind. Wohin es sich also entwicklen wird, ist noch nicht ganz klar. Im Moment besteht das Risiko, dass Q1 jetzt ziemlich unterhalb der Erwartungen ist.
Im Bereich des Eisenmangels bei Patienten mit Nierenproblemen gibt es zwei Märkte: der Markt der Patienten, die gleichzeitig eine Dialyse benötigen, ist schon ziemlich abgedeckt. Interessanter ist Feraheme für Patienten, die noch keine Dialyse benötigen, so dass sie eigentlich nicht regelmäßig zum Arzt müssen. Der Vorteil von Feraheme gegenüber den bereits etablierten Eisen-IV-Präparaten ist die schnellere Verabreichung... nur 2x eine kurze Injektion, während normalerweise 10x eine längere Injektion benötigt wird.
Die MK ist wegen einer KE von 170 Mio USD im Januar höher... ca. 850 Mio USD.
ein paar (teilweise ältere) Meinungen...
RW Baird, March 13
Reiterate Outperform rating, $53 price target.
Incrementally positive after CMS
published a Feraheme Q210 reimbursement rate of $817/gram, indicating that Q409
ASP was just 1% lower than list price – well within the 2% range guided to by
management. As such, we expect a strong finish to the quarter. Combining this with
what we see as the stock’s unwarranted pressure over the last several weeks, we
remain aggressive AMAG buyers.
Summary
• CMS’ Q210 reimbursement rates are out. CMS released payment allowance limits
over the weekend. Recall for Medicare Part B drugs, reimbursement is ASP-based
and set two quarters in arrears, so numbers derive from Q409 ASPs.
• Feraheme’s Q210 ASP in line. CMS provided Q210 reimbursement limits for both
Feraheme Q-codes (dialysis and pre-dialysis) of $0.817 per mg ($817 per gram),
inclusive of ASP and the 6% administration fee.
- As management indicated. AMAG has indicated total ASP for Q210 will be
within 2% of WAC. The ASP derived from this CMS update is ~$786 per
treatment, or just 1% shy of Feraheme’s $794 WAC.
- Next two weeks critical for the quarter. We expect significant buy-in through
month-end as providers now have visibility into the Q210 economics. With
December, January and February sales volume ~$6.8M,~ $2.6M, and ~$3.5M,
Feraheme will need a strong March (~$8.4M) in order to meet Q110 expectations.
• Still confident in our FY10 and longer-term estimates. We are confident in a
strong March, and we continue to model Q110-Q410 Feraheme revenue of $14.5M,
$17.8M, $20.5M and $23.5M, respectively.
• Remain buyers here. With the stock off 28% from its January 14 high of $51 (versus
the S&P 500 up ~1% in the same period), we think any questions on the progress of
the launch are more than priced in. With an enterprise value of $644M, yet
anticipated FY10 revenue of $77M, we remain aggressive buyers.
*********
http://www.thestreet.com/story/10532381/1/amags-got-room-to-…
AMAG's Got Room to Run
Adam Feuerstein
07/01/09 - 11:02 AM EDT
Feraheme -- approved at last.
The injectable iron replacement therapy from AMAG Pharmaceuticals(AMAG) was granted final marketing approval by U.S. regulators Tuesday night, ending a long-delayed process begun in December 2007.
Shares of AMAG were up 3.4% to $56.50 in recent trading.
AMAG will begin selling Feraheme later this month. The drug, given in two, 17-second injections about a week apart, will be used by doctors to correct anemia in patients with chronic kidney disease.
Sales of injectable iron products totals about $500 million in the U.S., primarily in kidney dialysis patients but also in patients not yet on dialysis. Feraheme's more convenient dosing and broader treatment label gives it an advantage over its competitors -- mainly Venofer, sold by Luitpold Pharmaceuticals and American Regent.
AMAG will announce Feraheme's price when it launches, but the Street is generally expecting the product to be priced at a 20-30% premium to its competitors. Consensus peak sales for Feraheme currently stands around $300 million, although that will increase significantly if/when AMAG expands Feraheme's use into other indications.
Is it too late to buy AMAG? Investors not in the stock today missed out, especially last fall when the skepticism over Feraheme's approval was at its peak and AMAG shares dipped to the low $20s. But even with the stock in the high $50s, AMAG still only carries an $800 million-plus enterprise value -- which means there's upside in the stock easily into the mid-$60s based on the current Feraheme commercial opportunity.
For those who owned AMAG going into Tuesday night's approval, well done! AMAG has not been an easy stock to own, especially late last year when Feraheme's initial approval decision date came and went without good news.
Full disclosure: I recommended AMAG as a long to readers of my biotech stock newsletter last October, and I've stuck with the stock since then, stubbornly believing in Feraheme's approval.
I also recommended to my newsletter readers to take some profits in AMAG with the stock in the low to mid $50s, just in case approval didn't happen. With victory at hand, some partial profit taking is still a good idea, especially for investors sitting on big gains.
As AMAG moves into Feraheme's launch phase, don't be surprised to see the stock's short interest tick up. Going long into a drug's approval and turning short for the drug's launch is a well-worn saw in the Street's biotech investing playbook.
Doubts will be raised about AMAG's ability to sell Feraheme effectively into what is a complicated and treacherously competitive chronic kidney disease treatment market. But just as I trusted AMAG management to get Feraheme approved, I also believe the company has the chops to launch the drug strongly.
Smart investors should be looking at any significant weakness in AMAG as a buying opportunity.
J.P. Morgan analyst Matt Roden, also an AMAG bull, believes AMAG might set Feraheme's price above Street expectations, which would drive upside to current analyst forecasts and move the stock higher.
Roden also believes there's upside to Feraheme's revenue potential if Davita(DVA), one of the two largest dialysis service providers, decides to bring Feraheme into its clinics in a big way. Fresenius(FMS), the other large dialysis service provider, isn't expected to utilize Feraheme to any significant degree since the company has an exclusive licensing deal for Venofer.
The easiest way to think about the dynamics of the kidney dialysis market is to split it into thirds - one third belongs to Fresenius, a third to Davita, and the remaining third is spread around independent dialysis providers.
Roden believes Feraheme can achieve 30% share of the overall iron replacement market by capturing a significant share of independent dialysis providers plus some small amount of use by Davita. His estimates are generally above other analysts. He has a buy rating on the stock and a $65 price target.
If Davita really embraces Feraheme, the drug has the potential to capture 50% of the overall market, which could be double what the Street is currently modeling, he says.
If you follow AMAG and the upcoming Feraheme launch, you're going to hear a lot of talk about Medicare bundling and what the means -- good or bad -- for Feraheme's commercial potential.
Under current Medicare guidelines, dialysis service providers are paid a set rate, or a bundled rate, for dialysis services, plus a separate rate for drugs used during dialysis, such as iron replacement therapy or widely used erythropoiesis-stimulating agents (ESAs) like Amgen's(AMGN) Epogen and Aranesp or Johnson & Johnson's(JNJ) Procrit.
But under reform proposals to lower Medicare costs, the agency is considering a plan to combine reimbursement for all dialysis services and drugs into one bundled rate. This may force dialysis service providers to lower their use of drugs, for instance, in order to maintain their profits.
This new "bundled environment" - which could start in 2011 - has the potential to be positive for AMAG because increased use of Feraheme would allow dialysis providers to reduce their use of the more costly (and therefore less profitable) ESAs like Epogen and Aranesp.
Feraheme, however, will be more expensive than other injectable iron replacement products, so the risk is that dialysis service providers rely more heavily on competing products such as Luitpold's Venofer in order to wring out even more profits from the Medicare dialysis bundle.
*************
http://www.zacks.com/stock/news/23004/AMAG+Pharma%92s+Ferahe…
AMAG Pharma’s Feraheme Holds Promise
July 30, 2009
Yesterday, AMAG Pharmaceuticals Inc. (AMAG - Snapshot Report) reported second-quarter net loss of $26.5 million, or $1.55 per share, which was slightly higher than the Zacks Consensus estimate of $1.53. The company had reported a net loss of $17.0 million, or $1.00 per share, in the year-ago period.
The higher quarterly net loss is attributable to a 39% increase in year-over-year operating expenses. Research and development expenses rose 43% as the company is working on expanding its manufacturing capabilities and development infrastructure. Selling, general and administrative expenses also increased 37% for commercialization of anti-anemia drug candidate, Feraheme.
Since Feraheme is a very important product for the company, we expect investor focus to remain on the initial sales ramp of the product.
As a reminder, the US Food and Drug Administration (FDA) granted marketing approval to Feraheme injection on June 30. AMAG officially launched Feraheme in the U.S. on July 13. It is an intravenous iron-replacement therapy used to fight anemia in patients with chronic kidney disease. The drug can be used by patients irrespective of dialysis.
Wholesalers and specialty distributors will primarily distribute Feraheme in the U.S. AMAG will use its commercial organization, consisting of approximately 150 seasoned professionals which includes an 80-person specialized sales force, to market the drug. For every 510 mg vial, the wholesale acquisition cost (WAC) of Feraheme is $396.78.
Iron deficiency anemia is a significant problem in patients with chronic kidney disease and often goes undiagnosed and undertreated. We believe peak sales of Feraheme could reach $500 million.
Although we are pleased to hear about Feraheme’s approval and subsequent launch, stiff competition awaits AMAG’s drug in the IV iron replacement therapy market. Fresenius Medical Care AG & Co.’s (FMS - Snapshot Report) Venofer and Watson Pharmaceuticals Inc. (WPI - Analyst Report)-Sanofi-Aventis’ (SNY - Analyst Report) Ferrlecit are AMAG’s main competitors in this market.
We believe that Feraheme's safety and efficacy profile should help it gain share in the market and advice investors to Buy the stock at the current price.
***************
http://seekingalpha.com/article/153347-amag-pharmaceuticals-…
AMAG Pharmaceuticals' Feraheme: The Next Billion Dollar Drug
On June 30, 2009, the FDA approved Feraheme for the treatment of anemia in chronic kidney disease (CKD), both in pre-dialysis patients and those undergoing dialysis. Feraheme, sold by AMAG Pharmaceuticals (AMAG), received a clean label without any black box warnings. Now that Feraheme has been approved, the key question for investors is what the market opportunity is and how quickly it will be achieved.
Background on Feraheme
Feraheme (generic name: ferumoxytol) was originally developed as an imaging agent. Iron, in order to be safely given, must be in a complex and not as a free metal. The free metal can cause a number of side effects including rash, wheezing, and anaphylaxis. Feraheme has the lowest free iron of any commercially available preparation (Balakrishnan, 2009). It is quite different from any other preparation because it is an iron oxide nanoparticle with a polyglucose sorbitol carboxymethylether coating.
Because of its low levels of free iron, Feraheme can be given much more rapidly than any other iron formulation. 510 mg can be given in a 17 second injection. (A standard dose of 1 gram of iron can thus be given in two office visits of a few minutes each.) In contrast, other iron preparations require 1 gram of iron to be given over 8-10 doses over 15-30 minutes each of injection time.
From an outpatient perspective, Feraheme is far more convenient and cost effective (when considering physician time, nursing time, and patient time) than any other iron preparation.
Competition
There are four other competing irons on the market: Venofer (iron sucrose), Ferrlecit (sodium ferric gluconate), INFeD (low molecular weight iron dextran), and Dexferrum (high molecular weight iron dextran). The dextrans have a statistically significant higher level of adverse events, including fatal anaphylatic reactions, and are used much less often than Venofer or Ferrlecit. According to IMS, 2008 sales were:
Venofer: $359M
Ferrlecit: $152M
Infed: $37M
Dexferrum: $10M
Before Feraheme's approval, Venofer was the only iron approved for pre-dialysis, one of the reasons for its higher sales.
Impact from bundling
Starting in January 2011, it is likely that the Centers for Medicare and Medicaid Services (CMS) will begin to pay for dialysis patients using a so called "bundling" payment system. In this system, dialysis centers are reimbursed on a per patient basis and are thus have the incentive to keep costs as low as possible. It should be noted that this only affects the dialysis market and not pre-dialysis (which as we will see is larger).
When one factors in nursing and technician time, even in a bundled environment it is likely that Feraheme will be at least cost neutral if not cost beneficial. This is largely because of these ease of administration and the fact that patients can become iron replete much more quickly (in as short as 3 days) with Feraheme than other iron preparations. As discussed above, in the outpatient setting it is unmatched because of dramatic time (and hence cost) savings to physicians, nurses, and patients. (See Leerink-Swann report on AMAG dated June 26, 2009 for more on cost savings.)
Market size estimates
There are two current markets for Feraheme: CKD pre-dialysis patients and CKD dialysis patients. There are three future markets for Feraheme: vascular-enhanced MRI (VE-MRI) imaging, pediatric anemia, and generalized adult iron-deficiency anemia (IDA) from causes like heavy uterine bleeding, cancer, or gastrointestinal bleeding.
The wholesale acquisition costs (WAC) of Feraheme was announced to be $396.78 per vial. (Each vial contains 510 mg of iron.) This is approximately 40% higher than other IV irons. For the purpose of estimating market opportunity, it will be assumed that AMAG will negotiate supplier deals at a discount. To err on the side of being conservative, we will assume that each vial will be sold at $300, which is likely lower than will occur in practice.
The average dialysis CKD patient receives about 2.5 grams of iron each year. There are 354,000 dialysis CKD patients who are currently treated for iron deficiency (USRDS 2007 Annual Data Report). Most of these patients receive Venofer today. Given the cost savings of labor and nursing time, Feraheme should attain 50% market share. This is likely an underestimate, especially because for the next 1.5 years, dialysis clinics that use Feraheme will receive greater reimbursement due to the ASP + 6% structure that CMS currently employs.
Under these assumptions, peak sales in dialysis CKD patients will be:
$300 / vial * (5 vials / patient ) * 354,000 patients / year *
50% penetration = $265.5 million / year
There are approximately 1.6 million pre-dialysis patients who have iron-deficiency anemia. While this is 4.5 fold more patients than dialysis CKD patients, they require less iron than dialysis patients, only about 1-1.5 grams per year (we will conservatively use 1 gram in our estimates). In this market, Feraheme is the clear winner because of the tremendous savings to patients and physicians regarding time and labor costs. From a cost perspective, it has no real competition in this clinical setting besides oral iron, which is less efficacious and more uncomfortable for patients. Hence in this market we will hence assume 70% penetration:
Under these assumptions, peak sales in pre-dialysis CKD patients will be:
$300 / vial * (2 vials / patient ) * 1,600,000 patients / year *
70% penetration = $672M
VE-MRI is an interesting market, but more difficult to estimate. For many years, it has been clear that superparamagnetic iron oxide has been useful as a contrast agent (Kent, 1990). Nearly 30% of the 31 million annual MRI examinations use gadolinium (Colletti, 2008), which imparts the risk of renal toxicity (in particular nephrogenic systemic fibrosis). This has led to the search for other superparamagnetic agents without the toxicities of gadolinium. Iron also is superparamagnetic, and is a natural metal present in the body. However giving iron as a bolus has traditionally been restricted because of high levels of free iron, which can have a number of side effects such as rash, wheezing, and anaphylaxis. Feraheme elegantly solves these issues. Yet few details have been provided around Feraheme dosing in their clinical trial development plan. Given the present uncertainties regarding market size and dosing, no contributions of VE-MRI to peak sales will be factored.
Similar to the adult market, the current pediatric market is dominated by CKD patients. Pediatric patients are usually not treated with sufficient iron (Horl, 2007). Pediatric renal transplant patients are also commonly iron deficient (Kausman, 2004). Venofer and Ferrlecit are standard of care in pediatric patients today (Horl, 2007). It is unclear to what extent AMAG plans to develop this market, so for the purposes of estimating market size, it will be assumed to be negligible.
AMAG is in discussion with the FDA about running a phase 3 trial for generalized adult iron-deficiency anemia. This would include cancer patients, gastrointestinal bleeding patients, and women with abnormal uterine bleeding. These are much larger markets, totalling at least 4 million people in the United States. Due to administration advantages and cost savings, Feraheme is again the clear winner. It will be assumed that 20% market penetration will be achieved here, the main competitor being oral iron. (Neither Venofer or Ferrlecit are competitive due to administration inconvenience.) Assuming smaller needs of 1 gram of iron per year, similar to pre-dialysis patients, this yields:
$300 / vial * (2 vials / patient ) * 4,000,000 patients / year * 20% penetration = $480M
Because 20% is likely a low penetration estimate, this could obviously be dramatically higher.
Putting this together we have:
$265.5M + $672M + $480M = $1.4 billion
Factoring in Japan, Europe, India, China, and Latin America, a rough rule of thumb for estimating market size is that the the non-US markets in total are about the size of the US market. It will be assumed that AMAG will partner Feraheme in these markets, and receive 25% royalties.
Royalties then represent another 25% * $1.4 billion = $350M.
All told, using conservative assumptions throughout, peak sales should be:
$1.4 billion + $350M = $1.75 billion.
Time to adoption
Given the ASP + 6% reimbursement system, it is likely that many dialysis chains will adopt Feraheme for strong financial reasons within the next 3-6 months. Pre-dialysis adoption should occur even faster due to the lack of requisite contract agreements. Regarding generalized IDA, trial design discussions are underway with the FDA. It is likely that phase III trial would begin in early 2010, with submission by late 2010 and approval in 2011. Given time for sales and marketing efforts to reach their intended audience, peak sales could occur as early as 4-5 years from the present, or 2013-2014.
One element that should accelerate adoption is the renown of the AMAG management team. Their CEO Brian Perreira is a world leading nephrologist and former president of the National Kidney Foundation. Louis Brenner, Senior Vice President at AMAG, is a nephrologist and former business development head of Genzyme's renal division. Robert Brenner, another nephrologist, spent nine years at Amgen (AMGN), arguably the leading renal disease company in the world.
Impact from generics?
Watson recently announced that it may develop a generic version of Ferrlecit. Most analysts are skeptical if this will occur given the high trial costs (AMAG performed four phase III trials of Feraheme) and already low sales of Ferrlecit. Even if Watson did pursue this route, it would not be of relevance until 2011, and probably never gain relevance for the largest markets of pre-dialysis CKD and generalized IDA. Ironically, Watson had filed a Citizen's petition trying to block the entrance of generic competitors into the market. It will be of interest to watch how it now shifts its position for presumably financial reasons.
Conclusions
For patient investors, AMAG (market cap $775M as of close of market July 30, 2009) represents one of the best risk/reward profiles in the biotechnology space given the $1-2B market for Feraheme, an already approved drug.
Disclosure: The author's firm is long AMAG.
**************
Advantages of Feraheme:• Avoids the Side Effects Associated with Oral Iron Replacement Therapies – Oral ironreplacement therapies have limited efficacy and they do not adequately increase the body's ironlevels in anemic CKD patients. Additionally, oral iron replacement therapies are poorly absorbedin anemic CKD patients, resulting in unwanted side effects such as diarrhea and cramping, oftenleading to the discontinuation of treatment. In contrast, the IV formulation of Feraheme providesgreater amounts of iron in anemic CKD patients, while avoiding the side effects linked with oraliron replacement therapies.• Rapid Administration and Less Frequent Dosing – The existing IV iron replacement therapies(like Venofer from FMS/Vifor International and Ferrlecit from WPI/SNY) need to be administeredslowly and in small dosages over several sessions to meet the body’s total iron requirement. Therecommended dosage of Venofer is 1,000mg, which cannot be administered at a single go. In non-dialysis dependent CKD patients, the total dosage (1,000mg) of Venofer is divided into5 sub-dosages (200mg each), with each sub-dosage requiring 5-10 minutes for administration. The 5 sub-dosages are given in 5 different occasions within a 14-day period. In dialysis dependent CKD patients, the total dosage (1,000mg) of Venofer is divided into 10sub-dosages (100mg each), with each sub-dosage administration requiring 5-10 minutes. Allthese sub-dosages are administered in 10 different occasions.
Ferrlecit, which is approved only for dialysis dependent CKD patients, has a recommended dosageof 1,000mg. Like Venofer, Ferrlecit cannot be given in single administration. The total dosage isbroken into 8 sub-dosages (125mg each), taking approximately 10 minutes (which sometimes canextend to an hour) for administration of each sub-dosage. These 8 sub-dosages are given over 8consecutive dialysis sessions.In contrast, Feraheme is rapidly administered in two 17-second injections, making it more patientfriendly.
******
mfg ipollit
Peak sales werden wohl so auf ein paar 100 Mio USD geschätzt... also es könnte mehr als ein Nischenprodukt werden. Anfang des Jahres gab es den Anstig von 40 auf 50 USD, weil die Q4 Umsätze über den Erwartungen lagen. Danach ging es aber wieder deutlich runter, weil andersherum die Umsätze in den ersten beiden Monaten eher schwach gewesen sind. Wohin es sich also entwicklen wird, ist noch nicht ganz klar. Im Moment besteht das Risiko, dass Q1 jetzt ziemlich unterhalb der Erwartungen ist.
Im Bereich des Eisenmangels bei Patienten mit Nierenproblemen gibt es zwei Märkte: der Markt der Patienten, die gleichzeitig eine Dialyse benötigen, ist schon ziemlich abgedeckt. Interessanter ist Feraheme für Patienten, die noch keine Dialyse benötigen, so dass sie eigentlich nicht regelmäßig zum Arzt müssen. Der Vorteil von Feraheme gegenüber den bereits etablierten Eisen-IV-Präparaten ist die schnellere Verabreichung... nur 2x eine kurze Injektion, während normalerweise 10x eine längere Injektion benötigt wird.
Die MK ist wegen einer KE von 170 Mio USD im Januar höher... ca. 850 Mio USD.
ein paar (teilweise ältere) Meinungen...
RW Baird, March 13
Reiterate Outperform rating, $53 price target.
Incrementally positive after CMS
published a Feraheme Q210 reimbursement rate of $817/gram, indicating that Q409
ASP was just 1% lower than list price – well within the 2% range guided to by
management. As such, we expect a strong finish to the quarter. Combining this with
what we see as the stock’s unwarranted pressure over the last several weeks, we
remain aggressive AMAG buyers.
Summary
• CMS’ Q210 reimbursement rates are out. CMS released payment allowance limits
over the weekend. Recall for Medicare Part B drugs, reimbursement is ASP-based
and set two quarters in arrears, so numbers derive from Q409 ASPs.
• Feraheme’s Q210 ASP in line. CMS provided Q210 reimbursement limits for both
Feraheme Q-codes (dialysis and pre-dialysis) of $0.817 per mg ($817 per gram),
inclusive of ASP and the 6% administration fee.
- As management indicated. AMAG has indicated total ASP for Q210 will be
within 2% of WAC. The ASP derived from this CMS update is ~$786 per
treatment, or just 1% shy of Feraheme’s $794 WAC.
- Next two weeks critical for the quarter. We expect significant buy-in through
month-end as providers now have visibility into the Q210 economics. With
December, January and February sales volume ~$6.8M,~ $2.6M, and ~$3.5M,
Feraheme will need a strong March (~$8.4M) in order to meet Q110 expectations.
• Still confident in our FY10 and longer-term estimates. We are confident in a
strong March, and we continue to model Q110-Q410 Feraheme revenue of $14.5M,
$17.8M, $20.5M and $23.5M, respectively.
• Remain buyers here. With the stock off 28% from its January 14 high of $51 (versus
the S&P 500 up ~1% in the same period), we think any questions on the progress of
the launch are more than priced in. With an enterprise value of $644M, yet
anticipated FY10 revenue of $77M, we remain aggressive buyers.
*********
http://www.thestreet.com/story/10532381/1/amags-got-room-to-…
AMAG's Got Room to Run
Adam Feuerstein
07/01/09 - 11:02 AM EDT
Feraheme -- approved at last.
The injectable iron replacement therapy from AMAG Pharmaceuticals(AMAG) was granted final marketing approval by U.S. regulators Tuesday night, ending a long-delayed process begun in December 2007.
Shares of AMAG were up 3.4% to $56.50 in recent trading.
AMAG will begin selling Feraheme later this month. The drug, given in two, 17-second injections about a week apart, will be used by doctors to correct anemia in patients with chronic kidney disease.
Sales of injectable iron products totals about $500 million in the U.S., primarily in kidney dialysis patients but also in patients not yet on dialysis. Feraheme's more convenient dosing and broader treatment label gives it an advantage over its competitors -- mainly Venofer, sold by Luitpold Pharmaceuticals and American Regent.
AMAG will announce Feraheme's price when it launches, but the Street is generally expecting the product to be priced at a 20-30% premium to its competitors. Consensus peak sales for Feraheme currently stands around $300 million, although that will increase significantly if/when AMAG expands Feraheme's use into other indications.
Is it too late to buy AMAG? Investors not in the stock today missed out, especially last fall when the skepticism over Feraheme's approval was at its peak and AMAG shares dipped to the low $20s. But even with the stock in the high $50s, AMAG still only carries an $800 million-plus enterprise value -- which means there's upside in the stock easily into the mid-$60s based on the current Feraheme commercial opportunity.
For those who owned AMAG going into Tuesday night's approval, well done! AMAG has not been an easy stock to own, especially late last year when Feraheme's initial approval decision date came and went without good news.
Full disclosure: I recommended AMAG as a long to readers of my biotech stock newsletter last October, and I've stuck with the stock since then, stubbornly believing in Feraheme's approval.
I also recommended to my newsletter readers to take some profits in AMAG with the stock in the low to mid $50s, just in case approval didn't happen. With victory at hand, some partial profit taking is still a good idea, especially for investors sitting on big gains.
As AMAG moves into Feraheme's launch phase, don't be surprised to see the stock's short interest tick up. Going long into a drug's approval and turning short for the drug's launch is a well-worn saw in the Street's biotech investing playbook.
Doubts will be raised about AMAG's ability to sell Feraheme effectively into what is a complicated and treacherously competitive chronic kidney disease treatment market. But just as I trusted AMAG management to get Feraheme approved, I also believe the company has the chops to launch the drug strongly.
Smart investors should be looking at any significant weakness in AMAG as a buying opportunity.
J.P. Morgan analyst Matt Roden, also an AMAG bull, believes AMAG might set Feraheme's price above Street expectations, which would drive upside to current analyst forecasts and move the stock higher.
Roden also believes there's upside to Feraheme's revenue potential if Davita(DVA), one of the two largest dialysis service providers, decides to bring Feraheme into its clinics in a big way. Fresenius(FMS), the other large dialysis service provider, isn't expected to utilize Feraheme to any significant degree since the company has an exclusive licensing deal for Venofer.
The easiest way to think about the dynamics of the kidney dialysis market is to split it into thirds - one third belongs to Fresenius, a third to Davita, and the remaining third is spread around independent dialysis providers.
Roden believes Feraheme can achieve 30% share of the overall iron replacement market by capturing a significant share of independent dialysis providers plus some small amount of use by Davita. His estimates are generally above other analysts. He has a buy rating on the stock and a $65 price target.
If Davita really embraces Feraheme, the drug has the potential to capture 50% of the overall market, which could be double what the Street is currently modeling, he says.
If you follow AMAG and the upcoming Feraheme launch, you're going to hear a lot of talk about Medicare bundling and what the means -- good or bad -- for Feraheme's commercial potential.
Under current Medicare guidelines, dialysis service providers are paid a set rate, or a bundled rate, for dialysis services, plus a separate rate for drugs used during dialysis, such as iron replacement therapy or widely used erythropoiesis-stimulating agents (ESAs) like Amgen's(AMGN) Epogen and Aranesp or Johnson & Johnson's(JNJ) Procrit.
But under reform proposals to lower Medicare costs, the agency is considering a plan to combine reimbursement for all dialysis services and drugs into one bundled rate. This may force dialysis service providers to lower their use of drugs, for instance, in order to maintain their profits.
This new "bundled environment" - which could start in 2011 - has the potential to be positive for AMAG because increased use of Feraheme would allow dialysis providers to reduce their use of the more costly (and therefore less profitable) ESAs like Epogen and Aranesp.
Feraheme, however, will be more expensive than other injectable iron replacement products, so the risk is that dialysis service providers rely more heavily on competing products such as Luitpold's Venofer in order to wring out even more profits from the Medicare dialysis bundle.
*************
http://www.zacks.com/stock/news/23004/AMAG+Pharma%92s+Ferahe…
AMAG Pharma’s Feraheme Holds Promise
July 30, 2009
Yesterday, AMAG Pharmaceuticals Inc. (AMAG - Snapshot Report) reported second-quarter net loss of $26.5 million, or $1.55 per share, which was slightly higher than the Zacks Consensus estimate of $1.53. The company had reported a net loss of $17.0 million, or $1.00 per share, in the year-ago period.
The higher quarterly net loss is attributable to a 39% increase in year-over-year operating expenses. Research and development expenses rose 43% as the company is working on expanding its manufacturing capabilities and development infrastructure. Selling, general and administrative expenses also increased 37% for commercialization of anti-anemia drug candidate, Feraheme.
Since Feraheme is a very important product for the company, we expect investor focus to remain on the initial sales ramp of the product.
As a reminder, the US Food and Drug Administration (FDA) granted marketing approval to Feraheme injection on June 30. AMAG officially launched Feraheme in the U.S. on July 13. It is an intravenous iron-replacement therapy used to fight anemia in patients with chronic kidney disease. The drug can be used by patients irrespective of dialysis.
Wholesalers and specialty distributors will primarily distribute Feraheme in the U.S. AMAG will use its commercial organization, consisting of approximately 150 seasoned professionals which includes an 80-person specialized sales force, to market the drug. For every 510 mg vial, the wholesale acquisition cost (WAC) of Feraheme is $396.78.
Iron deficiency anemia is a significant problem in patients with chronic kidney disease and often goes undiagnosed and undertreated. We believe peak sales of Feraheme could reach $500 million.
Although we are pleased to hear about Feraheme’s approval and subsequent launch, stiff competition awaits AMAG’s drug in the IV iron replacement therapy market. Fresenius Medical Care AG & Co.’s (FMS - Snapshot Report) Venofer and Watson Pharmaceuticals Inc. (WPI - Analyst Report)-Sanofi-Aventis’ (SNY - Analyst Report) Ferrlecit are AMAG’s main competitors in this market.
We believe that Feraheme's safety and efficacy profile should help it gain share in the market and advice investors to Buy the stock at the current price.
***************
http://seekingalpha.com/article/153347-amag-pharmaceuticals-…
AMAG Pharmaceuticals' Feraheme: The Next Billion Dollar Drug
On June 30, 2009, the FDA approved Feraheme for the treatment of anemia in chronic kidney disease (CKD), both in pre-dialysis patients and those undergoing dialysis. Feraheme, sold by AMAG Pharmaceuticals (AMAG), received a clean label without any black box warnings. Now that Feraheme has been approved, the key question for investors is what the market opportunity is and how quickly it will be achieved.
Background on Feraheme
Feraheme (generic name: ferumoxytol) was originally developed as an imaging agent. Iron, in order to be safely given, must be in a complex and not as a free metal. The free metal can cause a number of side effects including rash, wheezing, and anaphylaxis. Feraheme has the lowest free iron of any commercially available preparation (Balakrishnan, 2009). It is quite different from any other preparation because it is an iron oxide nanoparticle with a polyglucose sorbitol carboxymethylether coating.
Because of its low levels of free iron, Feraheme can be given much more rapidly than any other iron formulation. 510 mg can be given in a 17 second injection. (A standard dose of 1 gram of iron can thus be given in two office visits of a few minutes each.) In contrast, other iron preparations require 1 gram of iron to be given over 8-10 doses over 15-30 minutes each of injection time.
From an outpatient perspective, Feraheme is far more convenient and cost effective (when considering physician time, nursing time, and patient time) than any other iron preparation.
Competition
There are four other competing irons on the market: Venofer (iron sucrose), Ferrlecit (sodium ferric gluconate), INFeD (low molecular weight iron dextran), and Dexferrum (high molecular weight iron dextran). The dextrans have a statistically significant higher level of adverse events, including fatal anaphylatic reactions, and are used much less often than Venofer or Ferrlecit. According to IMS, 2008 sales were:
Venofer: $359M
Ferrlecit: $152M
Infed: $37M
Dexferrum: $10M
Before Feraheme's approval, Venofer was the only iron approved for pre-dialysis, one of the reasons for its higher sales.
Impact from bundling
Starting in January 2011, it is likely that the Centers for Medicare and Medicaid Services (CMS) will begin to pay for dialysis patients using a so called "bundling" payment system. In this system, dialysis centers are reimbursed on a per patient basis and are thus have the incentive to keep costs as low as possible. It should be noted that this only affects the dialysis market and not pre-dialysis (which as we will see is larger).
When one factors in nursing and technician time, even in a bundled environment it is likely that Feraheme will be at least cost neutral if not cost beneficial. This is largely because of these ease of administration and the fact that patients can become iron replete much more quickly (in as short as 3 days) with Feraheme than other iron preparations. As discussed above, in the outpatient setting it is unmatched because of dramatic time (and hence cost) savings to physicians, nurses, and patients. (See Leerink-Swann report on AMAG dated June 26, 2009 for more on cost savings.)
Market size estimates
There are two current markets for Feraheme: CKD pre-dialysis patients and CKD dialysis patients. There are three future markets for Feraheme: vascular-enhanced MRI (VE-MRI) imaging, pediatric anemia, and generalized adult iron-deficiency anemia (IDA) from causes like heavy uterine bleeding, cancer, or gastrointestinal bleeding.
The wholesale acquisition costs (WAC) of Feraheme was announced to be $396.78 per vial. (Each vial contains 510 mg of iron.) This is approximately 40% higher than other IV irons. For the purpose of estimating market opportunity, it will be assumed that AMAG will negotiate supplier deals at a discount. To err on the side of being conservative, we will assume that each vial will be sold at $300, which is likely lower than will occur in practice.
The average dialysis CKD patient receives about 2.5 grams of iron each year. There are 354,000 dialysis CKD patients who are currently treated for iron deficiency (USRDS 2007 Annual Data Report). Most of these patients receive Venofer today. Given the cost savings of labor and nursing time, Feraheme should attain 50% market share. This is likely an underestimate, especially because for the next 1.5 years, dialysis clinics that use Feraheme will receive greater reimbursement due to the ASP + 6% structure that CMS currently employs.
Under these assumptions, peak sales in dialysis CKD patients will be:
$300 / vial * (5 vials / patient ) * 354,000 patients / year *
50% penetration = $265.5 million / year
There are approximately 1.6 million pre-dialysis patients who have iron-deficiency anemia. While this is 4.5 fold more patients than dialysis CKD patients, they require less iron than dialysis patients, only about 1-1.5 grams per year (we will conservatively use 1 gram in our estimates). In this market, Feraheme is the clear winner because of the tremendous savings to patients and physicians regarding time and labor costs. From a cost perspective, it has no real competition in this clinical setting besides oral iron, which is less efficacious and more uncomfortable for patients. Hence in this market we will hence assume 70% penetration:
Under these assumptions, peak sales in pre-dialysis CKD patients will be:
$300 / vial * (2 vials / patient ) * 1,600,000 patients / year *
70% penetration = $672M
VE-MRI is an interesting market, but more difficult to estimate. For many years, it has been clear that superparamagnetic iron oxide has been useful as a contrast agent (Kent, 1990). Nearly 30% of the 31 million annual MRI examinations use gadolinium (Colletti, 2008), which imparts the risk of renal toxicity (in particular nephrogenic systemic fibrosis). This has led to the search for other superparamagnetic agents without the toxicities of gadolinium. Iron also is superparamagnetic, and is a natural metal present in the body. However giving iron as a bolus has traditionally been restricted because of high levels of free iron, which can have a number of side effects such as rash, wheezing, and anaphylaxis. Feraheme elegantly solves these issues. Yet few details have been provided around Feraheme dosing in their clinical trial development plan. Given the present uncertainties regarding market size and dosing, no contributions of VE-MRI to peak sales will be factored.
Similar to the adult market, the current pediatric market is dominated by CKD patients. Pediatric patients are usually not treated with sufficient iron (Horl, 2007). Pediatric renal transplant patients are also commonly iron deficient (Kausman, 2004). Venofer and Ferrlecit are standard of care in pediatric patients today (Horl, 2007). It is unclear to what extent AMAG plans to develop this market, so for the purposes of estimating market size, it will be assumed to be negligible.
AMAG is in discussion with the FDA about running a phase 3 trial for generalized adult iron-deficiency anemia. This would include cancer patients, gastrointestinal bleeding patients, and women with abnormal uterine bleeding. These are much larger markets, totalling at least 4 million people in the United States. Due to administration advantages and cost savings, Feraheme is again the clear winner. It will be assumed that 20% market penetration will be achieved here, the main competitor being oral iron. (Neither Venofer or Ferrlecit are competitive due to administration inconvenience.) Assuming smaller needs of 1 gram of iron per year, similar to pre-dialysis patients, this yields:
$300 / vial * (2 vials / patient ) * 4,000,000 patients / year * 20% penetration = $480M
Because 20% is likely a low penetration estimate, this could obviously be dramatically higher.
Putting this together we have:
$265.5M + $672M + $480M = $1.4 billion
Factoring in Japan, Europe, India, China, and Latin America, a rough rule of thumb for estimating market size is that the the non-US markets in total are about the size of the US market. It will be assumed that AMAG will partner Feraheme in these markets, and receive 25% royalties.
Royalties then represent another 25% * $1.4 billion = $350M.
All told, using conservative assumptions throughout, peak sales should be:
$1.4 billion + $350M = $1.75 billion.
Time to adoption
Given the ASP + 6% reimbursement system, it is likely that many dialysis chains will adopt Feraheme for strong financial reasons within the next 3-6 months. Pre-dialysis adoption should occur even faster due to the lack of requisite contract agreements. Regarding generalized IDA, trial design discussions are underway with the FDA. It is likely that phase III trial would begin in early 2010, with submission by late 2010 and approval in 2011. Given time for sales and marketing efforts to reach their intended audience, peak sales could occur as early as 4-5 years from the present, or 2013-2014.
One element that should accelerate adoption is the renown of the AMAG management team. Their CEO Brian Perreira is a world leading nephrologist and former president of the National Kidney Foundation. Louis Brenner, Senior Vice President at AMAG, is a nephrologist and former business development head of Genzyme's renal division. Robert Brenner, another nephrologist, spent nine years at Amgen (AMGN), arguably the leading renal disease company in the world.
Impact from generics?
Watson recently announced that it may develop a generic version of Ferrlecit. Most analysts are skeptical if this will occur given the high trial costs (AMAG performed four phase III trials of Feraheme) and already low sales of Ferrlecit. Even if Watson did pursue this route, it would not be of relevance until 2011, and probably never gain relevance for the largest markets of pre-dialysis CKD and generalized IDA. Ironically, Watson had filed a Citizen's petition trying to block the entrance of generic competitors into the market. It will be of interest to watch how it now shifts its position for presumably financial reasons.
Conclusions
For patient investors, AMAG (market cap $775M as of close of market July 30, 2009) represents one of the best risk/reward profiles in the biotechnology space given the $1-2B market for Feraheme, an already approved drug.
Disclosure: The author's firm is long AMAG.
**************
Advantages of Feraheme:• Avoids the Side Effects Associated with Oral Iron Replacement Therapies – Oral ironreplacement therapies have limited efficacy and they do not adequately increase the body's ironlevels in anemic CKD patients. Additionally, oral iron replacement therapies are poorly absorbedin anemic CKD patients, resulting in unwanted side effects such as diarrhea and cramping, oftenleading to the discontinuation of treatment. In contrast, the IV formulation of Feraheme providesgreater amounts of iron in anemic CKD patients, while avoiding the side effects linked with oraliron replacement therapies.• Rapid Administration and Less Frequent Dosing – The existing IV iron replacement therapies(like Venofer from FMS/Vifor International and Ferrlecit from WPI/SNY) need to be administeredslowly and in small dosages over several sessions to meet the body’s total iron requirement. Therecommended dosage of Venofer is 1,000mg, which cannot be administered at a single go. In non-dialysis dependent CKD patients, the total dosage (1,000mg) of Venofer is divided into5 sub-dosages (200mg each), with each sub-dosage requiring 5-10 minutes for administration. The 5 sub-dosages are given in 5 different occasions within a 14-day period. In dialysis dependent CKD patients, the total dosage (1,000mg) of Venofer is divided into 10sub-dosages (100mg each), with each sub-dosage administration requiring 5-10 minutes. Allthese sub-dosages are administered in 10 different occasions.
Ferrlecit, which is approved only for dialysis dependent CKD patients, has a recommended dosageof 1,000mg. Like Venofer, Ferrlecit cannot be given in single administration. The total dosage isbroken into 8 sub-dosages (125mg each), taking approximately 10 minutes (which sometimes canextend to an hour) for administration of each sub-dosage. These 8 sub-dosages are given over 8consecutive dialysis sessions.In contrast, Feraheme is rapidly administered in two 17-second injections, making it more patientfriendly.
******
mfg ipollit
Antwort auf Beitrag Nr.: 39.268.583 von ipollit am 02.04.10 12:59:23
Vielen Dank für die Infos!
Aus meiner Sicht ein interessanter (vielleicht in gewisser Weise auch etwas "schräger") Wert mit dem Potential für eine Verdopplung oder Verdreifachung binnen der nächsten 2 bis 5 Jahre (nur meine Meinung), falls alles optimal läuft - und das bei im Branchenvergleich relativ geringem Risiko.
Gruß
PS: Für irgendwelche Käufe müsste ich indes vorher erst etwas anderes verkaufen und nach Lage der Dinge könnte das nur ein Teil meiner MorphoSys-Position sein. Das bekomme ich aber irgendwie (noch?) nicht übers Herz. Die liegt jetzt nämlich seit fast genau 4 Jahren wie ein toter Fisch in meinem Depot und rührt sich nicht wirklich. Aber ich habe mich eben irgendwie an sie gewöhnt.
Vielen Dank für die Infos!
Aus meiner Sicht ein interessanter (vielleicht in gewisser Weise auch etwas "schräger") Wert mit dem Potential für eine Verdopplung oder Verdreifachung binnen der nächsten 2 bis 5 Jahre (nur meine Meinung), falls alles optimal läuft - und das bei im Branchenvergleich relativ geringem Risiko.
Gruß
PS: Für irgendwelche Käufe müsste ich indes vorher erst etwas anderes verkaufen und nach Lage der Dinge könnte das nur ein Teil meiner MorphoSys-Position sein. Das bekomme ich aber irgendwie (noch?) nicht übers Herz. Die liegt jetzt nämlich seit fast genau 4 Jahren wie ein toter Fisch in meinem Depot und rührt sich nicht wirklich. Aber ich habe mich eben irgendwie an sie gewöhnt.
Antwort auf Beitrag Nr.: 39.269.257 von SLGramann am 02.04.10 17:06:56"Potential für eine Verdopplung oder Verdreifachung binnen der nächsten 2 bis 5 Jahre"
im positiven Fall kann es etwas aufwärts gehen... allerdings besteht auch das Risiko, dass die Umsätze in diesem Quartal deutlich unter den Erwartungen liegen... damit würde vieles in Frage gestellt und mit dem Kurs könnte es deutlich abwärts gehen. Man wird erst in den nächsten Monaten ein wenig besser sehen können, wie Feraheme vom Markt angenommen wird. Zumindest scheint im Moment die Erwartung der Börse eher negativ zu sein... sonst wäre z.B. diese News über den Deal nicht einfach so verpufft bzw. abverkauft worden - ein eher schlechtes Zeichen.
mfg ipollit
im positiven Fall kann es etwas aufwärts gehen... allerdings besteht auch das Risiko, dass die Umsätze in diesem Quartal deutlich unter den Erwartungen liegen... damit würde vieles in Frage gestellt und mit dem Kurs könnte es deutlich abwärts gehen. Man wird erst in den nächsten Monaten ein wenig besser sehen können, wie Feraheme vom Markt angenommen wird. Zumindest scheint im Moment die Erwartung der Börse eher negativ zu sein... sonst wäre z.B. diese News über den Deal nicht einfach so verpufft bzw. abverkauft worden - ein eher schlechtes Zeichen.
mfg ipollit
Antwort auf Beitrag Nr.: 39.269.682 von ipollit am 02.04.10 19:42:28
Ganz klar, ein Erfolg im Markt ist noch nicht sicher, zumal bswp. Fresenius ja die Türen einfach teilweise zunagelt. Und ohne eine wirklich gute Marktdurchdringung wird das kein gutes Investment werden.
Ich denke auch, dass man erst in (vielen) Monaten klar sehen wird. Der Deal mit Takeda ist aber positiv und ein Zeichen des Vertrauens, auch wenn die Börse jetzt nichts draus machen wollte.
--------------
Noch was anderes, auch vor allem an Pathfinder, der vielleicht mitliest:
Der Erfolg von ARQL macht durchaus auch (noch mehr) Hoffnung für Exelixis.
Dazu diese Meinung:
Another piece of anecdotal data came from Arqule’s main competitor in the c-MET field, Exelixis (EXEL).
Exelixis has two c-MET inhibitors in the clinic, XL184 and XL880 that are partnered with BMS (BMY) and GSK (GSK), respectively. XL184 is also being evaluated in a combination with Tarceva (in a different, less robust trial design, though).
In a scientific meeting last November, one of the investigators of the XL184 trial presented a case study in which the drug was given in combination with Tarceva to a lung cancer patient who had relapsed on Tarceva. Interestingly, at diagnosis, the
patient’s tumor harbored a mutation rendering it sensitive to Tarceva. The patient had a profound response to Tarceva but after 8 months of treatment the tumor started growing back. The investigator found that the tumor started expressing c-MET and
postulated that the resistance was mediated by the c-MET pathway. Consequently, the patient was enrolled in the XL184 trial and achieved a partial response.
The association of Tarceva resistance with c-MET expression and the reversal of the resistance with a c-MET inhibitor is extremely encouraging, however, it is important to note that unlike Arqule’s c-MET inhibitor, XL184 hits additional targets besides c-MET. Therefore, the c-MET inhibition attribute of XL184 could be the reason for the clinical effect but other functions of the drug might be responsible for the tumor shrinkage.
Ganz klar, ein Erfolg im Markt ist noch nicht sicher, zumal bswp. Fresenius ja die Türen einfach teilweise zunagelt. Und ohne eine wirklich gute Marktdurchdringung wird das kein gutes Investment werden.
Ich denke auch, dass man erst in (vielen) Monaten klar sehen wird. Der Deal mit Takeda ist aber positiv und ein Zeichen des Vertrauens, auch wenn die Börse jetzt nichts draus machen wollte.
--------------
Noch was anderes, auch vor allem an Pathfinder, der vielleicht mitliest:
Der Erfolg von ARQL macht durchaus auch (noch mehr) Hoffnung für Exelixis.
Dazu diese Meinung:
Another piece of anecdotal data came from Arqule’s main competitor in the c-MET field, Exelixis (EXEL).
Exelixis has two c-MET inhibitors in the clinic, XL184 and XL880 that are partnered with BMS (BMY) and GSK (GSK), respectively. XL184 is also being evaluated in a combination with Tarceva (in a different, less robust trial design, though).
In a scientific meeting last November, one of the investigators of the XL184 trial presented a case study in which the drug was given in combination with Tarceva to a lung cancer patient who had relapsed on Tarceva. Interestingly, at diagnosis, the
patient’s tumor harbored a mutation rendering it sensitive to Tarceva. The patient had a profound response to Tarceva but after 8 months of treatment the tumor started growing back. The investigator found that the tumor started expressing c-MET and
postulated that the resistance was mediated by the c-MET pathway. Consequently, the patient was enrolled in the XL184 trial and achieved a partial response.
The association of Tarceva resistance with c-MET expression and the reversal of the resistance with a c-MET inhibitor is extremely encouraging, however, it is important to note that unlike Arqule’s c-MET inhibitor, XL184 hits additional targets besides c-MET. Therefore, the c-MET inhibition attribute of XL184 could be the reason for the clinical effect but other functions of the drug might be responsible for the tumor shrinkage.
Antwort auf Beitrag Nr.: 39.269.944 von SLGramann am 02.04.10 21:07:30Der Erfolg von ARQL macht durchaus auch (noch mehr) Hoffnung für Exelixis.
wobei allerdings gewisse Unterschiede zu beachten sind:
http://messages.finance.yahoo.com/Stocks_%28A_to_Z%29/Stocks…
As I understand it, there is not a real difference between MET and c-MET, both refer to a receptor tyrosine kinase for which the ligand is HGF(hepatocyte growth factor) and are just different terms for the same target (there may be some who would quibble about scientific convention and why one is technically more right, but in this setting, I believe they are the same). This HGF-MET combination has been found to be abnormally expressed or active in various tumors, hence the idea of inhibiting MET should be of value in treating these cancer. For NSCLC, MET amplification has been associated with the development of EGFR resistance, so a combination with erlotinib makes a lot of sense assuming they can both be given safely.
As far as the differences between ARQ197 and XL184, both ARQ197 and XL880 were actually presented several years ago at ASCO and if one assumes that XL880 and XL184 are similar (both target MET and VEGFR TKIs) the story was as follows:
XL880 targets both MET and VEGFR, whereas ARQ197 is MET specific,
XL880 at the time had more toxicity, while ARQ197 appeared to plateau in terms of exposure,
the data showing MET inhibition was more convincing for XL880, though ARQ197 has subsequently done some additional PD studies to shore up that story.
As far as the recent Arqule announcement, the ARQ197 data for NSCLC is only top line, is ahead of where XL184 is, and given that, might be seen as bad news for XL184, on the other hand, I would anticipate seeing XL184 + erlotinib data at ASCO this year (there has been some foreshadowing) which might allow a benchmark comparison. In theory, XL184 could be better by adding anti-VEGF activity, but bevacizumab didn;t add anything to erlotinib alone in a different trial, so if XL were unlucky, it could be that having additional anti-VEGF activity will only contribute toxicity. There's no way to really know that in advance, but from my perspective, seeing activity with a "pure" MET inhibitor is likely a good thing for XL184 as far as establishing that MET inhibition does add to EGFR inhibition with a positive clinical result. We'll need more data to see how this really plays out.
*****
http://seekingalpha.com/article/98214-licensing-deal-is-immi…
To date, only three c-MET kinase inhibitors managed to show some sort of activity in humans: XL184 (Exelixis), XL880 (GSK/Exelixis) and ARQ197 (ArQule). Although the data for these drugs is very preliminary, they clearly demonstrate the potential of c-MET inhibitors in various types of cancers, including thyroid, renal, ovarian, colorectal and pancreatic. Equally important, the three agents demonstrated their activity with no significant side effects. This proof of concept surely stimulates the rest of the market to launch or accelerate other c-MET projects.
ARQ-197 has two unique properties that may distinguish it from its competitors.
1) Selectivity
ARQ-197 is very selective for c-Met, in contrast to Exelixis’ compounds, which inhibit additional kinases as well. ARQ-197 and the Exelixis compounds represent two different approaches in the field of kinase inhibitors, as each approach has its merits. This issue as well as the promise of kinase inhibitor market are discussed in my previous article on Exelixis.
It is highly unlikely that one approach will be superior to the other in all cases and conditions, however, there are several reasons why a selective inhibitor might be more suitable some cases.
Based on clinical results from ongoing trials, c-MET inhibitors demonstrated signs of activity but they were not as potent as some had hoped. Coupled with the potential synergy with other drugs, this led many researchers to suggest that the way forward with c-MET inhibitors is in combination with other agents. Since anti-cancer drugs are often associated with strong side-effects, combining more than one drug increases the overall toxicity. Selective inhibitors are believed to be safer than multi-targeted inhibitors, so a benign safety profile can be a big advantage when several drugs are needed to be combined.
Another potential advantage for a compound that hits a single target is that there is a high likelihood that the clinical effect is a direct result of hitting that specific target. In the case of XL184 and XL880, there is no way of knowing which target contributes to the effect and to what extent. It is possible, for example, that the clinical effect seen with these agents has nothing to do with c-Met inhibition. Of course, when a drug leads to tumor shrinkage, it does not really matter to the patient what is the exact mechanism, but when a large pharma decides to license a compound and commit to a costly clinical program, such lack of clarity could be problematic, for example, in picking the right patients based on the targets expressed by their tumors.
2) Unique Mechanism of Action
An even more important differentiator ARQ-197 has over the rest of the compounds is its unique mechanism of action. Most kinase inhibitors in development are “ATP-competitive”, i.e. they disrupt signaling by binding to a specific region called “ATP-pocket” and prevent kinases from passing the signal. ARQ-197 disrupts c-MET signaling by binding a different region of the protein, therefore, as a non-ATP competitive kinase inhibitor, ARQ-197 is thought to be more specific and consequently safer than other agents. In addition, it may overcome common resistance mechanisms and may be used in combination with ATP-competitive c-MET inhibitors. In fact, this property can turn other c-MET kinase inhibitors from competing compounds into a combination opportunity for ARQ-197.
mfg ipollit
wobei allerdings gewisse Unterschiede zu beachten sind:
http://messages.finance.yahoo.com/Stocks_%28A_to_Z%29/Stocks…
As I understand it, there is not a real difference between MET and c-MET, both refer to a receptor tyrosine kinase for which the ligand is HGF(hepatocyte growth factor) and are just different terms for the same target (there may be some who would quibble about scientific convention and why one is technically more right, but in this setting, I believe they are the same). This HGF-MET combination has been found to be abnormally expressed or active in various tumors, hence the idea of inhibiting MET should be of value in treating these cancer. For NSCLC, MET amplification has been associated with the development of EGFR resistance, so a combination with erlotinib makes a lot of sense assuming they can both be given safely.
As far as the differences between ARQ197 and XL184, both ARQ197 and XL880 were actually presented several years ago at ASCO and if one assumes that XL880 and XL184 are similar (both target MET and VEGFR TKIs) the story was as follows:
XL880 targets both MET and VEGFR, whereas ARQ197 is MET specific,
XL880 at the time had more toxicity, while ARQ197 appeared to plateau in terms of exposure,
the data showing MET inhibition was more convincing for XL880, though ARQ197 has subsequently done some additional PD studies to shore up that story.
As far as the recent Arqule announcement, the ARQ197 data for NSCLC is only top line, is ahead of where XL184 is, and given that, might be seen as bad news for XL184, on the other hand, I would anticipate seeing XL184 + erlotinib data at ASCO this year (there has been some foreshadowing) which might allow a benchmark comparison. In theory, XL184 could be better by adding anti-VEGF activity, but bevacizumab didn;t add anything to erlotinib alone in a different trial, so if XL were unlucky, it could be that having additional anti-VEGF activity will only contribute toxicity. There's no way to really know that in advance, but from my perspective, seeing activity with a "pure" MET inhibitor is likely a good thing for XL184 as far as establishing that MET inhibition does add to EGFR inhibition with a positive clinical result. We'll need more data to see how this really plays out.
*****
http://seekingalpha.com/article/98214-licensing-deal-is-immi…
To date, only three c-MET kinase inhibitors managed to show some sort of activity in humans: XL184 (Exelixis), XL880 (GSK/Exelixis) and ARQ197 (ArQule). Although the data for these drugs is very preliminary, they clearly demonstrate the potential of c-MET inhibitors in various types of cancers, including thyroid, renal, ovarian, colorectal and pancreatic. Equally important, the three agents demonstrated their activity with no significant side effects. This proof of concept surely stimulates the rest of the market to launch or accelerate other c-MET projects.
ARQ-197 has two unique properties that may distinguish it from its competitors.
1) Selectivity
ARQ-197 is very selective for c-Met, in contrast to Exelixis’ compounds, which inhibit additional kinases as well. ARQ-197 and the Exelixis compounds represent two different approaches in the field of kinase inhibitors, as each approach has its merits. This issue as well as the promise of kinase inhibitor market are discussed in my previous article on Exelixis.
It is highly unlikely that one approach will be superior to the other in all cases and conditions, however, there are several reasons why a selective inhibitor might be more suitable some cases.
Based on clinical results from ongoing trials, c-MET inhibitors demonstrated signs of activity but they were not as potent as some had hoped. Coupled with the potential synergy with other drugs, this led many researchers to suggest that the way forward with c-MET inhibitors is in combination with other agents. Since anti-cancer drugs are often associated with strong side-effects, combining more than one drug increases the overall toxicity. Selective inhibitors are believed to be safer than multi-targeted inhibitors, so a benign safety profile can be a big advantage when several drugs are needed to be combined.
Another potential advantage for a compound that hits a single target is that there is a high likelihood that the clinical effect is a direct result of hitting that specific target. In the case of XL184 and XL880, there is no way of knowing which target contributes to the effect and to what extent. It is possible, for example, that the clinical effect seen with these agents has nothing to do with c-Met inhibition. Of course, when a drug leads to tumor shrinkage, it does not really matter to the patient what is the exact mechanism, but when a large pharma decides to license a compound and commit to a costly clinical program, such lack of clarity could be problematic, for example, in picking the right patients based on the targets expressed by their tumors.
2) Unique Mechanism of Action
An even more important differentiator ARQ-197 has over the rest of the compounds is its unique mechanism of action. Most kinase inhibitors in development are “ATP-competitive”, i.e. they disrupt signaling by binding to a specific region called “ATP-pocket” and prevent kinases from passing the signal. ARQ-197 disrupts c-MET signaling by binding a different region of the protein, therefore, as a non-ATP competitive kinase inhibitor, ARQ-197 is thought to be more specific and consequently safer than other agents. In addition, it may overcome common resistance mechanisms and may be used in combination with ATP-competitive c-MET inhibitors. In fact, this property can turn other c-MET kinase inhibitors from competing compounds into a combination opportunity for ARQ-197.
mfg ipollit
Antwort auf Beitrag Nr.: 39.254.233 von ipollit am 31.03.10 14:04:54Needham says $12
ArQule Inc. (ARQL) – Buy
ARQL: c-Met, c-Met Run – Robust Phase 2 ARQ197/Tarceva NSCLC Data Set the Stage for Phase 3
Development And Further ARQ197 Combination Studies; Reiterating BUY (3/31 @11:45AM $7.25)
This morning, ArQule reported top-line results from a RCT Phase 2 trial
evaluating ARQ197, its c-Met inhibitor, in combination with Tarceva vs.
Tarceva alone in 2nd line and 3rd line NSCLC patients who are EGFR inhibitor
naïve. The data showed that combination treatment of ARQ197 with Tarceva
improved PFS by 66% (16.1 wks for the combo vs. 9.7 wks for Tarceva
alone). The difference was not statistically significant by log-rank test
(HR=0.809), but statistical significance was reached after Cox
regression analysis that included pre-specified covariates (HR=0.675,
n=167 patients). A pre-specified subgroup analysis in patients with nonsquamous
NSCLC (n=117) showed a median PFS of 18.9 wks (combo)
versus 9.7 wks (Tarceva alone), and the Cox analysis demonstrated
statistical significance (HR=0.613). Regarding safety, the combination was
well tolerated with no clinically relevant differences in adverse events.
Further data are expected to be presented at the upcoming ASCO Meeting.
We believe the efficacy ARQ197/Tarceva results are robust in this RCT
Phase 2 trial, and the single agent data on Tarceva are consistent with
the previous literature. When adjusting for key prognostic factors as
well as key histology findings, the results showed almost a doubling in
PFS over the standard of care Tarceva in this setting. While we note the
modest sample size as well as the imbalance of patients, we believe the
most important takeaway from the data suggest a solid efficacy profile and a
safety profile without significant off-target toxicity and support Phase 3 testing
in a targeted population with favorable histology (non-squamous).
Furthermore, we believe the Phase 2 data presented here suggest the
potential for ARQ197 in combination with other SOC cancer therapeutics,
including gemcitabine (Phase 1 in pancreatic cancer) and Nexavar (Phase 1
in liver cancer).
At YE09, ArQule reported ~$117MM in cash, cash equivalents and longterm
marketable securities, net of the ~$46MM in notes payable. The
Company estimated a cash burn of ~$43-47MM for 2010, and expects to end
2010 with ~$70-74MM in cash and marketable securities. ArQule may have
sufficient cash well into 2012.
NEWSWORTHY EVENTS EXPECTED WITHIN THE NEXT 3-6 MONTHS
- Report further data on Phase 2 trial of Tarceva +/- ARQ 197 in
2nd /3rd line NSCLC (2010 ASCO Mtg)
- Report data on Phase 2 ARQ 197 in MiT tumors and plan Phase
3 program if Phase 2 endpoints met (1H10)
- Report data on Phase 2 PK study of ARQ 197 + gemcitabine in
solid tumors (mid-2010)
- Present data on Phase 1 dose escalation trial of ARQ 621 (2010)
mfg ipollit
ArQule Inc. (ARQL) – Buy
ARQL: c-Met, c-Met Run – Robust Phase 2 ARQ197/Tarceva NSCLC Data Set the Stage for Phase 3
Development And Further ARQ197 Combination Studies; Reiterating BUY (3/31 @11:45AM $7.25)
This morning, ArQule reported top-line results from a RCT Phase 2 trial
evaluating ARQ197, its c-Met inhibitor, in combination with Tarceva vs.
Tarceva alone in 2nd line and 3rd line NSCLC patients who are EGFR inhibitor
naïve. The data showed that combination treatment of ARQ197 with Tarceva
improved PFS by 66% (16.1 wks for the combo vs. 9.7 wks for Tarceva
alone). The difference was not statistically significant by log-rank test
(HR=0.809), but statistical significance was reached after Cox
regression analysis that included pre-specified covariates (HR=0.675,
n=167 patients). A pre-specified subgroup analysis in patients with nonsquamous
NSCLC (n=117) showed a median PFS of 18.9 wks (combo)
versus 9.7 wks (Tarceva alone), and the Cox analysis demonstrated
statistical significance (HR=0.613). Regarding safety, the combination was
well tolerated with no clinically relevant differences in adverse events.
Further data are expected to be presented at the upcoming ASCO Meeting.
We believe the efficacy ARQ197/Tarceva results are robust in this RCT
Phase 2 trial, and the single agent data on Tarceva are consistent with
the previous literature. When adjusting for key prognostic factors as
well as key histology findings, the results showed almost a doubling in
PFS over the standard of care Tarceva in this setting. While we note the
modest sample size as well as the imbalance of patients, we believe the
most important takeaway from the data suggest a solid efficacy profile and a
safety profile without significant off-target toxicity and support Phase 3 testing
in a targeted population with favorable histology (non-squamous).
Furthermore, we believe the Phase 2 data presented here suggest the
potential for ARQ197 in combination with other SOC cancer therapeutics,
including gemcitabine (Phase 1 in pancreatic cancer) and Nexavar (Phase 1
in liver cancer).
At YE09, ArQule reported ~$117MM in cash, cash equivalents and longterm
marketable securities, net of the ~$46MM in notes payable. The
Company estimated a cash burn of ~$43-47MM for 2010, and expects to end
2010 with ~$70-74MM in cash and marketable securities. ArQule may have
sufficient cash well into 2012.
NEWSWORTHY EVENTS EXPECTED WITHIN THE NEXT 3-6 MONTHS
- Report further data on Phase 2 trial of Tarceva +/- ARQ 197 in
2nd /3rd line NSCLC (2010 ASCO Mtg)
- Report data on Phase 2 ARQ 197 in MiT tumors and plan Phase
3 program if Phase 2 endpoints met (1H10)
- Report data on Phase 2 PK study of ARQ 197 + gemcitabine in
solid tumors (mid-2010)
- Present data on Phase 1 dose escalation trial of ARQ 621 (2010)
mfg ipollit
zu ISISs Mipomersen ein interessanter Artikel:
More data on mipomersen, agent that reduces Lp(a) as well as LDL
APRIL 5, 2010 | Lisa Nainggolan
Amsterdam, the Netherlands - A novel antisense agent that reduces LDL production by inhibiting
apolipoprotein B (apoB), mipomersen (Isis Pharmaceuticals/Genzyme), has shown some efficacy in a
phase 2 dose-escalation study in patients with hypercholesterolemia on stable statin therapy [1]. The
results of the study, by Dr Fatima Adkim (University of Amsterdam, the Netherlands) and colleagues,
are published in the April 13, 2010 issue of the Journal of the American College of Cardiology.
Interest in mipomersen is high because it lowers LDL in a different way from statins and it also
reduces lipoprotein(a) (Lp[a]), something statins are unable to do. Lp(a) has recently emerged as an
important independent risk factor for cardiovascular disease.
This study comes on the heels of a phase 3 trial with the same agent published in the Lancet last
month, as reported by heartwire, in which weekly subcutaneous injections of mipomersen lowered
LDL by a mean of 25% in patients with homozygous familial hypercholesterolemia (FH) who were
already receiving a variety of lipid-lowering drugs, including high-dose statins.
Large phase 3 safety trial would address ADR concerns
But there are concerns about possible hepatic problems with mipomersen. In the homozygous-FH trial,
four participants developed increases in the liver enzyme alanine aminotransferase, one of whom
showed a large increase in hepatic fat. In this new study, a substantial number of patients had
elevations in liver enzymes, particularly those taking the higher doses of mipomersen, so "additional
studies are needed to elucidate the mechanism as well as the clinical relevance of these transaminase
increases," say the researchers.
Senior author of the study, Dr John JP Kastelein (University of
Amsterdam), told heartwire: "When you want to know about
safety, phase 2 trials are never enough." He said he would like to
see Isis and Genzyme conduct a phase 3 safety trial with
mipomersen, similar to the 12- to 18-month one Merck is
conducting in 1500 patients with its new cholesteryl ester transfer protein inhibitor anacetrapib.
"This kind of phase 3 approach is meant to simply look and behold what happens in terms of safety
before embarking upon any real end-point study in which you are going to expose thousands of people
and [spend] $300 million. We don't want to put anyone at risk unnecessarily or spend that amount of
money for nothing, so I think it would be a very wise step to do that next."
Kastelein, for his part, does not think the side effects of mipomersen will turn out to be problematic.
"There will initially be some liver fat deposition in some people because you limit the export of a
lipoprotein called VLDL, but if you do this in animals, after a while the liver adapts and starts burning
up these fats. We have no idea whether this will happen in humans, but it's quite likely it will. So I
don't think that small increases in liver fat will have any clinical relevance."
Response compares favorably with other lipid-lowering agents
In the new study, 74 hypercholesterolemic subjects already taking statins were enrolled into one of six
dose cohorts at a 4:1 (active lacebo) ratio. Subjects in the first five cohorts received seven doses of
lacebo) ratio. Subjects in the first five cohorts received seven doses of
30- to 400-mg mipomersen subcutaneously over five weeks; those in the sixth cohort—the extendedtreatment
cohort—received 15 doses of 200 mg over 13 weeks. Prespecified end points included
percentage change from baseline in apoB and LDL cholesterol.
ApoB and LDL were reduced by 19% to 54% and 21% to 52%, respectively, at doses of 100-mg/week
mipomersen and higher in the five-week treatment cohorts. Efficacy seemed to increase in the
extended-treatment cohort treated with 200-mg/week mipomersen for 13 weeks.
"This response compares favorably with that reported for other lipid-lowering agents (eg, ezetimibe
and colesevelam) when added to stable doses of statins in subjects with primary
hypercholesterolemia," the researchers note.
But it was those who seemed to gain the most benefit—the subjects treated at the higher doses in the
extended-treatment cohort—who experienced the most adverse effects, with five of 10 subjects (50%)
having elevations of hepatic transaminases of more than three times the upper limit of normal.
Another adverse event was injection-site reaction.
Huge variation in response to mipomersen, but it's a "life-changer"
Kastelein told heartwire that there appears to be "huge variations" in individual response to
mipomersen in terms of LDL lowering, which so far cannot be explained in biological terms.
"Some people experience a <10% reduction in LDL, but others respond with LDL reductions of 60% to
70%," he explained. But as mipomersen is a subcutaneously administered drug, this range of efficacy
is not a huge problem, he said, because "after a certain amount of time . . . if it has reduced LDL by
less than 15%, you would probably discontinue it."
Kastelein stressed that for some patients, "mipomersen has the potential to be a life-changer." For
example, in a patient with heterozygous FH who has "had a big anterior MI at the age of 32, who is
taking 40 mg of Crestor (rosuvastatin) and 10 mg of ezetimibe, and whose LDL is still 3.6 mmol/L,
which is way above the target, there's nothing else that can help these patients other than this
therapy."
More data on mipomersen, agent that reduces Lp(a) as well as LDL
APRIL 5, 2010 | Lisa Nainggolan
Amsterdam, the Netherlands - A novel antisense agent that reduces LDL production by inhibiting
apolipoprotein B (apoB), mipomersen (Isis Pharmaceuticals/Genzyme), has shown some efficacy in a
phase 2 dose-escalation study in patients with hypercholesterolemia on stable statin therapy [1]. The
results of the study, by Dr Fatima Adkim (University of Amsterdam, the Netherlands) and colleagues,
are published in the April 13, 2010 issue of the Journal of the American College of Cardiology.
Interest in mipomersen is high because it lowers LDL in a different way from statins and it also
reduces lipoprotein(a) (Lp[a]), something statins are unable to do. Lp(a) has recently emerged as an
important independent risk factor for cardiovascular disease.
This study comes on the heels of a phase 3 trial with the same agent published in the Lancet last
month, as reported by heartwire, in which weekly subcutaneous injections of mipomersen lowered
LDL by a mean of 25% in patients with homozygous familial hypercholesterolemia (FH) who were
already receiving a variety of lipid-lowering drugs, including high-dose statins.
Large phase 3 safety trial would address ADR concerns
But there are concerns about possible hepatic problems with mipomersen. In the homozygous-FH trial,
four participants developed increases in the liver enzyme alanine aminotransferase, one of whom
showed a large increase in hepatic fat. In this new study, a substantial number of patients had
elevations in liver enzymes, particularly those taking the higher doses of mipomersen, so "additional
studies are needed to elucidate the mechanism as well as the clinical relevance of these transaminase
increases," say the researchers.
Senior author of the study, Dr John JP Kastelein (University of
Amsterdam), told heartwire: "When you want to know about
safety, phase 2 trials are never enough." He said he would like to
see Isis and Genzyme conduct a phase 3 safety trial with
mipomersen, similar to the 12- to 18-month one Merck is
conducting in 1500 patients with its new cholesteryl ester transfer protein inhibitor anacetrapib.
"This kind of phase 3 approach is meant to simply look and behold what happens in terms of safety
before embarking upon any real end-point study in which you are going to expose thousands of people
and [spend] $300 million. We don't want to put anyone at risk unnecessarily or spend that amount of
money for nothing, so I think it would be a very wise step to do that next."
Kastelein, for his part, does not think the side effects of mipomersen will turn out to be problematic.
"There will initially be some liver fat deposition in some people because you limit the export of a
lipoprotein called VLDL, but if you do this in animals, after a while the liver adapts and starts burning
up these fats. We have no idea whether this will happen in humans, but it's quite likely it will. So I
don't think that small increases in liver fat will have any clinical relevance."
Response compares favorably with other lipid-lowering agents
In the new study, 74 hypercholesterolemic subjects already taking statins were enrolled into one of six
dose cohorts at a 4:1 (active
 lacebo) ratio. Subjects in the first five cohorts received seven doses of
lacebo) ratio. Subjects in the first five cohorts received seven doses of30- to 400-mg mipomersen subcutaneously over five weeks; those in the sixth cohort—the extendedtreatment
cohort—received 15 doses of 200 mg over 13 weeks. Prespecified end points included
percentage change from baseline in apoB and LDL cholesterol.
ApoB and LDL were reduced by 19% to 54% and 21% to 52%, respectively, at doses of 100-mg/week
mipomersen and higher in the five-week treatment cohorts. Efficacy seemed to increase in the
extended-treatment cohort treated with 200-mg/week mipomersen for 13 weeks.
"This response compares favorably with that reported for other lipid-lowering agents (eg, ezetimibe
and colesevelam) when added to stable doses of statins in subjects with primary
hypercholesterolemia," the researchers note.
But it was those who seemed to gain the most benefit—the subjects treated at the higher doses in the
extended-treatment cohort—who experienced the most adverse effects, with five of 10 subjects (50%)
having elevations of hepatic transaminases of more than three times the upper limit of normal.
Another adverse event was injection-site reaction.
Huge variation in response to mipomersen, but it's a "life-changer"
Kastelein told heartwire that there appears to be "huge variations" in individual response to
mipomersen in terms of LDL lowering, which so far cannot be explained in biological terms.
"Some people experience a <10% reduction in LDL, but others respond with LDL reductions of 60% to
70%," he explained. But as mipomersen is a subcutaneously administered drug, this range of efficacy
is not a huge problem, he said, because "after a certain amount of time . . . if it has reduced LDL by
less than 15%, you would probably discontinue it."
Kastelein stressed that for some patients, "mipomersen has the potential to be a life-changer." For
example, in a patient with heterozygous FH who has "had a big anterior MI at the age of 32, who is
taking 40 mg of Crestor (rosuvastatin) and 10 mg of ezetimibe, and whose LDL is still 3.6 mmol/L,
which is way above the target, there's nothing else that can help these patients other than this
therapy."
Dem Mann kann man zuhören:
TWST: You mentioned the small- and mid-cap M And A rally. Would you talk about that for a minute?
Dr. Kantor: What I mean by that is in 2009 we saw Medarex and Genentech (DNA) taken out; we've already seen OSI Pharmaceuticals (OSIP) in play in the past weeks, and we've seen Facet (FACT), which was originally a target for Biogen (BIIB), ultimately getting taken out by Abbott (ABT) at a significant premium to where the Biogen deal was proposed. What we are seeing is a number of small- to mid-cap companies and, in some cases, large companies that are the targets of acquisition. Whenever that's the case and whenever you see companies paying 40%, 50%, 60% or more premium, you begin to see the other companies with similar profiles reflecting increased valuation.
TWST: Tell us about Pharmacyclics. What is your view on that company?
Dr. Kantor: Pharmacyclics (PCYC) is kind of a comeback story. It had been left for dead for a long time and it's really a remake. What I mean by that is its lead program, several years back, failed at the FDA. The company had acquired some preclinical early-stage projects from Celera (CRA) and has been diligently moving those forward. And it was not until late in 2009, at the American Society of Hematology meeting, where we saw the first clinical data for two of their drugs. The data in two different hard-to-treat lymphoma studies for two different drugs was actually quite impressive, and essentially put the company and the stock back on people's radar screen. And you can see the stock performance has been quite remarkable over the period from fall 2009 to today.
TWST: You mentioned OSI Pharma. Why is that company getting so much attention right now?
Dr. Kantor: There was an unsolicited hostile bid by Astellas (4503) to acquire the company for $52 a share, which was a significant premium to where the stock closed in the high $30s prior to that bid. That's the reason that OSI is in the news today. And the question is whether or not there will be another company bidding higher, or whether or not Astellas will need to raise their bid. Obviously with the stock trading in the high $50s, the overwhelming sentiment of investors is that OSI will get taken out at a higher price than $52 proposed in the unsolicited bid.
TWST: What about Celgene? What's happening with them?
Dr. Kantor: We highlighted Celgene (CELG) as our best idea, big-cap name for 2010. It is one of the top-growth stocks in biotech. It has a high p/e multiple, so it's certainly not without risk. But what's driving it here is the expansion globally of its multiple myeloma franchise and specifically REVLIMID. And what I like about it and what other folks like about it is the longevity of that growth curve. What I mean by that is there are significant new markets both geographically as well as in new disease indications for REVLIMID to enter over the next several years, such that we have good visibility on long-term, double-digit top-line growth. We also have very high margins in Celgene and in fact improving margins with product mix and geographic mix.
TWST: What about ImmunoGen?
Dr. Kantor: ImmunoGen (IMGN) is one of the few pure-play antibody technology companies, and this has been one of the very fruitful areas of M And A and deal-making that we've seen. For example, Genentech and Medarex, both antibody companies, Facet is another antibody company, and there are actually only a handful of remaining pure-play antibody companies. There is also a technology platform that's becoming very highly validated, called antibody-drug conjugation. This technology space is essentially controlled by an even smaller group of companies, including ImmunoGen and another one that I cover, Seattle Genetics (SGEN). There is scarcity value, that's one of the reasons we like ImmunoGen. The other reason of course is their lead program with Genentech/Roche (RHHBY) called T-DM1, which is a new drug for the treatment of breast cancer that has had some very exciting data and for which we expect Roche will file an NDA in the first half of 2010.
TWST: Who are your top picks right now and why?
Dr. Kantor: Our 2010 pick on the large-cap side is Celgene and on the small- to mid-cap side is Seattle Genetics, another pure-play antibody company with a deep pipeline of proprietary and partnered antibodies. And then I would add to that list, if you have a slightly longer time horizon, Cubist (CBST), which has proprietary antibiotics, which they sell directly with over $500 million in annual sales and growing. And we view this as the value play based on its low price-to-sales ratio.
------------
Dr. Jason Kantor, Ph.D., joined RBC Capital Markets in 2005 as a Managing Director and Senior Biotechnology Equity Research Analyst. In 2004 Fortune magazine ranked Dr. Kantor as an "All-Star Analyst," and The Wall Street Journal ranked him second in its "Best on the Street" survey. Dr. Kantor received his B.A. cum laude in biology from the University of California, San Diego, and his Ph.D. in cell and developmental biology from Harvard University.
----------------
Ich denke, dass, wenn Roche wirklich eine Zulassung für T-DM1 als third-line Therapy beantragt, Immunogen einen kleinen Satz nach oben machen wird. Kommerziell ist das nicht sehr bedeutend, aber das Vertrauen in T-DM1 würde weiter steigen und Immunogens ADC-Technologie wäre noch besser validiert.
Mittel- und langfristig kann T-DM1 vielleicht das nackte Herceptin auch in der first-line teilweise ersetzen. Und das wäre dann sehr bedeutend.
Außerdem ist IMGN eine gute Möglichkeit, im ADC-Bereich ein zweites Eisen im Feuer zu haben (neben SGEN). Kurz: Ich mag IMGN.
TWST: You mentioned the small- and mid-cap M And A rally. Would you talk about that for a minute?
Dr. Kantor: What I mean by that is in 2009 we saw Medarex and Genentech (DNA) taken out; we've already seen OSI Pharmaceuticals (OSIP) in play in the past weeks, and we've seen Facet (FACT), which was originally a target for Biogen (BIIB), ultimately getting taken out by Abbott (ABT) at a significant premium to where the Biogen deal was proposed. What we are seeing is a number of small- to mid-cap companies and, in some cases, large companies that are the targets of acquisition. Whenever that's the case and whenever you see companies paying 40%, 50%, 60% or more premium, you begin to see the other companies with similar profiles reflecting increased valuation.
TWST: Tell us about Pharmacyclics. What is your view on that company?
Dr. Kantor: Pharmacyclics (PCYC) is kind of a comeback story. It had been left for dead for a long time and it's really a remake. What I mean by that is its lead program, several years back, failed at the FDA. The company had acquired some preclinical early-stage projects from Celera (CRA) and has been diligently moving those forward. And it was not until late in 2009, at the American Society of Hematology meeting, where we saw the first clinical data for two of their drugs. The data in two different hard-to-treat lymphoma studies for two different drugs was actually quite impressive, and essentially put the company and the stock back on people's radar screen. And you can see the stock performance has been quite remarkable over the period from fall 2009 to today.
TWST: You mentioned OSI Pharma. Why is that company getting so much attention right now?
Dr. Kantor: There was an unsolicited hostile bid by Astellas (4503) to acquire the company for $52 a share, which was a significant premium to where the stock closed in the high $30s prior to that bid. That's the reason that OSI is in the news today. And the question is whether or not there will be another company bidding higher, or whether or not Astellas will need to raise their bid. Obviously with the stock trading in the high $50s, the overwhelming sentiment of investors is that OSI will get taken out at a higher price than $52 proposed in the unsolicited bid.
TWST: What about Celgene? What's happening with them?
Dr. Kantor: We highlighted Celgene (CELG) as our best idea, big-cap name for 2010. It is one of the top-growth stocks in biotech. It has a high p/e multiple, so it's certainly not without risk. But what's driving it here is the expansion globally of its multiple myeloma franchise and specifically REVLIMID. And what I like about it and what other folks like about it is the longevity of that growth curve. What I mean by that is there are significant new markets both geographically as well as in new disease indications for REVLIMID to enter over the next several years, such that we have good visibility on long-term, double-digit top-line growth. We also have very high margins in Celgene and in fact improving margins with product mix and geographic mix.
TWST: What about ImmunoGen?
Dr. Kantor: ImmunoGen (IMGN) is one of the few pure-play antibody technology companies, and this has been one of the very fruitful areas of M And A and deal-making that we've seen. For example, Genentech and Medarex, both antibody companies, Facet is another antibody company, and there are actually only a handful of remaining pure-play antibody companies. There is also a technology platform that's becoming very highly validated, called antibody-drug conjugation. This technology space is essentially controlled by an even smaller group of companies, including ImmunoGen and another one that I cover, Seattle Genetics (SGEN). There is scarcity value, that's one of the reasons we like ImmunoGen. The other reason of course is their lead program with Genentech/Roche (RHHBY) called T-DM1, which is a new drug for the treatment of breast cancer that has had some very exciting data and for which we expect Roche will file an NDA in the first half of 2010.
TWST: Who are your top picks right now and why?
Dr. Kantor: Our 2010 pick on the large-cap side is Celgene and on the small- to mid-cap side is Seattle Genetics, another pure-play antibody company with a deep pipeline of proprietary and partnered antibodies. And then I would add to that list, if you have a slightly longer time horizon, Cubist (CBST), which has proprietary antibiotics, which they sell directly with over $500 million in annual sales and growing. And we view this as the value play based on its low price-to-sales ratio.
------------
Dr. Jason Kantor, Ph.D., joined RBC Capital Markets in 2005 as a Managing Director and Senior Biotechnology Equity Research Analyst. In 2004 Fortune magazine ranked Dr. Kantor as an "All-Star Analyst," and The Wall Street Journal ranked him second in its "Best on the Street" survey. Dr. Kantor received his B.A. cum laude in biology from the University of California, San Diego, and his Ph.D. in cell and developmental biology from Harvard University.
----------------
Ich denke, dass, wenn Roche wirklich eine Zulassung für T-DM1 als third-line Therapy beantragt, Immunogen einen kleinen Satz nach oben machen wird. Kommerziell ist das nicht sehr bedeutend, aber das Vertrauen in T-DM1 würde weiter steigen und Immunogens ADC-Technologie wäre noch besser validiert.
Mittel- und langfristig kann T-DM1 vielleicht das nackte Herceptin auch in der first-line teilweise ersetzen. Und das wäre dann sehr bedeutend.
Außerdem ist IMGN eine gute Möglichkeit, im ADC-Bereich ein zweites Eisen im Feuer zu haben (neben SGEN). Kurz: Ich mag IMGN.
Hat jemand Lust, so etwas wie eine Newsflow-Erwartungsliste für die Depotwerte von ipollit zu erstellen?
Ich denke dabei an eine Übersicht über offiziell angekündigte Ergebnisse von Studien im zweiten Quartal 2010 oder vielleicht auch darüber hinaus.
ArQule hat ja sehr früh gemeldet.
Was mir fürs zweite Quartal spontan einfällt ist folgendes:
1.) REGN - Phase III-Ergebnisse für Arcalyst in der Indikation Gicht
2.) SGEN - Phase II b-Ergebnisse für Lintuzumab (Phase IIb randomized, double-blind, placebo-controlled trial of low-dose cytarabine with or without lintuzumab in patients with acute myeloid leukemia (AML)
Vielleicht können wir hier ja gemeinsam einiges zusammentragen?
Ich denke dabei an eine Übersicht über offiziell angekündigte Ergebnisse von Studien im zweiten Quartal 2010 oder vielleicht auch darüber hinaus.
ArQule hat ja sehr früh gemeldet.
Was mir fürs zweite Quartal spontan einfällt ist folgendes:
1.) REGN - Phase III-Ergebnisse für Arcalyst in der Indikation Gicht
2.) SGEN - Phase II b-Ergebnisse für Lintuzumab (Phase IIb randomized, double-blind, placebo-controlled trial of low-dose cytarabine with or without lintuzumab in patients with acute myeloid leukemia (AML)
Vielleicht können wir hier ja gemeinsam einiges zusammentragen?
Antwort auf Beitrag Nr.: 39.289.061 von SLGramann am 07.04.10 17:59:26
Ich habe mir gerade eine Präsentation von Regeneron vom März angeschaut und ergänze mal die Liste:
angekündigte Studienergebnisse in Q2/2010:
1.) REGN - Phase III-Ergebnisse für Arcalyst in der Indikation Gicht
2.) REGN - Phase II-Ergebnisse für REGN475 (REGN475 (NGF antibody) An antibody against nerve growth factor (NGF), a novel target for the treatment of pain.)
3.) REGN - Phase I-Ergebnisse für REGN88 (REGN88 (IL-6R antibody)
An antibody to the interleukin-6 receptor in development for the treatment of rheumatoid artritihs.)
4.) REGN - Phase I b-Ergebnisse (PoC) für REGN727 (REGN727 (PCSK9 antibody) An antibody to PCSK9, a novel target for LDL cholesterol reduction in patients with hyperlipidemia.)
5.) SGEN - Phase II b-Ergebnisse für Lintuzumab (Phase IIb randomized, double-blind, placebo-controlled trial of low-dose cytarabine with or without lintuzumab in patients with acute myeloid leukemia (AML)
Ich habe mir gerade eine Präsentation von Regeneron vom März angeschaut und ergänze mal die Liste:
angekündigte Studienergebnisse in Q2/2010:
1.) REGN - Phase III-Ergebnisse für Arcalyst in der Indikation Gicht
2.) REGN - Phase II-Ergebnisse für REGN475 (REGN475 (NGF antibody) An antibody against nerve growth factor (NGF), a novel target for the treatment of pain.)
3.) REGN - Phase I-Ergebnisse für REGN88 (REGN88 (IL-6R antibody)
An antibody to the interleukin-6 receptor in development for the treatment of rheumatoid artritihs.)
4.) REGN - Phase I b-Ergebnisse (PoC) für REGN727 (REGN727 (PCSK9 antibody) An antibody to PCSK9, a novel target for LDL cholesterol reduction in patients with hyperlipidemia.)
5.) SGEN - Phase II b-Ergebnisse für Lintuzumab (Phase IIb randomized, double-blind, placebo-controlled trial of low-dose cytarabine with or without lintuzumab in patients with acute myeloid leukemia (AML)
Antwort auf Beitrag Nr.: 39.289.331 von SLGramann am 07.04.10 18:33:53
So, letzter für heute (und keine Lust auf Formatierung)
angekündigte Studienergebnisse in Q2/2010:
1.) REGN - Phase III-Ergebnisse für Arcalyst in der Indikation Gicht
2.) REGN - Phase II-Ergebnisse für REGN475 (REGN475 (NGF antibody) An antibody against nerve growth factor (NGF), a novel target for the treatment of pain.)
3.) REGN - Phase I-Ergebnisse für REGN88 (REGN88 (IL-6R antibody)
An antibody to the interleukin-6 receptor in development for the treatment of rheumatoid artritihs.)
4.) REGN - Phase I b-Ergebnisse (PoC) für REGN727 (REGN727 (PCSK9 antibody) An antibody to PCSK9, a novel target for LDL cholesterol reduction in patients with hyperlipidemia.)
5.) SGEN - Phase II b-Ergebnisse für Lintuzumab (Phase IIb randomized, double-blind, placebo-controlled trial of low-dose cytarabine with or without lintuzumab in patients with acute myeloid leukemia (AML)
6.) INCY - Phase II-Ergebnisse für INCB18424 in der Indikation Psoriasis
7.) INCY - Phase II-Zwischenergebnisse für INCB28050 in der Indikation rheumatoid arthritis (Endergebnisse im 2. HJ 2010)
So, letzter für heute (und keine Lust auf Formatierung)
angekündigte Studienergebnisse in Q2/2010:
1.) REGN - Phase III-Ergebnisse für Arcalyst in der Indikation Gicht
2.) REGN - Phase II-Ergebnisse für REGN475 (REGN475 (NGF antibody) An antibody against nerve growth factor (NGF), a novel target for the treatment of pain.)
3.) REGN - Phase I-Ergebnisse für REGN88 (REGN88 (IL-6R antibody)
An antibody to the interleukin-6 receptor in development for the treatment of rheumatoid artritihs.)
4.) REGN - Phase I b-Ergebnisse (PoC) für REGN727 (REGN727 (PCSK9 antibody) An antibody to PCSK9, a novel target for LDL cholesterol reduction in patients with hyperlipidemia.)
5.) SGEN - Phase II b-Ergebnisse für Lintuzumab (Phase IIb randomized, double-blind, placebo-controlled trial of low-dose cytarabine with or without lintuzumab in patients with acute myeloid leukemia (AML)
6.) INCY - Phase II-Ergebnisse für INCB18424 in der Indikation Psoriasis
7.) INCY - Phase II-Zwischenergebnisse für INCB28050 in der Indikation rheumatoid arthritis (Endergebnisse im 2. HJ 2010)
ARQL... neues Update von Ohad Hammer
http://www.hammerstockblog.com/arqule-joins-the-phase-iii-cl…
Arqule Joins The Phase III Club
No matter how diversified or well managed a biotech company is, eventually there is no escape from binary events. These events usually come in the form of data readout from clinical trials that can make or break the stock. Two weeks ago, Arqule (ARQL) announced topline results from a much anticipated phase II trial of its lead agent, ARQ 197, in lung cancer. The trial is evaluating ARQ 197 in combination with the approved drug, Tarceva, in patients with non-small cell lung cancer (NSCLC) who have been previously treated with chemotherapy. Although the data set is not a clear slam dunk, it generated an impressive efficacy signal that warrants advancing ARQ 197 into phase III. This, coupled with the progress the company has made on other fronts in the past year makes Arqule one of most attractive biotech stocks in the market.
Designed for success
Arqule’s study is an excellent example of a properly designed clinical program, especially when it comes to targeted therapies. From the get go, the trial had the right ingredients, including a robust trial design, the right indication and a combination regimen supported by solid scientific rationale.
Doing a combination trial with Tarceva in NSCLC looks like the ideal place to start with ARQ 197 for several reasons. ARQ 197 inhibits c-MET, a protein associated with multiple cancer-related functions such as invasion and metastasis. The expression and involvement of c-MET in lung cancer is well established, based on numerous reports evaluating tumors from patients. The past years saw a flood of data with respect to c-MET and lung cancer, including a study which found that more than 70% of lung cancer tissues had some level of c-MET expression. Some studies also point to c-MET as a potential marker of aggressive disease and poor prognosis.
ARQ 197 and Tarceva have distinct yet intertwined mechanisms of action, so combining the two drugs is expected to result in at least additive, if not synergistic effect. c-MET activation is believed to account for 20-25% of cases of Tarceva resistance, so by adding ARQ 197, investigators hope to delay Tarceva resistance at least in some of the patients. Interestingly, recent experimental data shows that cells can become resistant to c-MET inhibitors by activating the EGFR pathway, which is inhibited by Tarceva.
From a commercial standpoint, it is always easier to add a new drug to an existing treatment line rather than replacing it. Tarceva is a commonly used drug in NSCLC with annual sales of $1.2B, so by adding ARQ 197, Arqule can build on Tarceva’s market penetration and create a new standard of care rather quickly. In addition, by the time ARQ 197 reaches the market, Tarceva will probably be generic, which will make the combination cheaper for patients and reimbursers. Lastly, both drugs are oral, so adding ARQ 197 does not lead to compliance or logistic issues.
The design of the trial is also noteworthy, as it is a fairly large (~170 patients) randomized study in which patients receive a combination of Tarceva with either ARQ 197 or placebo. Large randomized phase II trials are becoming ever more popular in oncology due to their ability to identify ineffective drugs and avoid unnecessary phase III studies. Their main disadvantage is cost, however that is a price the industry has learned to accept, especially in the case of combining targeted agents that often have a subtle effect in terms of tumor shrinkage.
Naturally, evaluating a drug in a randomized phase II increases the risk of failure, as results are compared head to head with a control arm. Non randomized trials are more likely to generate a “positive” signal in comparison with historical data, but results are less reliable. Therefore, the robust design of the ARQ 197 trial renders it more challenging but also more reliable for deciding whether to advance this combination into a registration study. In other words, if the trial is positive, it serves as a strong basis for a phase III study with a higher probability of success.
Results
Results from the phase II trial included 167 patients who received ARQ 197+Tarceva or Tarceva+placebo. On the safety front, the combination regimen was well tolerated with no meaningful safety issues. The primary endpoint was progression free survival (PFS), which is the time until disease progression (usually tumor growth or appearance of new lesions). Although patients in the ARQ 197+Tarceva arm had a PFS of 16.1 weeks, compared with 9.7 weeks for the patients on the Tarceva+placebo arm, the difference was not statistically significant. Nevertheless, two additional analyses were extremely encouraging.
It turned out that there were imbalances between treatment arms in terms of mutations in two genes (EGFR and KRAS) that are known to be linked to response to Tarceva. The imbalances were in favor of the control arm, which means that the control arm had more patients with a high likelihood of responding to Tarceva. When this difference was factored in, the difference between the combination and control arms became statistically significant. Another analysis was done in a subset of patients classified as non-squamous (histological classification). In this subset of patients, which accounted for ~70% of patients in the trial, the difference between the two arms was more pronounced (18.9 vs. 9.7 weeks) and reached statistical significance.
Positive signals
Although the trial did not meet its pre-defined primary endpoint, the efficacy signal is definitely there. Retrospective and subset analyses should always be treated with skepticism, but in this case, both analyses seem reasonable and reliable. Obviously, these signals will have to be validated prospectively in a large phase III trial.
Distinguishing between squamous and non-squamous NSCLC is a common analysis that had originally been part of the trial design. It is conducted on a regular basis in clinical trials, as the difference between the two subpopulations in terms of response to certain drugs is well recognized. The best example for that is Alimta, which is extremely effective in non-squamous NSCLC but ineffective and potentially deleterious in squamous NSCLC. Importantly, as non-squamous NSCLC accounts for 65-70% of NSCLC cases, it still represents a huge opportunity. Alimta, which is approved only for non-squamous NSCLC generated sales of $1.7B in 2009.
The imbalances between the two arms in EGFR and KRAS mutations are not surprising, as these factors were not included in the randomization process. EGFR and KRAS mutations are considered validated predictors of Tarceva response and are not always available at the time of treatment initiation. Patients with EGFR mutations tend to derive meaningful benefit from Tarceva whereas KRAS mutant tumors are considered relatively resistant to the drug. Therefore, factoring in these imbalances seems reasonable in trials that involve Tarceva.
Comparison with other trials
Comparing Arqule’s study to previous studies (see table below) in a similar patient population may provide additional insights. It is important to understand that no definitive conclusions can be drawn from comparing results from different trials, and usually there is an inverse correlation between a drug’s performance and trial size. Nevertheless, one can get a sense of where ARQ 197 stands in terms of PFS from such analysis. Even in comparison with established chemotherapy agents such as Taxotere and Alimta, the combination of ARQ 197 and Tarceva looks promising. The mild safety profile of the regimen is definitely a major advantage over chemo regimens, but cost might be an issue.

In its registration trial, Tarceva’s improvement in PFS was minimal yet statistically significant (2.2 vs. 1.8 months), but the real benefit was in overall survival (a 2 month difference). In the phase II trial, ARQ 197’s increase in PFS was more profound, especially in the non-squamous subgroup, despite the imbalances between the two arms. Alimta’s benefit in non-squamous patients also seems limited to overall survival, with no real difference in PFS. On the one hand, this makes Arqule’s data even more promising. On the other, it might imply that PFS is not necessarily indicative of survival, but phase II trials cannot evaluate overall survival in a reliable manner, especially in lung cancer where there are so many treatment lines.
Although the overall survival data is still unavailable, it is highly unlikely to see a clear statistically significant signal there as the trial was not powered to demonstrate that. In addition, the trial employed a cross-over design, where patients on the control arm can receive the combination of ARQ 197 and Tarceva after progressing on Tarceva alone, which may offset any potential effect of ARQ 197. Therefore, there is actually a third arm of patients who receive ARQ 197 and Tarceva following progressing on Tarceva. What investors can hope for is a trend of improved survival in patients on the ARQ 197 arm and those on the control arm who did not cross over after progression. According to the company, the majority of patients in the control arm did not crossover to the combination arm, so numbers might be sufficient to show a survival trend.
The control arm in the ARQ 197 trial had a median PFS of 9.7 weeks (2.2 months), which is similar to what Tarceva achieved in its registration trial. Although one could expect the control arm to do slightly better than historic controls, this further validates the trial design and implies the control arm in Arqule’s study is more or less reflective of a typical phase III population.
Next steps
Armed with such a promising data set, Arqule’s next obvious step is a global phase III trial in lung cancer. Although the formal decision has not been made, Arqule and its partner, Daiichi Sankyo, are likely to launch a phase III trial towards year end. Given the doubling of PFS in non-squamous patients, the trial will probably be limited to this subpopulation. Although the phase II was conducted primarily in the US and EU, the phase III program will probably include Japan, the second largest oncology market and an important Tarceva market. The commercialization rights for ARQ 197 in Japan belong to Kyowa-Hakko, which licensed the drug in April 2007. Kyowa Hakko recently initiated a phase I study of ARQ 197 and Tarceva which will probably serve as a basis for larger studies in Japan.
The companies have no time to waste as the field of c-MET inhibitors is becoming very crowded, with over 10 programs at various stages of development. Although Arqule is leading the pack in lung cancer, Exelixis (EXEL) is already in phase III in medullary thyroid cancer with one of its c-MET inhibitors, XL184 (licensed to BMS). XL184 is also in a phase II study in lung cancer where it is being given with Tarceva. Although the trial design is not as robust as Arqule’s trial, there is anecdotal evidence that it can reverse Tarceva resistance. Exelxis’ second c-MET inhibitor, XL880 (licensed to GSK) is also in a large clinical development program with good activity in a subtype of renal cancer. Most of the other c-MET inhibitors are still at an earlier stage of development with no clear proof of concept.
Designing a proper phase III trial in lung cancer has become a daunting task due to the increasing number of approved and experimental agents and the realization that tumors can be classified into multiple subtypes based on histology and tumor biology. This is another place where a robust phase II trial comes in handy.
The phase II study generated a database of biomarkers and correlations related to c-MET inhibition, which is accessible only to Arqule and Daiichi. This database, probably the most reliable and extensive of its kind (since it is derived of a randomized placebo controlled trial), will enable the companies to optimally design the phase III trial. Therefore, the value of the study is not only in the increased likelihood of success of the pivotal trial, but also in its ability to guide the teams towards the most suitable trial design.
Even when assuming all goes well with the regulators and trial design, data from the pivotal study will take at least two years to emerge (depending on size, design, patient population etc.). In the meantime, ARQ 197 will face multiple additional binary events, each representing a value creation opportunity as well as a potential point of failure.
The collaboration with Daiichi Sankyo enabled Arqule to launch multiple phase II studies, including 3 randomized phase II studies in liver, pancreatic and colon cancer. These studies are similar to the lung cancer trial in that they are large randomized controlled trials where ARQ 197 is given in combination with standard of care. Data from the studies is expected to be available next year, even though each of these studies also includes a run in phase I portion that could generate data already in 2010. The value of such data is limited, as it will be very hard to distinguish between ARQ 197’s contribution and that of the combined drugs.
Last February, Arqule submitted a protocol for a trial in gastric cancer but withdrew it the following day. Based on clinicaltrials.gov website, the trial’s goal was comparing ARQ 197 alone to chemotherapy in second line gastric cancer patients. The trial size as appears on the site was 300 patients, which is surprising due to the lack of experience with ARQ 197 in this indication. In addition, although c-MET activity has been found in many gastric cancer samples, so far results from Exelixis’ XL880 as a single agent were disappointing in this indication.
ARQ 197 is also being evaluated as a single agent in several trials, including phase II trials in two niche indications in MiT tumors and germ cell cancer. Although these opportunities represent fast routes to market, the commercial opportunity is low and so are expectations, at least in the MiT trial. Another study that could have interesting data is a forgotten phase I in Japan, which was initiated by Kyowa Hakko more than two years ago. There is currently no visibility on if and when data from this study, which should be completed by now, will be published. Neither Arqule nor its partner have disclosed anything about this trial in a long time.
A growing pipeline
ARQ 197 is clearly Arqule’s most advanced asset, but the company has other shots on goal. Arqule has a wholly owned agent in phase I, with results due in the second half of the year and may put two additional wholly owned drugs in the clinic this year. Therefore, by year end, Arqule may have 4 drugs in the clinic. The market rightfully ascribes minimal value to Arqule’s early stage programs as they have not reached proof of concept in humans. In addition, the three agents hit targets that are already pursued by other companies (Eg5, BRAF and FGFR), even though Arqule believes its molecules possess certain disclosed and undisclosed advantages, especially the BRAF inhibitor.
Arqule’s most interesting, and potentially most valuable asset is its kinase inhibitor discovery platform. The platform, AKIP (Arqule Kinase Inhibitor Platform), appears to be unique in its ability to generate a specific type of kinase inhibitors usually referred to as “non-ATP competitive” or “allosteric” kinase inhibitors. With this platform, Arqule might be able to differentiate itself in a highly competitive field that has experienced a surge in activity in recent years.
Allosteric inhibitors are believed to be extremely specific, which may result in fewer side effects. As the industry is moving towards combining targeted therapies, this attribute may prove very useful. In addition, as kinase inhibitors migrate from oncology to conditions where safety profile is more important (inflammation and pain), having selective kinase inhibitors could be a huge differentiator. The amount of value that can be extracted from AKIP is immense, however, it represents more of a longer term opportunity.
Summary
Since my previous write up on Arqule 16 months ago, the company has been consistently diversifying and de-risking its business. Its lead agent, ARQ 197, a phase III ready drug with blockbuster potential and favorable chances of success, is part of a broad clinical program including 3 randomized trials, each representing a substantial value creation opportunity. The company also complemented its lead program with multiple early stage programs and could have 3 additional programs in clinical testing this year. Finally, the company’s most undervalued asset is a unique discovery platform which could expand the pipeline and provide the company with non-dilutive funds through licensing deals.
*******
mfg ipollit
http://www.hammerstockblog.com/arqule-joins-the-phase-iii-cl…
Arqule Joins The Phase III Club
No matter how diversified or well managed a biotech company is, eventually there is no escape from binary events. These events usually come in the form of data readout from clinical trials that can make or break the stock. Two weeks ago, Arqule (ARQL) announced topline results from a much anticipated phase II trial of its lead agent, ARQ 197, in lung cancer. The trial is evaluating ARQ 197 in combination with the approved drug, Tarceva, in patients with non-small cell lung cancer (NSCLC) who have been previously treated with chemotherapy. Although the data set is not a clear slam dunk, it generated an impressive efficacy signal that warrants advancing ARQ 197 into phase III. This, coupled with the progress the company has made on other fronts in the past year makes Arqule one of most attractive biotech stocks in the market.
Designed for success
Arqule’s study is an excellent example of a properly designed clinical program, especially when it comes to targeted therapies. From the get go, the trial had the right ingredients, including a robust trial design, the right indication and a combination regimen supported by solid scientific rationale.
Doing a combination trial with Tarceva in NSCLC looks like the ideal place to start with ARQ 197 for several reasons. ARQ 197 inhibits c-MET, a protein associated with multiple cancer-related functions such as invasion and metastasis. The expression and involvement of c-MET in lung cancer is well established, based on numerous reports evaluating tumors from patients. The past years saw a flood of data with respect to c-MET and lung cancer, including a study which found that more than 70% of lung cancer tissues had some level of c-MET expression. Some studies also point to c-MET as a potential marker of aggressive disease and poor prognosis.
ARQ 197 and Tarceva have distinct yet intertwined mechanisms of action, so combining the two drugs is expected to result in at least additive, if not synergistic effect. c-MET activation is believed to account for 20-25% of cases of Tarceva resistance, so by adding ARQ 197, investigators hope to delay Tarceva resistance at least in some of the patients. Interestingly, recent experimental data shows that cells can become resistant to c-MET inhibitors by activating the EGFR pathway, which is inhibited by Tarceva.
From a commercial standpoint, it is always easier to add a new drug to an existing treatment line rather than replacing it. Tarceva is a commonly used drug in NSCLC with annual sales of $1.2B, so by adding ARQ 197, Arqule can build on Tarceva’s market penetration and create a new standard of care rather quickly. In addition, by the time ARQ 197 reaches the market, Tarceva will probably be generic, which will make the combination cheaper for patients and reimbursers. Lastly, both drugs are oral, so adding ARQ 197 does not lead to compliance or logistic issues.
The design of the trial is also noteworthy, as it is a fairly large (~170 patients) randomized study in which patients receive a combination of Tarceva with either ARQ 197 or placebo. Large randomized phase II trials are becoming ever more popular in oncology due to their ability to identify ineffective drugs and avoid unnecessary phase III studies. Their main disadvantage is cost, however that is a price the industry has learned to accept, especially in the case of combining targeted agents that often have a subtle effect in terms of tumor shrinkage.
Naturally, evaluating a drug in a randomized phase II increases the risk of failure, as results are compared head to head with a control arm. Non randomized trials are more likely to generate a “positive” signal in comparison with historical data, but results are less reliable. Therefore, the robust design of the ARQ 197 trial renders it more challenging but also more reliable for deciding whether to advance this combination into a registration study. In other words, if the trial is positive, it serves as a strong basis for a phase III study with a higher probability of success.
Results
Results from the phase II trial included 167 patients who received ARQ 197+Tarceva or Tarceva+placebo. On the safety front, the combination regimen was well tolerated with no meaningful safety issues. The primary endpoint was progression free survival (PFS), which is the time until disease progression (usually tumor growth or appearance of new lesions). Although patients in the ARQ 197+Tarceva arm had a PFS of 16.1 weeks, compared with 9.7 weeks for the patients on the Tarceva+placebo arm, the difference was not statistically significant. Nevertheless, two additional analyses were extremely encouraging.
It turned out that there were imbalances between treatment arms in terms of mutations in two genes (EGFR and KRAS) that are known to be linked to response to Tarceva. The imbalances were in favor of the control arm, which means that the control arm had more patients with a high likelihood of responding to Tarceva. When this difference was factored in, the difference between the combination and control arms became statistically significant. Another analysis was done in a subset of patients classified as non-squamous (histological classification). In this subset of patients, which accounted for ~70% of patients in the trial, the difference between the two arms was more pronounced (18.9 vs. 9.7 weeks) and reached statistical significance.
Positive signals
Although the trial did not meet its pre-defined primary endpoint, the efficacy signal is definitely there. Retrospective and subset analyses should always be treated with skepticism, but in this case, both analyses seem reasonable and reliable. Obviously, these signals will have to be validated prospectively in a large phase III trial.
Distinguishing between squamous and non-squamous NSCLC is a common analysis that had originally been part of the trial design. It is conducted on a regular basis in clinical trials, as the difference between the two subpopulations in terms of response to certain drugs is well recognized. The best example for that is Alimta, which is extremely effective in non-squamous NSCLC but ineffective and potentially deleterious in squamous NSCLC. Importantly, as non-squamous NSCLC accounts for 65-70% of NSCLC cases, it still represents a huge opportunity. Alimta, which is approved only for non-squamous NSCLC generated sales of $1.7B in 2009.
The imbalances between the two arms in EGFR and KRAS mutations are not surprising, as these factors were not included in the randomization process. EGFR and KRAS mutations are considered validated predictors of Tarceva response and are not always available at the time of treatment initiation. Patients with EGFR mutations tend to derive meaningful benefit from Tarceva whereas KRAS mutant tumors are considered relatively resistant to the drug. Therefore, factoring in these imbalances seems reasonable in trials that involve Tarceva.
Comparison with other trials
Comparing Arqule’s study to previous studies (see table below) in a similar patient population may provide additional insights. It is important to understand that no definitive conclusions can be drawn from comparing results from different trials, and usually there is an inverse correlation between a drug’s performance and trial size. Nevertheless, one can get a sense of where ARQ 197 stands in terms of PFS from such analysis. Even in comparison with established chemotherapy agents such as Taxotere and Alimta, the combination of ARQ 197 and Tarceva looks promising. The mild safety profile of the regimen is definitely a major advantage over chemo regimens, but cost might be an issue.
In its registration trial, Tarceva’s improvement in PFS was minimal yet statistically significant (2.2 vs. 1.8 months), but the real benefit was in overall survival (a 2 month difference). In the phase II trial, ARQ 197’s increase in PFS was more profound, especially in the non-squamous subgroup, despite the imbalances between the two arms. Alimta’s benefit in non-squamous patients also seems limited to overall survival, with no real difference in PFS. On the one hand, this makes Arqule’s data even more promising. On the other, it might imply that PFS is not necessarily indicative of survival, but phase II trials cannot evaluate overall survival in a reliable manner, especially in lung cancer where there are so many treatment lines.
Although the overall survival data is still unavailable, it is highly unlikely to see a clear statistically significant signal there as the trial was not powered to demonstrate that. In addition, the trial employed a cross-over design, where patients on the control arm can receive the combination of ARQ 197 and Tarceva after progressing on Tarceva alone, which may offset any potential effect of ARQ 197. Therefore, there is actually a third arm of patients who receive ARQ 197 and Tarceva following progressing on Tarceva. What investors can hope for is a trend of improved survival in patients on the ARQ 197 arm and those on the control arm who did not cross over after progression. According to the company, the majority of patients in the control arm did not crossover to the combination arm, so numbers might be sufficient to show a survival trend.
The control arm in the ARQ 197 trial had a median PFS of 9.7 weeks (2.2 months), which is similar to what Tarceva achieved in its registration trial. Although one could expect the control arm to do slightly better than historic controls, this further validates the trial design and implies the control arm in Arqule’s study is more or less reflective of a typical phase III population.
Next steps
Armed with such a promising data set, Arqule’s next obvious step is a global phase III trial in lung cancer. Although the formal decision has not been made, Arqule and its partner, Daiichi Sankyo, are likely to launch a phase III trial towards year end. Given the doubling of PFS in non-squamous patients, the trial will probably be limited to this subpopulation. Although the phase II was conducted primarily in the US and EU, the phase III program will probably include Japan, the second largest oncology market and an important Tarceva market. The commercialization rights for ARQ 197 in Japan belong to Kyowa-Hakko, which licensed the drug in April 2007. Kyowa Hakko recently initiated a phase I study of ARQ 197 and Tarceva which will probably serve as a basis for larger studies in Japan.
The companies have no time to waste as the field of c-MET inhibitors is becoming very crowded, with over 10 programs at various stages of development. Although Arqule is leading the pack in lung cancer, Exelixis (EXEL) is already in phase III in medullary thyroid cancer with one of its c-MET inhibitors, XL184 (licensed to BMS). XL184 is also in a phase II study in lung cancer where it is being given with Tarceva. Although the trial design is not as robust as Arqule’s trial, there is anecdotal evidence that it can reverse Tarceva resistance. Exelxis’ second c-MET inhibitor, XL880 (licensed to GSK) is also in a large clinical development program with good activity in a subtype of renal cancer. Most of the other c-MET inhibitors are still at an earlier stage of development with no clear proof of concept.
Designing a proper phase III trial in lung cancer has become a daunting task due to the increasing number of approved and experimental agents and the realization that tumors can be classified into multiple subtypes based on histology and tumor biology. This is another place where a robust phase II trial comes in handy.
The phase II study generated a database of biomarkers and correlations related to c-MET inhibition, which is accessible only to Arqule and Daiichi. This database, probably the most reliable and extensive of its kind (since it is derived of a randomized placebo controlled trial), will enable the companies to optimally design the phase III trial. Therefore, the value of the study is not only in the increased likelihood of success of the pivotal trial, but also in its ability to guide the teams towards the most suitable trial design.
Even when assuming all goes well with the regulators and trial design, data from the pivotal study will take at least two years to emerge (depending on size, design, patient population etc.). In the meantime, ARQ 197 will face multiple additional binary events, each representing a value creation opportunity as well as a potential point of failure.
The collaboration with Daiichi Sankyo enabled Arqule to launch multiple phase II studies, including 3 randomized phase II studies in liver, pancreatic and colon cancer. These studies are similar to the lung cancer trial in that they are large randomized controlled trials where ARQ 197 is given in combination with standard of care. Data from the studies is expected to be available next year, even though each of these studies also includes a run in phase I portion that could generate data already in 2010. The value of such data is limited, as it will be very hard to distinguish between ARQ 197’s contribution and that of the combined drugs.
Last February, Arqule submitted a protocol for a trial in gastric cancer but withdrew it the following day. Based on clinicaltrials.gov website, the trial’s goal was comparing ARQ 197 alone to chemotherapy in second line gastric cancer patients. The trial size as appears on the site was 300 patients, which is surprising due to the lack of experience with ARQ 197 in this indication. In addition, although c-MET activity has been found in many gastric cancer samples, so far results from Exelixis’ XL880 as a single agent were disappointing in this indication.
ARQ 197 is also being evaluated as a single agent in several trials, including phase II trials in two niche indications in MiT tumors and germ cell cancer. Although these opportunities represent fast routes to market, the commercial opportunity is low and so are expectations, at least in the MiT trial. Another study that could have interesting data is a forgotten phase I in Japan, which was initiated by Kyowa Hakko more than two years ago. There is currently no visibility on if and when data from this study, which should be completed by now, will be published. Neither Arqule nor its partner have disclosed anything about this trial in a long time.
A growing pipeline
ARQ 197 is clearly Arqule’s most advanced asset, but the company has other shots on goal. Arqule has a wholly owned agent in phase I, with results due in the second half of the year and may put two additional wholly owned drugs in the clinic this year. Therefore, by year end, Arqule may have 4 drugs in the clinic. The market rightfully ascribes minimal value to Arqule’s early stage programs as they have not reached proof of concept in humans. In addition, the three agents hit targets that are already pursued by other companies (Eg5, BRAF and FGFR), even though Arqule believes its molecules possess certain disclosed and undisclosed advantages, especially the BRAF inhibitor.
Arqule’s most interesting, and potentially most valuable asset is its kinase inhibitor discovery platform. The platform, AKIP (Arqule Kinase Inhibitor Platform), appears to be unique in its ability to generate a specific type of kinase inhibitors usually referred to as “non-ATP competitive” or “allosteric” kinase inhibitors. With this platform, Arqule might be able to differentiate itself in a highly competitive field that has experienced a surge in activity in recent years.
Allosteric inhibitors are believed to be extremely specific, which may result in fewer side effects. As the industry is moving towards combining targeted therapies, this attribute may prove very useful. In addition, as kinase inhibitors migrate from oncology to conditions where safety profile is more important (inflammation and pain), having selective kinase inhibitors could be a huge differentiator. The amount of value that can be extracted from AKIP is immense, however, it represents more of a longer term opportunity.
Summary
Since my previous write up on Arqule 16 months ago, the company has been consistently diversifying and de-risking its business. Its lead agent, ARQ 197, a phase III ready drug with blockbuster potential and favorable chances of success, is part of a broad clinical program including 3 randomized trials, each representing a substantial value creation opportunity. The company also complemented its lead program with multiple early stage programs and could have 3 additional programs in clinical testing this year. Finally, the company’s most undervalued asset is a unique discovery platform which could expand the pipeline and provide the company with non-dilutive funds through licensing deals.
*******
mfg ipollit
Antwort auf Beitrag Nr.: 39.310.278 von ipollit am 11.04.10 14:41:34ARQL...
Oppenheimer
ArQule, Inc.
Update from Management Meeting; Expect Detailed Ph.II '197 Data at ASCO
PT: $11
SUMMARY
On 4/8, we met with ARQL management. Based on our discussion, we expect
detailed ph.II data for ARQ 197+Tarceva vs. Tarceva alone in 2nd/3rd-line NSCLC
will be presented at ASCO in June. We believe the detailed results will be
compelling, especially in non-squamous NSCLC, opening a clear path toward future
ph.III success. Considering the competitive landscape, we expect ARQL to move
forward rapidly with ph.III NSCLC plans, and see a good probability the company
will initiate ph.III testing of '197 in non-squamous NSCLC before YE10. Although
ARQL has appreciated substantially on the top-line '197 NSCLC results, we expect
meaningful additional upside in the stock pre- and post-ASCO. We believe the
detailed ph.II results will be an ASCO highlight.
KEY POINTS
* We anticipate detailed ph.II '197 data at ASCO. ARQL met the ASCO
late-breaker deadline, and we believe strong top-line PFS results for
'197+Tarceva should lead to an acceptance. We believe subset analyses from
the trial, notably in non-squamous vs. squamous and EGFR/KRAS mutant pts
will clarify the top-line results and drive physician enthusiasm.
* We see a high probability ARQL to begin a ph.III trial in non-squamous
NSCLC by YE10. While ARQL has not yet formulated a go-forward ph.III plan,
based on the ph.II results, we believe a trial of '197+Tarceva in non-squamous
NSCLC would be relatively quick ('12 results, or earlier) and have a high
likelihood of success.
* The emerging profile of '197+Tarceva in 2nd/3rd-line NSCLC appears
compelling. If the ~4 mo. absolute PFS benefit seen with '197+Tarceva in ph.II
is replicated in ph.III testing, we believe this would position the combo ahead of
single-agent Tarceva or Alimta, which showed 2-3 mo. absolute PFS benefits in
pivotal data.
* ARQL's economics on '197 are likely under-appreciated. Although terms of
ARQL's tiered royalty from Daiichi from '197 sales are undisclosed, we believe
the rates substantially exceed the 15-20% KyowaHakko Asian royalty, given
ARQL/Daiichi's ~50/50 cost sharing in '197's development. Based on this, we
believe our, and Street, estimates for ARQL's '197 revenues are conservative.
mfg ipollit
Oppenheimer
ArQule, Inc.
Update from Management Meeting; Expect Detailed Ph.II '197 Data at ASCO
PT: $11
SUMMARY
On 4/8, we met with ARQL management. Based on our discussion, we expect
detailed ph.II data for ARQ 197+Tarceva vs. Tarceva alone in 2nd/3rd-line NSCLC
will be presented at ASCO in June. We believe the detailed results will be
compelling, especially in non-squamous NSCLC, opening a clear path toward future
ph.III success. Considering the competitive landscape, we expect ARQL to move
forward rapidly with ph.III NSCLC plans, and see a good probability the company
will initiate ph.III testing of '197 in non-squamous NSCLC before YE10. Although
ARQL has appreciated substantially on the top-line '197 NSCLC results, we expect
meaningful additional upside in the stock pre- and post-ASCO. We believe the
detailed ph.II results will be an ASCO highlight.
KEY POINTS
* We anticipate detailed ph.II '197 data at ASCO. ARQL met the ASCO
late-breaker deadline, and we believe strong top-line PFS results for
'197+Tarceva should lead to an acceptance. We believe subset analyses from
the trial, notably in non-squamous vs. squamous and EGFR/KRAS mutant pts
will clarify the top-line results and drive physician enthusiasm.
* We see a high probability ARQL to begin a ph.III trial in non-squamous
NSCLC by YE10. While ARQL has not yet formulated a go-forward ph.III plan,
based on the ph.II results, we believe a trial of '197+Tarceva in non-squamous
NSCLC would be relatively quick ('12 results, or earlier) and have a high
likelihood of success.
* The emerging profile of '197+Tarceva in 2nd/3rd-line NSCLC appears
compelling. If the ~4 mo. absolute PFS benefit seen with '197+Tarceva in ph.II
is replicated in ph.III testing, we believe this would position the combo ahead of
single-agent Tarceva or Alimta, which showed 2-3 mo. absolute PFS benefits in
pivotal data.
* ARQL's economics on '197 are likely under-appreciated. Although terms of
ARQL's tiered royalty from Daiichi from '197 sales are undisclosed, we believe
the rates substantially exceed the 15-20% KyowaHakko Asian royalty, given
ARQL/Daiichi's ~50/50 cost sharing in '197's development. Based on this, we
believe our, and Street, estimates for ARQL's '197 revenues are conservative.
mfg ipollit
Antwort auf Beitrag Nr.: 39.310.278 von ipollit am 11.04.10 14:41:34
Hi ipollit,
danke für den Hinweis. Du musst den Artikel sofort entdeckt haben, als er erschienen ist...
Nach langer Schaffenspause mal wieder ein echter "Hammer". Man fühlt sich einfach umfassend informiert. Der Mann hat echt die Gabe, Sachverhalte extrem gut und verständlich zu analysieren und zu bewerten.
Gruß
PS: Was ich allerdings immer noch nicht so ganz verstehe, ist, warum die Ergebnisse grundsätzlich statistisch nicht relevant gewesen sind.
Although patients in the ARQ 197+Tarceva arm had a PFS of 16.1 weeks, compared with 9.7 weeks for the patients on the Tarceva+placebo arm, the difference was not statistically significant.
Sind mehr als 6 Wochen Unterschie noch immer zu wenig, um statistisch signifikant zu sein, während dann 9 Wochen ausreichen?
In this subset of patients, which accounted for ~70% of patients in the trial, the difference between the two arms was more pronounced (18.9 vs. 9.7 weeks) and reached statistical significance.
Oder was genau meint "statistisch signifikant" eigentlich?
Hi ipollit,
danke für den Hinweis. Du musst den Artikel sofort entdeckt haben, als er erschienen ist...
Nach langer Schaffenspause mal wieder ein echter "Hammer". Man fühlt sich einfach umfassend informiert.
Der Mann hat echt die Gabe, Sachverhalte extrem gut und verständlich zu analysieren und zu bewerten.Gruß
PS: Was ich allerdings immer noch nicht so ganz verstehe, ist, warum die Ergebnisse grundsätzlich statistisch nicht relevant gewesen sind.
Although patients in the ARQ 197+Tarceva arm had a PFS of 16.1 weeks, compared with 9.7 weeks for the patients on the Tarceva+placebo arm, the difference was not statistically significant.
Sind mehr als 6 Wochen Unterschie noch immer zu wenig, um statistisch signifikant zu sein, während dann 9 Wochen ausreichen?
In this subset of patients, which accounted for ~70% of patients in the trial, the difference between the two arms was more pronounced (18.9 vs. 9.7 weeks) and reached statistical significance.
Oder was genau meint "statistisch signifikant" eigentlich?
Antwort auf Beitrag Nr.: 39.194.451 von SLGramann am 23.03.10 07:52:01
Man hatte gehofft, dass für das 3rd-line Treatment bereits P II-Daten für die Zulassung reichen würden, aber Roche scheint zu zögern, damit auf die FDA zuzugehen. Jedenfalls ist der Status da nicht ganz klar, was den Kurs seit Dezember belastet hatte.
Dennoch spricht einiges dafür, dass T-DM1 sehr werthaltig ist.
So, der nächste Schritt wird getan werden. Roche beantragt für T-DM1 die Zulassung bei 3rd-line:
In its quarterly release issued earlier today, Roche disclosed that company representatives have had discussions with the US Food and Drug Administration (FDA) and that, based on these discussions, Genentech/Roche plans to submit a marketing application to the FDA in 2010 for use of T-DM1 in the treatment of advanced HER2-positive metastatic breast cancer.
http://www.businesswire.com/portal/site/home/permalink/?ndmV…
Damit ist
1.) die ADC-Technologie von IMGN bereits weitgehend validiert,
2.) vielleicht(!) nicht nur die Ergänzung, sondern der allgemeine Nachfolger von Herceptin endgültig auf dem Weg.
Ausgezeichnet!
Man hatte gehofft, dass für das 3rd-line Treatment bereits P II-Daten für die Zulassung reichen würden, aber Roche scheint zu zögern, damit auf die FDA zuzugehen. Jedenfalls ist der Status da nicht ganz klar, was den Kurs seit Dezember belastet hatte.
Dennoch spricht einiges dafür, dass T-DM1 sehr werthaltig ist.
So, der nächste Schritt wird getan werden. Roche beantragt für T-DM1 die Zulassung bei 3rd-line:
In its quarterly release issued earlier today, Roche disclosed that company representatives have had discussions with the US Food and Drug Administration (FDA) and that, based on these discussions, Genentech/Roche plans to submit a marketing application to the FDA in 2010 for use of T-DM1 in the treatment of advanced HER2-positive metastatic breast cancer.
http://www.businesswire.com/portal/site/home/permalink/?ndmV…
Damit ist
1.) die ADC-Technologie von IMGN bereits weitgehend validiert,
2.) vielleicht(!) nicht nur die Ergänzung, sondern der allgemeine Nachfolger von Herceptin endgültig auf dem Weg.
Ausgezeichnet!
Antwort auf Beitrag Nr.: 39.338.083 von SLGramann am 15.04.10 08:13:28
Ohad Hammer hat die Lage bei ImmunoGen in einem aktuellen Artikel sehr umfassend und kompetent dargestellt. Jeder, der ein gewisses Interesse an diesem Sachverhalt hat, kann hier mal lesen. Wer das Thema ADCs allgemein vertiefen will, sollte den drei Verlinkungen folgen, die Hammer im Text anbietet.
Earlier this week, Immunogen (IMGN) got closer than ever to having a commercial product in the market. During its quarterly call, Roche removed the regulatory overhang on T-DM1’s near term fate after it disclosed plans to file for T-DM1’s approval already this year. The submission will be based on results from a recently announced phase II trial where T-DM1 demonstrated overwhelming activity in late stage breast cancer patients. Until now, it was unclear whether the FDA would be willing to consider approval based on a single arm phase II trial. Roche’s decision implies the FDA gave its unofficial and non-binding blessing for the accelerated approval.
High likelihood of approval
As a way of background, T-DM1 is based on Immunogen’s technology and the company retains financial interest in the drug in the form of mid single digit royalties. T-DM1 is Immunogen’s most important opportunity for generating commercial revenues as well as validating its antibody drug conjugate (ADC) platform.
hier gehts weiter:
http://www.hammerstockblog.com/immunogen-%E2%80%93-another-s…
------------------
Außerdem verspreche ich, jetzt nicht mehr mit IMGN rumzunerven, die ja gar nicht in ipollits Portfolio sind, um das es hier geht, wie mir durchaus klar ist.
Ohad Hammer hat die Lage bei ImmunoGen in einem aktuellen Artikel sehr umfassend und kompetent dargestellt. Jeder, der ein gewisses Interesse an diesem Sachverhalt hat, kann hier mal lesen. Wer das Thema ADCs allgemein vertiefen will, sollte den drei Verlinkungen folgen, die Hammer im Text anbietet.
Earlier this week, Immunogen (IMGN) got closer than ever to having a commercial product in the market. During its quarterly call, Roche removed the regulatory overhang on T-DM1’s near term fate after it disclosed plans to file for T-DM1’s approval already this year. The submission will be based on results from a recently announced phase II trial where T-DM1 demonstrated overwhelming activity in late stage breast cancer patients. Until now, it was unclear whether the FDA would be willing to consider approval based on a single arm phase II trial. Roche’s decision implies the FDA gave its unofficial and non-binding blessing for the accelerated approval.
High likelihood of approval
As a way of background, T-DM1 is based on Immunogen’s technology and the company retains financial interest in the drug in the form of mid single digit royalties. T-DM1 is Immunogen’s most important opportunity for generating commercial revenues as well as validating its antibody drug conjugate (ADC) platform.
hier gehts weiter:
http://www.hammerstockblog.com/immunogen-%E2%80%93-another-s…
------------------
Außerdem verspreche ich, jetzt nicht mehr mit IMGN rumzunerven, die ja gar nicht in ipollits Portfolio sind, um das es hier geht, wie mir durchaus klar ist.
Antwort auf Beitrag Nr.: 39.357.943 von SLGramann am 18.04.10 19:43:25IMGN... bin leider nicht dabei, lese aber interessiert. Du kannst hier gerne weiter dazu schreiben, danke!

im Hammer Stock Blog wurde ein neuer Wert aufgenommen... AVEO Pharmaceuticals http://finance.yahoo.com/q?s=AVEO. Was ist davon zu halten? Eines der wenigen IPOs in der letzten Zeit.

mfg ipollit
im Hammer Stock Blog wurde ein neuer Wert aufgenommen... AVEO Pharmaceuticals http://finance.yahoo.com/q?s=AVEO. Was ist davon zu halten? Eines der wenigen IPOs in der letzten Zeit.
mfg ipollit
ARRY... findet einen weiteren Partner, diesmal für die MEK-Hemmer. Weitere Deals sind kurz- oder mittelfristig möglich.

UPDATE 1-Array BioPharma inks $467 mln deal with Novartis
* Signs deal on cancer drugs with Novartis
* To get $45 mln in upfront payment
* To get double-digit royalties on sales outside U.S.
* Array shares up 26 percent after the bell
April 19 (Reuters) - Array BioPharma Inc (ARRY.O) said it signed a licensing deal worth up to $467 million for its experimental cancer drug candidates with Swiss drugmaker Novartis AG (NOVN.VX), sending its shares up 26 percent in extended trade.
The deal, which includes an early-stage candidate ARRY-162 and other MEK inhibitors, entitles Array to $45 million in upfront payments and $422 million in milestones.
Array, which is eligible to double-digit royalties on sales of the approved products outside the United States, plans to co-develop ARRY-162 and fund part of the development costs.
If Array meets its co-funding obligations, it becomes eligible for a "significantly higher" royalty rate on sales in the United States.
The company, which focuses on developing drugs to treat cancer and inflammatory diseases, continues to be successful in partnering its pipeline. It partnered its experimental diabetes drug with Amgen Inc (AMGN.O) last December. Shares of Array were up at $3.80 in post-market trade.
mfg ipollit
UPDATE 1-Array BioPharma inks $467 mln deal with Novartis
* Signs deal on cancer drugs with Novartis
* To get $45 mln in upfront payment
* To get double-digit royalties on sales outside U.S.
* Array shares up 26 percent after the bell
April 19 (Reuters) - Array BioPharma Inc (ARRY.O) said it signed a licensing deal worth up to $467 million for its experimental cancer drug candidates with Swiss drugmaker Novartis AG (NOVN.VX), sending its shares up 26 percent in extended trade.
The deal, which includes an early-stage candidate ARRY-162 and other MEK inhibitors, entitles Array to $45 million in upfront payments and $422 million in milestones.
Array, which is eligible to double-digit royalties on sales of the approved products outside the United States, plans to co-develop ARRY-162 and fund part of the development costs.
If Array meets its co-funding obligations, it becomes eligible for a "significantly higher" royalty rate on sales in the United States.
The company, which focuses on developing drugs to treat cancer and inflammatory diseases, continues to be successful in partnering its pipeline. It partnered its experimental diabetes drug with Amgen Inc (AMGN.O) last December. Shares of Array were up at $3.80 in post-market trade.
mfg ipollit
Wo ist bei diesem Wert der Haken? JAZZ... http://finance.yahoo.com/q?s=JAZZ

erst stark abgestürzt wegen geringem Cash, hohem Cashburn und vielen Schulden, dann wieder nach starker Reduzierung des Cashburn auf das alte Niveau gestiegen. Aktuelle MK bei ca. 300 Mio USD. Interessant ist, dass zwei Produkte am Markt sind, die dieses Jahr ca. 150 Mio USD einbringen sollen... eps10e von 0,49 USD (vom Unternehmen sind 0,80 bis 0,95 USD angepeilt), eps11e 1,36 USD... und das ganze bei einem aktuellen Kurs von 9,81 USD. Zudem steht im Oktober die Zulassung von JZP-6 an, das alleine in den USA einen Umsatz von 500 Mio USD einbringen könnte. Wo ist der Haken?
mfg ipollit
erst stark abgestürzt wegen geringem Cash, hohem Cashburn und vielen Schulden, dann wieder nach starker Reduzierung des Cashburn auf das alte Niveau gestiegen. Aktuelle MK bei ca. 300 Mio USD. Interessant ist, dass zwei Produkte am Markt sind, die dieses Jahr ca. 150 Mio USD einbringen sollen... eps10e von 0,49 USD (vom Unternehmen sind 0,80 bis 0,95 USD angepeilt), eps11e 1,36 USD... und das ganze bei einem aktuellen Kurs von 9,81 USD. Zudem steht im Oktober die Zulassung von JZP-6 an, das alleine in den USA einen Umsatz von 500 Mio USD einbringen könnte. Wo ist der Haken?
mfg ipollit
Antwort auf Beitrag Nr.: 39.364.546 von ipollit am 19.04.10 23:15:00
Okay, dann berichte ich hier gern weiter zu IMGN, wenn es relevante Neuigkeiten gibt.
Zu AVEO:
Das klingt natürlich zunächst mal hochinteressant. Hammer verfolgt ja grundsätzlich den Ansatz, Unternehmen mit einer Platform-Technologie zu favorisieren (Beispiele wären SGEN, IMGN, MITI, ISIS, EXEL, inzwischen wohl auch ARQL).
AVEO würde in diese Reihe passen und die Platform, die sie haben, könnte in der Tat sehr relevant sein, falls sie hält, was AVEO verspricht.
Auf der AVEO-Site sind einige Artikel verlinkt, die das Prinzip und ihren Denkansatz gut erklären.
Ich verlinke mal drei davon direkt:
http://www.boston.com/news/science/articles/2007/07/02/aveos…
http://aveopharma.com/files/InTheNews-Xconomy.pdf
http://aveopharma.com/files/InTheNews-GEN.pdf
Natürlich kann ich absolut nicht beurteilen, ob AVEO hier auf einem Erfolgspfad wandelt, aber es gibt einige Anzeichen, dass es so sein könnte (Partnerschaften insbesondere).
Auf kürzere Sicht geht es aber sicher vor allem erst mal um Tivozanib und vielleicht trauen sich ipollit oder Pathfinder hier eine Beurteilung zu.
Auf jeden Fall denke ich, dass es lohnend sein könnte, an AVEO dran zu bleiben.
Okay, dann berichte ich hier gern weiter zu IMGN, wenn es relevante Neuigkeiten gibt.
Zu AVEO:
Das klingt natürlich zunächst mal hochinteressant. Hammer verfolgt ja grundsätzlich den Ansatz, Unternehmen mit einer Platform-Technologie zu favorisieren (Beispiele wären SGEN, IMGN, MITI, ISIS, EXEL, inzwischen wohl auch ARQL).
AVEO würde in diese Reihe passen und die Platform, die sie haben, könnte in der Tat sehr relevant sein, falls sie hält, was AVEO verspricht.
Auf der AVEO-Site sind einige Artikel verlinkt, die das Prinzip und ihren Denkansatz gut erklären.
Ich verlinke mal drei davon direkt:
http://www.boston.com/news/science/articles/2007/07/02/aveos…
http://aveopharma.com/files/InTheNews-Xconomy.pdf
http://aveopharma.com/files/InTheNews-GEN.pdf
Natürlich kann ich absolut nicht beurteilen, ob AVEO hier auf einem Erfolgspfad wandelt, aber es gibt einige Anzeichen, dass es so sein könnte (Partnerschaften insbesondere).
Auf kürzere Sicht geht es aber sicher vor allem erst mal um Tivozanib und vielleicht trauen sich ipollit oder Pathfinder hier eine Beurteilung zu.
Auf jeden Fall denke ich, dass es lohnend sein könnte, an AVEO dran zu bleiben.
Antwort auf Beitrag Nr.: 39.365.254 von SLGramann am 20.04.10 09:07:45
nochmals zu AVEO:
Hier mal ein paar wesentliche Eckpunkte zu Tivozanib, wie ich sie aus einem SEC-File entnommen und wie ich sie verstanden habe.
Für mich klingt das durchaus hoffnungsvoll, allerdings kann ich mir in diesen Dingen einfach keine wirkliche Meinung erlauben. Vielleicht möchte das ein anderer weiter kommentieren.
Tivozanib ist ein oraler small-molecul VEGF-Rezeptor-Inhibitor, der eine P II in der Indikation Nierenzellkarzinom abgeschlossen hat. Als VEGF-Inhibitor ist der Wirkstoff grundsätzlich sehr breit einsetzbar (bspw. ist Avastin auch gegen VEGF gerichtet).
In der P II mit 272 Patienten wurde ein durchschnittliches progression-free survival (PFS) von 11,8 Monaten erreicht. In einer Subgruppe mit 176 Patienten wurden 14,8 Monate PFS erreicht.
AVEO schreibt, dass Nexavar in seiner P III in der von AVEO favorisierten Subgruppe eine median-PFS von 5,5 Monaten erreicht habe.
Die gerade gestartete P III (500 Patienten) hat als primären Endpunkt PFS im direkten Vergleich zu Nexavar und ist so designt, dass ein 3-monatiger Vorteil beim PFS von Tivozanib gegenüber Nexavar als statistisch signifikant gilt. Ich gehe davon aus, dass AVEO im Erfolgsfall auf der Basis dieser Studie die Zulassung beantragen wird (Ende 2012?).
AVEO hebt weiterhin hervor, dass Tivozanib relativ nebenwirkungsarm und sehr spezifisch ist und deshalb gut mit anderen Präparaten kombiniert werden kann. Es laufen eine Reihe P I b –Studien für andere Indikationen bzw. Kombitherapien.
nochmals zu AVEO:
Hier mal ein paar wesentliche Eckpunkte zu Tivozanib, wie ich sie aus einem SEC-File entnommen und wie ich sie verstanden habe.
Für mich klingt das durchaus hoffnungsvoll, allerdings kann ich mir in diesen Dingen einfach keine wirkliche Meinung erlauben. Vielleicht möchte das ein anderer weiter kommentieren.
Tivozanib ist ein oraler small-molecul VEGF-Rezeptor-Inhibitor, der eine P II in der Indikation Nierenzellkarzinom abgeschlossen hat. Als VEGF-Inhibitor ist der Wirkstoff grundsätzlich sehr breit einsetzbar (bspw. ist Avastin auch gegen VEGF gerichtet).
In der P II mit 272 Patienten wurde ein durchschnittliches progression-free survival (PFS) von 11,8 Monaten erreicht. In einer Subgruppe mit 176 Patienten wurden 14,8 Monate PFS erreicht.
AVEO schreibt, dass Nexavar in seiner P III in der von AVEO favorisierten Subgruppe eine median-PFS von 5,5 Monaten erreicht habe.
Die gerade gestartete P III (500 Patienten) hat als primären Endpunkt PFS im direkten Vergleich zu Nexavar und ist so designt, dass ein 3-monatiger Vorteil beim PFS von Tivozanib gegenüber Nexavar als statistisch signifikant gilt. Ich gehe davon aus, dass AVEO im Erfolgsfall auf der Basis dieser Studie die Zulassung beantragen wird (Ende 2012?).
AVEO hebt weiterhin hervor, dass Tivozanib relativ nebenwirkungsarm und sehr spezifisch ist und deshalb gut mit anderen Präparaten kombiniert werden kann. Es laufen eine Reihe P I b –Studien für andere Indikationen bzw. Kombitherapien.
Hallo,
bin hier seid einiger Zeit stiller Leser und finde Eure Postings sehr interessant.
Mich würde Eure Meinung zu Celldex interessieren.
Siehe hierzu folgender Link.
http://www.wallstreet-online.de/diskussion/1157067-1-10/cell…
Celldex hat wie Roche auch einen Antikörper gegen Brustkrebs in der Entwicklung. Hierzu wird die Technologie von SGEN genutzt.
Im Vergleich zu T-DM1 von Roche noch folgende Info aus dem Yahoo board.
The best benchmark for assessing CR-011 is Roche’s T-DM1, which is also an antibody drug conjugate (comprises of Herceptin and a drug payload).
The two agents have a lot in common. Both utilize validated ADC technologies; CR-011 employs Seattle Genetics’ (SGEN) technology whereas T-DM1 employs Immunogen’s (IMGN) technology. Furthermore, the targets for CR-011 and T-DM1 (GPNMB and Her2, respectively) are found on a minority of breast cancer patients and are associated with disease aggressiveness and poor outcome. Lastly, according to Timothy Shannon, Curagen’s CEO, the company patented GPNMB as a target, similarly to what Genentech/Roche did with Her2. This strategy enabled Roche to be the sole company who can sell anti-Her2 antibodies
GPNMB is in fact expressed in ~70-75% of all breast cancers, regardless of subtype. It is expressed in 2 different cell types within the tumors: epithelial cells (the actual cancer cells) and stromal cells (cells that support the tumor). The vast majority (85%) of GPNMB-positive breast tumors only have GPNMB expression in stromal cells - and these GPNMB-positive tumors are equally represented across subtypes (Her2+, ER/PR+, triple negative).
However, the remaining 15% of GPNMB-pos tumors have GPNMB expressed in the epithelium (cancer cells) - and this group of GPNMB-positive tumors are highly over-represented in the triple negative subtype. Within triple negative tumors, those that have GPNMB expression in the epithelium are much more likely to metastasize and need targeted therapies.
The thing is, CDX-011 will target and kill stromal and epithelial cells. In fact much of its efficacy may lie in the fact that it targets stromal cells (for example, all of the anti-angiogenic therapies out there target the stromal cells).
So, yes CDX-011 represents an exciting therapy for triple negative breast cancer, but it will likely also be effective against the other subtypes, including Her2+, though in this subtype it may not be as effective as T-DM1.
Absolutely, it is still very early on in the CDX-011 trials; in fact as a single agent therapy in heavily pre-treated metastatic breast cancer - the results from CDX-011 are really quite striking in GPNMB-positive patients. But still not as striking as T-DM1 in Her2+ patients. However there is room for improvement with CDX-011, (ie. give once per wk vs. once every 3 weeks; try combining it with traditional chemotherapies). And for that we will have to wait and see.
Meine Frage: Wie schätzt Ihr Celldex mittel- und langfristig ein.
Gruß
wachholder
bin hier seid einiger Zeit stiller Leser und finde Eure Postings sehr interessant.
Mich würde Eure Meinung zu Celldex interessieren.
Siehe hierzu folgender Link.
http://www.wallstreet-online.de/diskussion/1157067-1-10/cell…
Celldex hat wie Roche auch einen Antikörper gegen Brustkrebs in der Entwicklung. Hierzu wird die Technologie von SGEN genutzt.
Im Vergleich zu T-DM1 von Roche noch folgende Info aus dem Yahoo board.
The best benchmark for assessing CR-011 is Roche’s T-DM1, which is also an antibody drug conjugate (comprises of Herceptin and a drug payload).
The two agents have a lot in common. Both utilize validated ADC technologies; CR-011 employs Seattle Genetics’ (SGEN) technology whereas T-DM1 employs Immunogen’s (IMGN) technology. Furthermore, the targets for CR-011 and T-DM1 (GPNMB and Her2, respectively) are found on a minority of breast cancer patients and are associated with disease aggressiveness and poor outcome. Lastly, according to Timothy Shannon, Curagen’s CEO, the company patented GPNMB as a target, similarly to what Genentech/Roche did with Her2. This strategy enabled Roche to be the sole company who can sell anti-Her2 antibodies
GPNMB is in fact expressed in ~70-75% of all breast cancers, regardless of subtype. It is expressed in 2 different cell types within the tumors: epithelial cells (the actual cancer cells) and stromal cells (cells that support the tumor). The vast majority (85%) of GPNMB-positive breast tumors only have GPNMB expression in stromal cells - and these GPNMB-positive tumors are equally represented across subtypes (Her2+, ER/PR+, triple negative).
However, the remaining 15% of GPNMB-pos tumors have GPNMB expressed in the epithelium (cancer cells) - and this group of GPNMB-positive tumors are highly over-represented in the triple negative subtype. Within triple negative tumors, those that have GPNMB expression in the epithelium are much more likely to metastasize and need targeted therapies.
The thing is, CDX-011 will target and kill stromal and epithelial cells. In fact much of its efficacy may lie in the fact that it targets stromal cells (for example, all of the anti-angiogenic therapies out there target the stromal cells).
So, yes CDX-011 represents an exciting therapy for triple negative breast cancer, but it will likely also be effective against the other subtypes, including Her2+, though in this subtype it may not be as effective as T-DM1.
Absolutely, it is still very early on in the CDX-011 trials; in fact as a single agent therapy in heavily pre-treated metastatic breast cancer - the results from CDX-011 are really quite striking in GPNMB-positive patients. But still not as striking as T-DM1 in Her2+ patients. However there is room for improvement with CDX-011, (ie. give once per wk vs. once every 3 weeks; try combining it with traditional chemotherapies). And for that we will have to wait and see.
Meine Frage: Wie schätzt Ihr Celldex mittel- und langfristig ein.
Gruß
wachholder
zu SGEN:
BOTHELL, Wash.--(BUSINESS WIRE)--Seattle Genetics, Inc. (Nasdaq: SGEN) today announced that Genentech, Inc., a wholly owned member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), will pay $9.5 million to renew exclusive licenses to specific targets and extend the research term under the parties’ existing antibody-drug conjugate (ADC) collaboration agreement. Under the terms of the agreement, Genentech has rights to use Seattle Genetics’ ADC technology with antibodies against targets selected by Genentech. Genentech is responsible for research, product development, manufacturing and commercialization. Seattle Genetics is entitled to receive fees, progress-dependent milestone payments and royalties on net sales of any resulting ADC products.
.“We believe this collaboration, coupled with our multiple other ADC collaborations, reinforces the value of our industry-leading technology,” said Eric L. Dobmeier, Chief Business Officer of Seattle Genetics. “For Seattle Genetics, these collaborations broaden the reach of our technology into diverse disease settings where empowered antibodies may be able to address significant unmet medical needs. In addition, these deals provide a substantial source of capital as we continue to advance our own pipeline of proprietary antibodies and ADCs for cancer.”
Seattle Genetics has generated approximately $120 million through ADC technology license agreements with leading biotechnology and pharmaceutical companies. ADCs utilize the targeting ability of monoclonal antibodies to deliver potent, cell-killing payloads to specific cells. Seattle Genetics’ proprietary technology employs synthetic, highly potent drugs attached to antibodies through stable linker systems. The linkers are designed to release the drug payload only under specific conditions once inside target cells, thereby sparing non-target cells many of the toxic effects of traditional chemotherapy.
Solche Nachrichten sind immer gern gesehen. Es zeigt ein mal mehr, dass SGEN über eine echte Plattform-Technik verfügt, auf der stabile Partnerschaften begründet sind:
BOTHELL, Wash.--(BUSINESS WIRE)--Seattle Genetics, Inc. (Nasdaq: SGEN) today announced that Genentech, Inc., a wholly owned member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), will pay $9.5 million to renew exclusive licenses to specific targets and extend the research term under the parties’ existing antibody-drug conjugate (ADC) collaboration agreement. Under the terms of the agreement, Genentech has rights to use Seattle Genetics’ ADC technology with antibodies against targets selected by Genentech. Genentech is responsible for research, product development, manufacturing and commercialization. Seattle Genetics is entitled to receive fees, progress-dependent milestone payments and royalties on net sales of any resulting ADC products.
.“We believe this collaboration, coupled with our multiple other ADC collaborations, reinforces the value of our industry-leading technology,” said Eric L. Dobmeier, Chief Business Officer of Seattle Genetics. “For Seattle Genetics, these collaborations broaden the reach of our technology into diverse disease settings where empowered antibodies may be able to address significant unmet medical needs. In addition, these deals provide a substantial source of capital as we continue to advance our own pipeline of proprietary antibodies and ADCs for cancer.”
Seattle Genetics has generated approximately $120 million through ADC technology license agreements with leading biotechnology and pharmaceutical companies. ADCs utilize the targeting ability of monoclonal antibodies to deliver potent, cell-killing payloads to specific cells. Seattle Genetics’ proprietary technology employs synthetic, highly potent drugs attached to antibodies through stable linker systems. The linkers are designed to release the drug payload only under specific conditions once inside target cells, thereby sparing non-target cells many of the toxic effects of traditional chemotherapy.
Solche Nachrichten sind immer gern gesehen. Es zeigt ein mal mehr, dass SGEN über eine echte Plattform-Technik verfügt, auf der stabile Partnerschaften begründet sind:
meine kleinsten Positionen habe ich glattgestellt... Addex, SCMP und PGNX verkauft. Dafür neu eine kleine Position JAZZ.

11,6% Incyte http://finance.yahoo.com/q?s=INCY
8,9% Regeneron http://finance.yahoo.com/q?s=REGN
7,3% Cubist http://finance.yahoo.com/q?s=CBST
6,2% Allos http://finance.yahoo.com/q?s=ALTH
5,7% NeuroSearch http://finance.yahoo.com/q?s=NEUR.CO
5,3% ArQule http://finance.yahoo.com/q?s=ARQL
5,2% Genmab http://finance.yahoo.com/q?s=GEN.CO
5,1% Micromet http://finance.yahoo.com/q?s=MITI
5,1% Onyx http://finance.yahoo.com/q?s=ONXX
4,4% Rigel http://finance.yahoo.com/q?s=RIGL
4,4% Isis http://finance.yahoo.com/q?s=ISIS
4,4% Exelixis http://finance.yahoo.com/q?s=EXEL
4,0% Array http://finance.yahoo.com/q?s=ARRY
3,9% NeurogesX http://finance.yahoo.com/q?s=NGSX
3,7% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,5% AMAG http://finance.yahoo.com/q?s=AMAG
2,9% JAZZ http://finance.yahoo.com/q?s=JAZZ
2,9% ViroPharma http://finance.yahoo.com/q?s=VPHM
2,3% Medigene http://finance.yahoo.com/q?s=MDG.DE
1,7% Arena http://finance.yahoo.com/q?s=ARNA
1,5% NicOx http://finance.yahoo.com/q?s=COX.PA
Veränderung seit Anfang des Jahres auf EUR-Basis...
23,5% Depot
21,0% NBI
5,1% Dax
7,2% USD
+131,2% NeuroSearch ( 5,7% )
+80,6% ArQule ( 5,3% )
+76,0% ViroPharma ( 2,9% )
+64,5% Incyte ( 11,6% )
+53,3% Array ( 4,0% )
+36,5% Allos ( 6,2% )
+35,7% JAZZ ( 2,9% )
+29,7% Cubist ( 7,3% )
+29,2% Micromet ( 5,1% )
+26,5% NeurogesX ( 3,9% )
+21,1% Seattle Genetics ( 3,7% )
+11,4% Regeneron ( 8,9% )
+9,8% NicOx ( 1,5% )
+9,7% Onyx ( 5,1% )
+4,2% Isis ( 4,4% )
-1,4% AMAG ( 3,5% )
-4,0% Arena ( 1,7% )
-6,8% Rigel ( 4,4% )
-10,7% Exelixis ( 4,4% )
-15,9% Genmab ( 5,2% )
-16,8% Medigene ( 2,3% )
mfg ipollit
11,6% Incyte http://finance.yahoo.com/q?s=INCY
8,9% Regeneron http://finance.yahoo.com/q?s=REGN
7,3% Cubist http://finance.yahoo.com/q?s=CBST
6,2% Allos http://finance.yahoo.com/q?s=ALTH
5,7% NeuroSearch http://finance.yahoo.com/q?s=NEUR.CO
5,3% ArQule http://finance.yahoo.com/q?s=ARQL
5,2% Genmab http://finance.yahoo.com/q?s=GEN.CO
5,1% Micromet http://finance.yahoo.com/q?s=MITI
5,1% Onyx http://finance.yahoo.com/q?s=ONXX
4,4% Rigel http://finance.yahoo.com/q?s=RIGL
4,4% Isis http://finance.yahoo.com/q?s=ISIS
4,4% Exelixis http://finance.yahoo.com/q?s=EXEL
4,0% Array http://finance.yahoo.com/q?s=ARRY
3,9% NeurogesX http://finance.yahoo.com/q?s=NGSX
3,7% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
3,5% AMAG http://finance.yahoo.com/q?s=AMAG
2,9% JAZZ http://finance.yahoo.com/q?s=JAZZ
2,9% ViroPharma http://finance.yahoo.com/q?s=VPHM
2,3% Medigene http://finance.yahoo.com/q?s=MDG.DE
1,7% Arena http://finance.yahoo.com/q?s=ARNA
1,5% NicOx http://finance.yahoo.com/q?s=COX.PA
Veränderung seit Anfang des Jahres auf EUR-Basis...
23,5% Depot
21,0% NBI
5,1% Dax
7,2% USD
+131,2% NeuroSearch ( 5,7% )
+80,6% ArQule ( 5,3% )
+76,0% ViroPharma ( 2,9% )
+64,5% Incyte ( 11,6% )
+53,3% Array ( 4,0% )
+36,5% Allos ( 6,2% )
+35,7% JAZZ ( 2,9% )
+29,7% Cubist ( 7,3% )
+29,2% Micromet ( 5,1% )
+26,5% NeurogesX ( 3,9% )
+21,1% Seattle Genetics ( 3,7% )
+11,4% Regeneron ( 8,9% )
+9,8% NicOx ( 1,5% )
+9,7% Onyx ( 5,1% )
+4,2% Isis ( 4,4% )
-1,4% AMAG ( 3,5% )
-4,0% Arena ( 1,7% )
-6,8% Rigel ( 4,4% )
-10,7% Exelixis ( 4,4% )
-15,9% Genmab ( 5,2% )
-16,8% Medigene ( 2,3% )
mfg ipollit
Antwort auf Beitrag Nr.: 39.366.609 von wachholder am 20.04.10 12:28:38Celldex...
ich habe mich bisher von Immuntherapien gegen Krebs ferngehalten, da schon viele gescheitert sind. Bei Celldex sind diese eine wesentlicher Teil der Entwicklung, weshalb ich auch hier vorsichtig bin. Allerdings scheint laut dem Hammer Blog CR-011 interessant zu sein: http://www.hammerstockblog.com/curagen-%e2%80%93-positive-re… Vielleicht wird das ja was. Vorerst bin ich allerdings nicht dabei.
mfg ipollit
ich habe mich bisher von Immuntherapien gegen Krebs ferngehalten, da schon viele gescheitert sind. Bei Celldex sind diese eine wesentlicher Teil der Entwicklung, weshalb ich auch hier vorsichtig bin. Allerdings scheint laut dem Hammer Blog CR-011 interessant zu sein: http://www.hammerstockblog.com/curagen-%e2%80%93-positive-re… Vielleicht wird das ja was. Vorerst bin ich allerdings nicht dabei.
mfg ipollit
ipollit,
ja, da gebe ich Dir Recht. Bei dem Versuch einen Krebsimpfstoff durch alle klinischen Phasen zu bringen und das OK von der FDA zu bekommen, sind schon viele Firmen gescheitert.
Wenn es allerdings gelingt, siehe Dendreon, kann man als Aktionär sehr profitieren.
Celldex verwendet eine spezifische Technologie und man konzentriert sich dabei auf ganz bestimmte Patientengruppen.
Bei Brustkrebs nämlich nur auf Patienten mit GPNMB expression.
Ähnlich geht man bei CDX-110 gegen Gerhirntumor vor.
Celldex hat 12 Produkte in der Pipeline und cash bis 2012.
Wenn CDX-110, welches in Zusammenarbeit mit Pfizer entwickelt wird, den Markteintritt schaffen sollte, fließen weitere 390 Mio EUR von Pfizer zu.
Ich denke man hat hier einen Biotechwert mit einem guten Chancen-Risken Profil.
Gruß
wachholder
ja, da gebe ich Dir Recht. Bei dem Versuch einen Krebsimpfstoff durch alle klinischen Phasen zu bringen und das OK von der FDA zu bekommen, sind schon viele Firmen gescheitert.
Wenn es allerdings gelingt, siehe Dendreon, kann man als Aktionär sehr profitieren.
Celldex verwendet eine spezifische Technologie und man konzentriert sich dabei auf ganz bestimmte Patientengruppen.
Bei Brustkrebs nämlich nur auf Patienten mit GPNMB expression.
Ähnlich geht man bei CDX-110 gegen Gerhirntumor vor.
Celldex hat 12 Produkte in der Pipeline und cash bis 2012.
Wenn CDX-110, welches in Zusammenarbeit mit Pfizer entwickelt wird, den Markteintritt schaffen sollte, fließen weitere 390 Mio EUR von Pfizer zu.
Ich denke man hat hier einen Biotechwert mit einem guten Chancen-Risken Profil.
Gruß
wachholder
@ipollit, wachholder,
wärt ihr bereit, die Besonderheit bzw. das Wirkprinzip dieser Immuntherapie zu beschreiben?
Nehmen wir den CR-011 von Celldex (bzw. früher von Curagen) - das ist ein ADC - wie das geht und wirkt, habe ich verstanden.
Der CDX 110 ist nun ja aber wohl eine "Immuntherapie", also doch offensichtlich ein ganz anderes Wirkprinzip. Die Frage ist nun also: Wie funktioniert das? Und insbesondere, inwiefern unterscheidet sich das von normalen mabs oder ADCs?
wärt ihr bereit, die Besonderheit bzw. das Wirkprinzip dieser Immuntherapie zu beschreiben?
Nehmen wir den CR-011 von Celldex (bzw. früher von Curagen) - das ist ein ADC - wie das geht und wirkt, habe ich verstanden.
Der CDX 110 ist nun ja aber wohl eine "Immuntherapie", also doch offensichtlich ein ganz anderes Wirkprinzip. Die Frage ist nun also: Wie funktioniert das? Und insbesondere, inwiefern unterscheidet sich das von normalen mabs oder ADCs?
Antwort auf Beitrag Nr.: 39.372.387 von SLGramann am 21.04.10 09:01:46@SL Gramann,
folgendes zu CDX-110
All the diseases that attack brain have very frightful effects. Not only they could cause permanent disabilities, they also could cause death. For example, brain cancer, which attacks the controlling function of the body nerves.
A specific vaccine has been developed to prevent brain cancer. This vaccine is
in use in an experiment on Karen Vaneman, a brain cancer patient from Preston Robert Tisch Brain Tumor Center at Duke University. She had to give 21 small bottles of her blood during the first week of every month.
Vaneman was diagnosed with brain cancer two years ago. She suffered symptoms such as weakness and paralyzed left hand and was brought to the emergency room. At that time, the doctor diagnosed her as having a stroke
and heart attack. Later they found out that she had Glioblastoma or GBM which is one of most common brain cancers.
Despite GBM brain cancer being very deathly, a team from Preston Robert Tisch had a different way to cure Vaneman. They operated on her and now receives intensive treatments after the operation.
“Other doctors might have told her that she only has six or nine months, or maybe one year to live, since GBM had no cure. But we told her that she could heal, with hard working and her self-belief,” said Dr. Henry Friedman, head of Preston Robert Tisch Brain Tumor Center.
Vaneman received new vaccine treatment that was produced by Pfizer. The new vaccine is called CDX-110. It has been produced not to prevent the cancer but to trigger body-immune responses, which could hit the cancer. The vaccine itself is extracted from EGFRviii (third factor of EGFR)—the protein
found in 40 percent of tumor cells.
Dr. John Sampson, a surgeon who helped to develop the vaccine, explained that the team made the vaccine immune response to be more specific.
\"Unlike chemotherapy or radiation which makes the patient felt very sick because it divides cells in the body, this vaccine is effective and has more appropriate precision,\" he said.
The vaccine could make brain cancer patient to live longer. It also could reduce or diminish the cancer cells in less painful way, comparing to chemotherapy.
Treatment of brain cancer is very complex. The treatment for a brain tumor should be individual to the patient, based on the age and general health, and the size, location and type of the tumor. Most treatment usually involves a team of doctors and includes neurosurgeons (specialists in the brain and
nervous system), oncologists, radiation oncologists (doctors who practice radiation therapy). The team will also include a dietitian, a social worker, a physiotherapist and, probably, other specialists.
Our lead clinical development program, CDX-110, is an immunotherapy that targets the tumor specific molecule called EGFRvIII, a functional variant of the epidermal growth factor receptor (EGFR). EGFRvIII is a common mutated form of the natural EGFR and is present in multiple cancer types. Unlike EGFR, EGFRvIII has not been detected at a significant level in normal tissues, and it can directly lead to cancer through its well documented oncogenic properties that provide a constant growth signal to tumor cells which bear it. Consequently, cells producing EGFRvIII have an enhanced capacity for unregulated growth. CDX-110 is designed to activate the patient’s immune response against tumor cells specifically expressing the EGFRvIII mutation.
Gruß
wachholder
folgendes zu CDX-110
All the diseases that attack brain have very frightful effects. Not only they could cause permanent disabilities, they also could cause death. For example, brain cancer, which attacks the controlling function of the body nerves.
A specific vaccine has been developed to prevent brain cancer. This vaccine is
in use in an experiment on Karen Vaneman, a brain cancer patient from Preston Robert Tisch Brain Tumor Center at Duke University. She had to give 21 small bottles of her blood during the first week of every month.
Vaneman was diagnosed with brain cancer two years ago. She suffered symptoms such as weakness and paralyzed left hand and was brought to the emergency room. At that time, the doctor diagnosed her as having a stroke
and heart attack. Later they found out that she had Glioblastoma or GBM which is one of most common brain cancers.
Despite GBM brain cancer being very deathly, a team from Preston Robert Tisch had a different way to cure Vaneman. They operated on her and now receives intensive treatments after the operation.
“Other doctors might have told her that she only has six or nine months, or maybe one year to live, since GBM had no cure. But we told her that she could heal, with hard working and her self-belief,” said Dr. Henry Friedman, head of Preston Robert Tisch Brain Tumor Center.
Vaneman received new vaccine treatment that was produced by Pfizer. The new vaccine is called CDX-110. It has been produced not to prevent the cancer but to trigger body-immune responses, which could hit the cancer. The vaccine itself is extracted from EGFRviii (third factor of EGFR)—the protein
found in 40 percent of tumor cells.
Dr. John Sampson, a surgeon who helped to develop the vaccine, explained that the team made the vaccine immune response to be more specific.
\"Unlike chemotherapy or radiation which makes the patient felt very sick because it divides cells in the body, this vaccine is effective and has more appropriate precision,\" he said.
The vaccine could make brain cancer patient to live longer. It also could reduce or diminish the cancer cells in less painful way, comparing to chemotherapy.
Treatment of brain cancer is very complex. The treatment for a brain tumor should be individual to the patient, based on the age and general health, and the size, location and type of the tumor. Most treatment usually involves a team of doctors and includes neurosurgeons (specialists in the brain and
nervous system), oncologists, radiation oncologists (doctors who practice radiation therapy). The team will also include a dietitian, a social worker, a physiotherapist and, probably, other specialists.
Our lead clinical development program, CDX-110, is an immunotherapy that targets the tumor specific molecule called EGFRvIII, a functional variant of the epidermal growth factor receptor (EGFR). EGFRvIII is a common mutated form of the natural EGFR and is present in multiple cancer types. Unlike EGFR, EGFRvIII has not been detected at a significant level in normal tissues, and it can directly lead to cancer through its well documented oncogenic properties that provide a constant growth signal to tumor cells which bear it. Consequently, cells producing EGFRvIII have an enhanced capacity for unregulated growth. CDX-110 is designed to activate the patient’s immune response against tumor cells specifically expressing the EGFRvIII mutation.
Gruß
wachholder
Celldex mit News!
http://finance.yahoo.com/news/Celldex-Presents-Data-for-bw-3…
Ein weiterer wichtiger Schritt für die Firma Celldex.
Gruß
wachholder
http://finance.yahoo.com/news/Celldex-Presents-Data-for-bw-3…
Ein weiterer wichtiger Schritt für die Firma Celldex.
Gruß
wachholder
CBST... mittelfristig sollen die Umsätze mit Cubicin weiter steigen. Interessant ist, wie sich die Pipeline in der nächsten Zeit entwickelt
Cubist Pharmaceuticals' 1Q profit nearly triples
The Associated Press April 15, 2010, 6:26PM ET
Cubist Pharmaceuticals Inc. said Thursday that its first-quarter profit nearly tripled amid strong sales of its antibiotic Cubicin.
Net income for the first three months of 2010 was $20.4 million, or 34 cents per share, compared with $7.8 million, or 13 cents per share, for the same period a year ago, when the company recorded a $20 million charge for research and development.
Excluding one-time items, Cubist reported income of 61 cents per share, up from 53 cents per share in the 2009 quarter.
Revenue climbed to $144.1 million from $121.1 million. That consisted almost entirely of Cubicin sales, which rose 20 percent to $135.3 million.
Analysts predicted income of 35 cents per share per share on revenue of $150.3 million, according to a survey by Thomson Reuters.
The company confirmed its guidance for 2010 revenue of $627 million to $652 million.
*********
Needham & Company Reiterates a 'Buy' on Cubist Pharmaceuticals (CBST); Focused on Upcoming Pipeline News
April 16, 2010 9:59 AM EDT
Needham & Company reiterates a 'Buy' rating on Cubist Pharmaceuticals (Nasdaq: CBST), price target $27.
Needham analyst says, "We continue to believe that investors should focus on the pipeline over the next several months. We expect news flow from the CXA and CB-804 programs to provide perspective as to how well Cubist can build a revenue stream behind Cubicin. Results from the CB-804 Phase 1 trial (a significant de-risking event for antibiotics) and from the Phase 2 cUTI CXA-101 trial are expected by mid-2010. Both programs have peak sales potential comparable to that of Cubicin and are only minimally reflected in the stock.
mfg ipollit
Cubist Pharmaceuticals' 1Q profit nearly triples
The Associated Press April 15, 2010, 6:26PM ET
Cubist Pharmaceuticals Inc. said Thursday that its first-quarter profit nearly tripled amid strong sales of its antibiotic Cubicin.
Net income for the first three months of 2010 was $20.4 million, or 34 cents per share, compared with $7.8 million, or 13 cents per share, for the same period a year ago, when the company recorded a $20 million charge for research and development.
Excluding one-time items, Cubist reported income of 61 cents per share, up from 53 cents per share in the 2009 quarter.
Revenue climbed to $144.1 million from $121.1 million. That consisted almost entirely of Cubicin sales, which rose 20 percent to $135.3 million.
Analysts predicted income of 35 cents per share per share on revenue of $150.3 million, according to a survey by Thomson Reuters.
The company confirmed its guidance for 2010 revenue of $627 million to $652 million.
*********
Needham & Company Reiterates a 'Buy' on Cubist Pharmaceuticals (CBST); Focused on Upcoming Pipeline News
April 16, 2010 9:59 AM EDT
Needham & Company reiterates a 'Buy' rating on Cubist Pharmaceuticals (Nasdaq: CBST), price target $27.
Needham analyst says, "We continue to believe that investors should focus on the pipeline over the next several months. We expect news flow from the CXA and CB-804 programs to provide perspective as to how well Cubist can build a revenue stream behind Cubicin. Results from the CB-804 Phase 1 trial (a significant de-risking event for antibiotics) and from the Phase 2 cUTI CXA-101 trial are expected by mid-2010. Both programs have peak sales potential comparable to that of Cubicin and are only minimally reflected in the stock.
mfg ipollit
NicOx... vor dem 12. Mai wird man erfahren, was die FDA von Naproxcinod hält. Ende Juli könnte es dann die Zulassung geben.
http://www.businessweek.com/news/2010-04-21/nicox-shares-ris…" target="_blank" rel="nofollow ugc noopener">http://www.businessweek.com/news/2010-04-21/nicox-shares-ris…
NicOx Rises; Natixis Sees Promise in Arthritis Drug (Update1)
April 21, 2010, 12:02 PM EDT
By Phil Serafino
April 21 (Bloomberg) -- NicOx SA rose the most in 13 months in Paris trading after Natixis Securities said investors are underestimating the sales potential of the French company’s arthritis drug candidate.
NicOx rose 93 cents, or 15 percent, to close at 7.33 euros, giving the company a market value of 529 million euros ($707.7 million). The gain was the biggest since March 26, 2009.
An advisory committee on May 12 will give a recommendation to the U.S. Food and Drug Administration on NicOx’s naproxcinod, and the agency will decide whether to approve the treatment by July 24, Sebastien Malafosse, a Paris-based analyst for Natixis, wrote in a report today. A favorable recommendation by the panel is tantamount to approval and would boost the shares, he said.
“The market does not think that the NicOx product will become a blockbuster,” the analyst wrote, referring to the industry term for a medicine that generates at least $1 billion a year in revenue. Malafosse predicts peak sales of 1.3 billion euros by 2017 if the FDA allows the company to promote the drug as safe for the heart, Malafosse wrote.
He raised his rating on NicOx’s stock to “buy” from “reduce.” The stock may reach 13 euros, higher than his previous prediction of 10.50 euros, Malafosse wrote.
mfg ipollit
http://www.businessweek.com/news/2010-04-21/nicox-shares-ris…" target="_blank" rel="nofollow ugc noopener">http://www.businessweek.com/news/2010-04-21/nicox-shares-ris…
NicOx Rises; Natixis Sees Promise in Arthritis Drug (Update1)
April 21, 2010, 12:02 PM EDT
By Phil Serafino
April 21 (Bloomberg) -- NicOx SA rose the most in 13 months in Paris trading after Natixis Securities said investors are underestimating the sales potential of the French company’s arthritis drug candidate.
NicOx rose 93 cents, or 15 percent, to close at 7.33 euros, giving the company a market value of 529 million euros ($707.7 million). The gain was the biggest since March 26, 2009.
An advisory committee on May 12 will give a recommendation to the U.S. Food and Drug Administration on NicOx’s naproxcinod, and the agency will decide whether to approve the treatment by July 24, Sebastien Malafosse, a Paris-based analyst for Natixis, wrote in a report today. A favorable recommendation by the panel is tantamount to approval and would boost the shares, he said.
“The market does not think that the NicOx product will become a blockbuster,” the analyst wrote, referring to the industry term for a medicine that generates at least $1 billion a year in revenue. Malafosse predicts peak sales of 1.3 billion euros by 2017 if the FDA allows the company to promote the drug as safe for the heart, Malafosse wrote.
He raised his rating on NicOx’s stock to “buy” from “reduce.” The stock may reach 13 euros, higher than his previous prediction of 10.50 euros, Malafosse wrote.
mfg ipollit
JAZZ... etwas zu Xyrem bei Fibromyalgie, bei dem am 11.10. die Zulassungsentscheidung der FDA ansteht (bisher nur gegen Narkolepsie zugelassen)
http://chronic-illness-treatments.suite101.com/article.cfm/x…
Xyrem for Fibromyalgia
Sodium Oxybate is a Sleep Aid that Increases Growth Hormone
Mar 10, 2010 Maija Haavisto
Fibromyalgia is strongly associated with poor sleep. Some doctors even believe it is the cause of fibromyalgia pain, as sleep deprivation is associated with increased secretion of inflammatory cytokines like IL-6, which can cause pain and other symptoms. IL-6 in particular is associated with hyperalgesia, or increased sensitivity to pain.
Whether or not fibromyalgia is essentially a sleep disorder, impaired sleep can certainly worsen pain, fatigue and other symptoms like depression. Jazz Pharmaceuticals has applied for FDA approval for a drug called Xyrem (JZP-6) in fibromyalgia. It is a novel kind of sleep aid with some interesting properties.
What Is Xyrem?
Sodium oxybate is the sodium salt of gammahydroxybutyric acid or GHB, a substance naturally present in the body. It works through GHB receptors and possibly GABAB receptors (many sleep aids like benzodiazepines bind to GABAA receptors). It has been used in medicine for decades, but recreational use and other abuse has slowed its development.
Xyrem is currently indicated in the treatment of narcolepsy. It is also used off-label as a sleep aid and has helped people who have been refractory to all other treatments. It is known to have muscle relaxant and antidepressant properties.
Xyrem has been shown to increase secretion of growth hormone, which may explain why it is so effective in fibromyalgia. Many studies have shown that growth hormone secretion is low in fibromyalgia and that growth hormone supplementation can reduce symptoms. Growth hormone therapy, while safe and effective, is extremely expensive and has to be given by injection.
Several studies have been done of Xyrem in fibromyalgia, showing that not only it improves sleep, but also pain and fatigue levels. It is also being studied in chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME), which is similarly associated with a growth hormone deficiency.
Currently Xyrem is only available through a special program in the United States and is a Schedule III drug, but the company behind it, Jazz Pharmaceuticals, has applied for extending its indication to fibromyalgia. The FDA will have to make a decision by the end of 2010. In some countries Xyrem is available without any restrictions, but tends to be extremely expensive.
Xyrem Side Effects and Drug Interactions
Unlike e.g. benzodiazepines, sodium oxybate does not cause dependency or tolerance when used according to the instructions. It also does not usually produce morning grogginess or other after effects. It can cause nausea, confusion, headache, tremor and dizziness, but is usually well-tolerated.
Sodium oxybate can dangerously potentiate the effects of other sedative drugs, herbs and alcohol. Other than that it has no known drug interactions. It could thus be combined with other fibromyalgia treatments, such as Savella, Lyrica, Cymbalta or low dose naltrexone.
Alternatives to Xyrem
Those who cannot get sodium oxybate may have a similar alternative in baclofen (Lioresal). It is classified as a muscle relaxant, but also works well to improve sleep. Baclofen appears to have somewhat similar mode of action to sodium oxybate and has also been shown to increase growth hormone secretion.
Baclofen has never been studied in fibromyalgia, but many doctors prescribe it for this use. It has also been used in the treatment of e.g. neurological symptoms, acid reflux (GERD), trigeminal neuralgia, tinnitus, hyperacusis and nystagmus. It may also improve tolerance to heat and cold.
mfg ipollit
http://chronic-illness-treatments.suite101.com/article.cfm/x…
Xyrem for Fibromyalgia
Sodium Oxybate is a Sleep Aid that Increases Growth Hormone
Mar 10, 2010 Maija Haavisto
Fibromyalgia is strongly associated with poor sleep. Some doctors even believe it is the cause of fibromyalgia pain, as sleep deprivation is associated with increased secretion of inflammatory cytokines like IL-6, which can cause pain and other symptoms. IL-6 in particular is associated with hyperalgesia, or increased sensitivity to pain.
Whether or not fibromyalgia is essentially a sleep disorder, impaired sleep can certainly worsen pain, fatigue and other symptoms like depression. Jazz Pharmaceuticals has applied for FDA approval for a drug called Xyrem (JZP-6) in fibromyalgia. It is a novel kind of sleep aid with some interesting properties.
What Is Xyrem?
Sodium oxybate is the sodium salt of gammahydroxybutyric acid or GHB, a substance naturally present in the body. It works through GHB receptors and possibly GABAB receptors (many sleep aids like benzodiazepines bind to GABAA receptors). It has been used in medicine for decades, but recreational use and other abuse has slowed its development.
Xyrem is currently indicated in the treatment of narcolepsy. It is also used off-label as a sleep aid and has helped people who have been refractory to all other treatments. It is known to have muscle relaxant and antidepressant properties.
Xyrem has been shown to increase secretion of growth hormone, which may explain why it is so effective in fibromyalgia. Many studies have shown that growth hormone secretion is low in fibromyalgia and that growth hormone supplementation can reduce symptoms. Growth hormone therapy, while safe and effective, is extremely expensive and has to be given by injection.
Several studies have been done of Xyrem in fibromyalgia, showing that not only it improves sleep, but also pain and fatigue levels. It is also being studied in chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME), which is similarly associated with a growth hormone deficiency.
Currently Xyrem is only available through a special program in the United States and is a Schedule III drug, but the company behind it, Jazz Pharmaceuticals, has applied for extending its indication to fibromyalgia. The FDA will have to make a decision by the end of 2010. In some countries Xyrem is available without any restrictions, but tends to be extremely expensive.
Xyrem Side Effects and Drug Interactions
Unlike e.g. benzodiazepines, sodium oxybate does not cause dependency or tolerance when used according to the instructions. It also does not usually produce morning grogginess or other after effects. It can cause nausea, confusion, headache, tremor and dizziness, but is usually well-tolerated.
Sodium oxybate can dangerously potentiate the effects of other sedative drugs, herbs and alcohol. Other than that it has no known drug interactions. It could thus be combined with other fibromyalgia treatments, such as Savella, Lyrica, Cymbalta or low dose naltrexone.
Alternatives to Xyrem
Those who cannot get sodium oxybate may have a similar alternative in baclofen (Lioresal). It is classified as a muscle relaxant, but also works well to improve sleep. Baclofen appears to have somewhat similar mode of action to sodium oxybate and has also been shown to increase growth hormone secretion.
Baclofen has never been studied in fibromyalgia, but many doctors prescribe it for this use. It has also been used in the treatment of e.g. neurological symptoms, acid reflux (GERD), trigeminal neuralgia, tinnitus, hyperacusis and nystagmus. It may also improve tolerance to heat and cold.
mfg ipollit
ONXX profitiert davon, dass Pfizers Sutent als direkter Konkurrent zu Nexavar offensichtlich Nexavar in der wichtigen Indikation Leberkrebs unterlegen ist...
Nexavar könnte vielleicht demnächst bei Lungenkrebs erfolgreich sein, wo die Erwartungen bisher bei Null liegen. Außerdem gibt es Anzeichen dafür, dass Nexavar bei Brustkrebs wirksam ist.
Pfizer ends trial of Sutent in liver cancer
Pfizer says Sutent was not working better than Bayer's Nexavar as a treatment for liver cancer
On Thursday April 22, 2010, 9:10 pm EDT
NEW YORK (AP) -- Pfizer Inc. said Thursday it stopped a late-stage clinical study of its drug Sutent as a treatment for liver cancer.
Pfizer said patients who were treated with Sutent were not surviving as long as patients who were treated with another drug, Bayer Healthcare's and Onyx Pharmaceuticals Inc.'s Nexavar. Patients on Sutent were also more likely to suffer serious side effects, the company said.
The study was designed to measure if Sutent was more effective than Nexavar, or as effective, in treating the most common type of liver cancer, hepatocellular carcinoma. Pfizer said Sutent was not meeting either goal. It said the side effects were not new or unexpected.
In March, Pfizer said Sutent did not meet its main goals in two late-stage studies that tested it as a treatment for advanced breast cancer. Sutent, or sunitinib, is currently on the market as a treatment for advanced kidney cancer and gastrointestinal stromal cancer.
Pfizer said it is running late stage trials of Sutent as a treatment for advanced non-small cell lung cancer, advanced castration-resistant prostate cancer, and as an adjuvant treatment in kidney cancer.
mfg ipollit
Nexavar könnte vielleicht demnächst bei Lungenkrebs erfolgreich sein, wo die Erwartungen bisher bei Null liegen. Außerdem gibt es Anzeichen dafür, dass Nexavar bei Brustkrebs wirksam ist.
Pfizer ends trial of Sutent in liver cancer
Pfizer says Sutent was not working better than Bayer's Nexavar as a treatment for liver cancer
On Thursday April 22, 2010, 9:10 pm EDT
NEW YORK (AP) -- Pfizer Inc. said Thursday it stopped a late-stage clinical study of its drug Sutent as a treatment for liver cancer.
Pfizer said patients who were treated with Sutent were not surviving as long as patients who were treated with another drug, Bayer Healthcare's and Onyx Pharmaceuticals Inc.'s Nexavar. Patients on Sutent were also more likely to suffer serious side effects, the company said.
The study was designed to measure if Sutent was more effective than Nexavar, or as effective, in treating the most common type of liver cancer, hepatocellular carcinoma. Pfizer said Sutent was not meeting either goal. It said the side effects were not new or unexpected.
In March, Pfizer said Sutent did not meet its main goals in two late-stage studies that tested it as a treatment for advanced breast cancer. Sutent, or sunitinib, is currently on the market as a treatment for advanced kidney cancer and gastrointestinal stromal cancer.
Pfizer said it is running late stage trials of Sutent as a treatment for advanced non-small cell lung cancer, advanced castration-resistant prostate cancer, and as an adjuvant treatment in kidney cancer.
mfg ipollit
Antwort auf Beitrag Nr.: 39.392.571 von ipollit am 23.04.10 16:21:16ONXX... hier auch noch etwas zu Nexavar und Lungenkrebs. Gerade bei Vorhandensein einer KRAS-Mutation scheint Nexavar eine Wirkung zu haben.
http://www.businessweek.com/news/2010-04-19/onyx-anti-tumor-…
Onyx Drug Shows Broader Potential in Cancer Studies (Update3)
April 19, 2010, 4:18 PM EDT
By Ellen Gibson
April 19 (Bloomberg) -- Onyx Pharmaceuticals Inc. and Bayer AG’s Nexavar for liver and kidney tumors may have broader usefulness, according to a study testing how gene mutations affect drug effectiveness.
The study found that Nexavar prevented lung-cancer from progressing in 61 percent of patients with KRAS mutations. The research, presented yesterday at the meeting of the American Association for Cancer Research in Washington, was designed to show how gene testing of tumors can improve treatment when drugs are matched with biomarkers that predict their success.
Nexavar, not currently approved for lung tumors, showed the highest disease control of the four drugs used in the study of lung-cancer patients. The results from the study, dubbed BATTLE, were surprising because Nexavar had not worked in a previous trial of lung-cancer patients.
“BATTLE arguably does indicate Nexavar has activity in this disease,” said Christopher Raymond, an analyst at Robert W. Baird & Co., in a note to investors.
Not only does this data provide more evidence that tailored treatments work better, it helps answer the question, “What drugs can we move forward with?” said thoracic oncologist Edward Kim, who led the study at the University of Texas M.D. Anderson Center. Nexavar “is approved in two tumor types already. It gives us some nice hypotheses for further trials.”
Onyx fell 51 cents, or 1.7 percent, to $29.62 at 4 p.m. in Nasdaq Stock Market composite trading.
Cancer Biomarkers
In the lung-cancer study, patients who had been treated with chemotherapy in the past underwent another biopsy. Tumor samples were tested for cancer biomarkers, including mutations to a gene called KRAS and EGFR, or epidermal growth factor receptor, a cell-signaling protein that causes cancer cells to grow and divide.
Patients were placed into five marker groups. A first subset of patients was assigned to one of four drugs without regard to their biomarkers. Then patients in a second wave were assigned to medicines based on their particular tumor biomarkers, taking into consideration how people in the first group with similar biomarkers were faring.
Overall disease control -- defined as “non-progression” of the cancer -- among all participants was 46 percent after eight weeks, Kim said. However, about 61 percent of patients with KRAS mutations had non-progression of their cancer when given Nexavar, Kim said in an Apr. 13 phone interview.
By comparison, patients with KRAS mutations assigned to Tarceva, a treatment designed to inhibit EGFR, had just 22 percent disease control. Tarceva is jointly sold by Roche Holding AG, based in Basel, Switzerland, and OSI Pharmaceuticals Inc., of Melville, New York.
Nexavar Surprise
Nexavar also showed the highest overall disease control of all the treatments in the study at 58 percent of patients. Other drugs used in the study were AstraZeneca Plc’s experimental Zactima and Eisai Co.’s Targretin.
The Nexavar results were a surprise, according to the M.D. Anderson researchers. Two years ago, Bayer and Onyx halted a trial of Nexavar because the medicine failed to help lung cancer patients live longer than standard treatment.
The KRAS mutation is found in 20 percent of lung-cancer patients and “nothing works,” said Roy Herbst, chief of thoracic oncology and co-author of the study.
“When we treated them with Nexavar, we saw they did very well,” said Kim, director of clinical research in thoracic oncology.
Last-Stage Testing
Nexavar, co-marketed by Leverkusen, Germany-based Bayer and Emeryville, California-based Onyx, is in the last stage of testing required for U.S. approval as a third-line monotherapy for lung cancer and as a first-line treatment in combination with chemotherapy, according to Onyx spokeswoman Lori Murray.
“We’re very pleased that KRAS-mutated patients responded favorably,” Murray said of today’s study. “Onyx and Bayer are evaluating to determine potential next steps in our lung cancer development program.”
Nexavar is approved for use in patients with kidney cancer and inoperable liver cancer, and worldwide sales of the drug were $844 million in 2009.
While this study shows Nexavar can work in lung-cancer patients as a third or fourth line of treatment, winning approval as a first-line treatment in combination with chemotherapy “would be more meaningful commercially,” said Howard Liang, an analyst at Leerink Swann in Boston.
Some doctors say they would like to see independent follow- up studies. “The results are surprisingly good,” said Anil Potti, an associate professor of medicine at Duke University, “but I hesitate to commit to the fact that this is a valuable drug in lung cancer yet.”
*******
https://commerce.us.reuters.com/purchase/showReportDetail.do…
ONXX: Analysis Ahead of NExUS Data – Favorable Risk/Reward
Brean Murray & Co., Inc
Summary:
We expect top-line data from the Phase 3 NExUS trial of Nexavar + cisplatin and gemcitabine in front-line NSCLC patients in mid-2010. Given investors’ very low expectations and the Street’s uniformly bearish view on the trial outcome (we believe no analyst currently assigns any value to the lung program), we believe the potential downside should be very limited, while potentially a positive outcome could provide significant upside given the huge unmet medical need in NSCLC (~1.2 million new cases and 1.1 million deaths worldwide in 2007), creating a favorable risk/reward heading into the data reporting. In our view, the outcome of the NExUS trial is hard to handicap. The bearish view on the NExUS trial (and the Nexavar lung program) is due to the failure and premature termination of a previous Phase 3 ESCAPE trial (Nexavar + carboplatin and paclitaxel, also in front-line NSCLC patients) in February 2008, due to the subset of squamous-cell patients showing a higher mortality rate in the triple combination arm than the chemo doublet arm. However, we remind investors that the NExUS trial outcome may differ because of the different chemo doublet used and the different patient mix in terms of histology types. In this note, we review the trial design of the NExUS trial in comparison with the ESCAPE trial and analyze the updated interim data of the ESCAPE trial. We believe there is a strong rationale for developing Nexavar in combination with chemotherapy for treating NSCLC based on the single agent activity and no increase in toxicity when in combination with chemotherapy. Additionally, although the NExUS and ESCAPE trials are similar in many ways, we view the exclusion of squamous-cell patients in the late half of the NExUS trial as a potentially positive factor, as squamous-cell histology has been proven to be a poor diagnostic factor for NSCLC in several trials, including a previous Phase 2 trial of Avastin and the Phase 3 ESCAPE trial.
mfg ipollit
http://www.businessweek.com/news/2010-04-19/onyx-anti-tumor-…
Onyx Drug Shows Broader Potential in Cancer Studies (Update3)
April 19, 2010, 4:18 PM EDT
By Ellen Gibson
April 19 (Bloomberg) -- Onyx Pharmaceuticals Inc. and Bayer AG’s Nexavar for liver and kidney tumors may have broader usefulness, according to a study testing how gene mutations affect drug effectiveness.
The study found that Nexavar prevented lung-cancer from progressing in 61 percent of patients with KRAS mutations. The research, presented yesterday at the meeting of the American Association for Cancer Research in Washington, was designed to show how gene testing of tumors can improve treatment when drugs are matched with biomarkers that predict their success.
Nexavar, not currently approved for lung tumors, showed the highest disease control of the four drugs used in the study of lung-cancer patients. The results from the study, dubbed BATTLE, were surprising because Nexavar had not worked in a previous trial of lung-cancer patients.
“BATTLE arguably does indicate Nexavar has activity in this disease,” said Christopher Raymond, an analyst at Robert W. Baird & Co., in a note to investors.
Not only does this data provide more evidence that tailored treatments work better, it helps answer the question, “What drugs can we move forward with?” said thoracic oncologist Edward Kim, who led the study at the University of Texas M.D. Anderson Center. Nexavar “is approved in two tumor types already. It gives us some nice hypotheses for further trials.”
Onyx fell 51 cents, or 1.7 percent, to $29.62 at 4 p.m. in Nasdaq Stock Market composite trading.
Cancer Biomarkers
In the lung-cancer study, patients who had been treated with chemotherapy in the past underwent another biopsy. Tumor samples were tested for cancer biomarkers, including mutations to a gene called KRAS and EGFR, or epidermal growth factor receptor, a cell-signaling protein that causes cancer cells to grow and divide.
Patients were placed into five marker groups. A first subset of patients was assigned to one of four drugs without regard to their biomarkers. Then patients in a second wave were assigned to medicines based on their particular tumor biomarkers, taking into consideration how people in the first group with similar biomarkers were faring.
Overall disease control -- defined as “non-progression” of the cancer -- among all participants was 46 percent after eight weeks, Kim said. However, about 61 percent of patients with KRAS mutations had non-progression of their cancer when given Nexavar, Kim said in an Apr. 13 phone interview.
By comparison, patients with KRAS mutations assigned to Tarceva, a treatment designed to inhibit EGFR, had just 22 percent disease control. Tarceva is jointly sold by Roche Holding AG, based in Basel, Switzerland, and OSI Pharmaceuticals Inc., of Melville, New York.
Nexavar Surprise
Nexavar also showed the highest overall disease control of all the treatments in the study at 58 percent of patients. Other drugs used in the study were AstraZeneca Plc’s experimental Zactima and Eisai Co.’s Targretin.
The Nexavar results were a surprise, according to the M.D. Anderson researchers. Two years ago, Bayer and Onyx halted a trial of Nexavar because the medicine failed to help lung cancer patients live longer than standard treatment.
The KRAS mutation is found in 20 percent of lung-cancer patients and “nothing works,” said Roy Herbst, chief of thoracic oncology and co-author of the study.
“When we treated them with Nexavar, we saw they did very well,” said Kim, director of clinical research in thoracic oncology.
Last-Stage Testing
Nexavar, co-marketed by Leverkusen, Germany-based Bayer and Emeryville, California-based Onyx, is in the last stage of testing required for U.S. approval as a third-line monotherapy for lung cancer and as a first-line treatment in combination with chemotherapy, according to Onyx spokeswoman Lori Murray.
“We’re very pleased that KRAS-mutated patients responded favorably,” Murray said of today’s study. “Onyx and Bayer are evaluating to determine potential next steps in our lung cancer development program.”
Nexavar is approved for use in patients with kidney cancer and inoperable liver cancer, and worldwide sales of the drug were $844 million in 2009.
While this study shows Nexavar can work in lung-cancer patients as a third or fourth line of treatment, winning approval as a first-line treatment in combination with chemotherapy “would be more meaningful commercially,” said Howard Liang, an analyst at Leerink Swann in Boston.
Some doctors say they would like to see independent follow- up studies. “The results are surprisingly good,” said Anil Potti, an associate professor of medicine at Duke University, “but I hesitate to commit to the fact that this is a valuable drug in lung cancer yet.”
*******
https://commerce.us.reuters.com/purchase/showReportDetail.do…
ONXX: Analysis Ahead of NExUS Data – Favorable Risk/Reward
Brean Murray & Co., Inc
Summary:
We expect top-line data from the Phase 3 NExUS trial of Nexavar + cisplatin and gemcitabine in front-line NSCLC patients in mid-2010. Given investors’ very low expectations and the Street’s uniformly bearish view on the trial outcome (we believe no analyst currently assigns any value to the lung program), we believe the potential downside should be very limited, while potentially a positive outcome could provide significant upside given the huge unmet medical need in NSCLC (~1.2 million new cases and 1.1 million deaths worldwide in 2007), creating a favorable risk/reward heading into the data reporting. In our view, the outcome of the NExUS trial is hard to handicap. The bearish view on the NExUS trial (and the Nexavar lung program) is due to the failure and premature termination of a previous Phase 3 ESCAPE trial (Nexavar + carboplatin and paclitaxel, also in front-line NSCLC patients) in February 2008, due to the subset of squamous-cell patients showing a higher mortality rate in the triple combination arm than the chemo doublet arm. However, we remind investors that the NExUS trial outcome may differ because of the different chemo doublet used and the different patient mix in terms of histology types. In this note, we review the trial design of the NExUS trial in comparison with the ESCAPE trial and analyze the updated interim data of the ESCAPE trial. We believe there is a strong rationale for developing Nexavar in combination with chemotherapy for treating NSCLC based on the single agent activity and no increase in toxicity when in combination with chemotherapy. Additionally, although the NExUS and ESCAPE trials are similar in many ways, we view the exclusion of squamous-cell patients in the late half of the NExUS trial as a potentially positive factor, as squamous-cell histology has been proven to be a poor diagnostic factor for NSCLC in several trials, including a previous Phase 2 trial of Avastin and the Phase 3 ESCAPE trial.
mfg ipollit
NGSX... nichts Neues, nur ein paar Analystenkommentare
http://www.dailyfinance.com/story/investing/inside-wall-stre…
Inside Wall Street: This Little Pharma May Have a Big Pain Reliever
By GENE MARCIAL Posted 7:30 AM 04/23/10
Pain management is a much sought-after therapy, so just imagine if it came in the form of a convenient skin patch that provides three months of relief. That's just what tiny, little-known pharmaceutical company NeurogesX (NGSX) has come up with, and which the Food and Drug Administration approved last November. The pain treatment has also gotten approvals in Europe and Hong Kong.
But even as the patch, called Qutenza, is now being marketed in the U.S., NeurogesX has yet to catch much attention. True, its stock bumped up from $7.70 a share in February to a 52-week high of $10.57 on Apr. 5 when Qutenza was launched. But it has since eased to around $9. However, the four Wall Street analysts who follow the company are all high on the stock. And some pros believe NeurogesX could double in a year or so.
Dr. Juan Sanchez, an analyst at Punk Ziegel Healthcare Group, says Qutenza could be widely adopted as a treatment for chronic peripheral neuropathic pain conditions, and as such it has an opportunity to be a $300 million product in the U.S. and generate sales of $175 million in Europe. The FDA's approval of Qutenza was the first in 10 years for a prescription-strength topical treatment of pain from post-herpetic neuralgia (PHN). Neuralgia is the nerve pain that occurs with shingles. Every year about 1 million Americans develop shingles, a painful viral infection caused by reactivation of a virus that causes chickenpox.
"An Unmet Medical Need"
In the European Union, Qutenza was approved in May 2009 for treatment of chronic neuropathic pain in nondiabetic adults. It's scheduled to be marketed there later this year. NeurogesX has signed a licensing agreement with Astellas Pharma to market Qutenza in Europe, the Middle East and Africa. Sanchez says the partnership serves to validate the patch's commercial potential.
Demand for Qutenza is expected to be high. Sanchez notes that chronic neuropathic pain "remains an unmet medical need, with over 50% of patients not achieving appropriate pain control." Qutenza can make an important contribution, he says, because it has a long lasting analgesic effect with just a single application. Qutenza is the only product that contains a synthetic form of capsaicin, a naturally occurring compound found in chili pepper that delivers heat sensation.
Astellas paid NeurogesX an upfront fee of $42 million, plus $7 million for an option to license a follow-up product, NGX 1998, a liquid formulation of Qutenza. Clinical trials for the liquid product are scheduled for later this year. In all, NeurogesX is entitled to additional milestone and option payments related to the liquid version totaling $97 million.
Tough Market to Enter
Sanchez, who rates Neurogesx a buy with a 12-month price target of $16 a share, says the company could generate revenues of $300 million by 2015. He doesn't worry much about competition. "Potential generic competitors would find it challenging to demonstrate bioequivalence to a nonabsorbable drug," he says. That, he adds, is a big a potential barrier to entry into the skin-patch pain relief market.
Analyst Gregory R. Wade of Wedbush Securities, who rates the stock as outperform, is more bullish. Although he has a 12-month target of $13 a share, he argues that once it's clear that Qutenza is gaining traction in the U.S. and Europe, the stock could trade closer to $20 in the next 12 months. The higher target is based on his earnings forecast for 2012. He expects the company to be in the red through 2011, but in 2012, Wade sees NeurogesX in the black with earnings of about $50 million, or $2.19 a share.
The next immediate catalyst for the stock would be getting a major U.S pharmaceutical company to partner with NeurogesX for the U.S. market. Without a deep-pocketed U.S. ally, it'll have to raise additional funds.
Some big U.S. institutional investors have already put in early stakes in NeurogesX, including BlackRock Fund Advisers, State Street, and Northern Trust. But the major shareholders are less known, such as Arch Venture Partners, which owns a 16.4% stake, and SV Life Sciences, with 14.2%.
Buying in early on NeurogesX could be one painless investment.
mfg ipollit
http://www.dailyfinance.com/story/investing/inside-wall-stre…
Inside Wall Street: This Little Pharma May Have a Big Pain Reliever
By GENE MARCIAL Posted 7:30 AM 04/23/10
Pain management is a much sought-after therapy, so just imagine if it came in the form of a convenient skin patch that provides three months of relief. That's just what tiny, little-known pharmaceutical company NeurogesX (NGSX) has come up with, and which the Food and Drug Administration approved last November. The pain treatment has also gotten approvals in Europe and Hong Kong.
But even as the patch, called Qutenza, is now being marketed in the U.S., NeurogesX has yet to catch much attention. True, its stock bumped up from $7.70 a share in February to a 52-week high of $10.57 on Apr. 5 when Qutenza was launched. But it has since eased to around $9. However, the four Wall Street analysts who follow the company are all high on the stock. And some pros believe NeurogesX could double in a year or so.
Dr. Juan Sanchez, an analyst at Punk Ziegel Healthcare Group, says Qutenza could be widely adopted as a treatment for chronic peripheral neuropathic pain conditions, and as such it has an opportunity to be a $300 million product in the U.S. and generate sales of $175 million in Europe. The FDA's approval of Qutenza was the first in 10 years for a prescription-strength topical treatment of pain from post-herpetic neuralgia (PHN). Neuralgia is the nerve pain that occurs with shingles. Every year about 1 million Americans develop shingles, a painful viral infection caused by reactivation of a virus that causes chickenpox.
"An Unmet Medical Need"
In the European Union, Qutenza was approved in May 2009 for treatment of chronic neuropathic pain in nondiabetic adults. It's scheduled to be marketed there later this year. NeurogesX has signed a licensing agreement with Astellas Pharma to market Qutenza in Europe, the Middle East and Africa. Sanchez says the partnership serves to validate the patch's commercial potential.
Demand for Qutenza is expected to be high. Sanchez notes that chronic neuropathic pain "remains an unmet medical need, with over 50% of patients not achieving appropriate pain control." Qutenza can make an important contribution, he says, because it has a long lasting analgesic effect with just a single application. Qutenza is the only product that contains a synthetic form of capsaicin, a naturally occurring compound found in chili pepper that delivers heat sensation.
Astellas paid NeurogesX an upfront fee of $42 million, plus $7 million for an option to license a follow-up product, NGX 1998, a liquid formulation of Qutenza. Clinical trials for the liquid product are scheduled for later this year. In all, NeurogesX is entitled to additional milestone and option payments related to the liquid version totaling $97 million.
Tough Market to Enter
Sanchez, who rates Neurogesx a buy with a 12-month price target of $16 a share, says the company could generate revenues of $300 million by 2015. He doesn't worry much about competition. "Potential generic competitors would find it challenging to demonstrate bioequivalence to a nonabsorbable drug," he says. That, he adds, is a big a potential barrier to entry into the skin-patch pain relief market.
Analyst Gregory R. Wade of Wedbush Securities, who rates the stock as outperform, is more bullish. Although he has a 12-month target of $13 a share, he argues that once it's clear that Qutenza is gaining traction in the U.S. and Europe, the stock could trade closer to $20 in the next 12 months. The higher target is based on his earnings forecast for 2012. He expects the company to be in the red through 2011, but in 2012, Wade sees NeurogesX in the black with earnings of about $50 million, or $2.19 a share.
The next immediate catalyst for the stock would be getting a major U.S pharmaceutical company to partner with NeurogesX for the U.S. market. Without a deep-pocketed U.S. ally, it'll have to raise additional funds.
Some big U.S. institutional investors have already put in early stakes in NeurogesX, including BlackRock Fund Advisers, State Street, and Northern Trust. But the major shareholders are less known, such as Arch Venture Partners, which owns a 16.4% stake, and SV Life Sciences, with 14.2%.
Buying in early on NeurogesX could be one painless investment.
mfg ipollit
ARRY... Einschätzung des letzten ARRY-162 Deals von Ohad Hammer aus seinem Blog
http://www.hammerstockblog.com/how-array-got-its-groove-back…
How Array Got its Groove Back
Array’s (ARRY) recent licensing deal with Novartis (NVS) is another evidence of pharma’s appetite for new oncology compounds, especially for targeted agents. Facing a patent cliff and dwindling internal pipelines, pharmaceutical companies are willing to pay a generous price for promising early stage compounds.
This is why companies with broad platform technologies that can feed the industry with new compounds represent an attractive investment opportunity. These companies include (in alphabetical order) Arqule (ARQL), Array, Exelixis (EXEL), Immunogen (IMGN), Micromet (MITI) and Seattle Genetics (SGEN). From that list, Array has been the worst performer in 2009 due to liquidity fears as well as lack of exciting clinical data for its proprietary compounds. The two recent deals with Amgen (AMGN) and Novartis helped Array strengthen its balance sheet, but more importantly, they prove that the company’s discovery and early stage development capabilities have been underestimated by the market.
Novartis in-licensed Array’s MEK inhibitors, including ARRY-162, currently in phase I in cancer patients. The deal included an upfront payment of $45M as well as double digit royalties on international sales and opt-in rights for the US. Although large licensing deals for early stage oncology compounds with limited efficacy data are not a rare event, Array’s deal with Novartis stands out as it has already out licensed a similar compound to AstraZeneca (AZN) several years ago. The AstraZeneca deal, which was announced in late 2003, covered AZD6244 (a preclinical stage compound nearing IND at the time) and backup compounds for an upfront payment of $10M.
The interesting part of the Astra agreement was that it permitted Array to develop other MEK inhibitors but only for non-oncology indications. This restriction was set to expire in April 2009. During that period, Array advanced two additional MEK inhibitors to clinical trials as part of its inflammation pipeline, one of which, ARRY-162, failed in a phase II trial in rheumatoid arthritis last year. Despite the failure, ARRY-162’s value kept going up, as the interest in MEK as a target for cancer therapy reached unprecedented levels in 2009.
MEK is part of the MAPK pathway, one of the two major signaling axes in cancer, on which multiple signals converge and lead to cell growth. The other axis is the PI3K pathway. The current approach with targeted therapies such as MEK inhibitors is to combine them with other drugs in order to block several cascades in parallel. The hottest trend right now is modulating both the MAPK and the PI3K pathways simultaneously, as cancer cells often activate both of these pathways. A good example for this trend is an ongoing phase I trial combining Astra’s MEK inhibitor (AZD6244) and Merck’s Akt inhibitor (MK-2206). This is probably the first time in history two pharmas decide to combine two drugs at such an early stage of development.
Array was not the first to strike a large MEK inhibitor deal. In April of last year, Ardea (RDEA) sold rights for a phase I MEK inhibitor to Bayer in a deal that included an upfront payment of $35M and low double-digit royalties. Array managed to get better terms, as its drug has a comprehensive safety and pharmacokinetics data package thanks to the inflammation program. In addition, ARRY-162 was the last unpartnered clinical stage MEK inhibitor in the market, which probably helped Array in the negotiation table.
On the clinical front there were mainly two factors that positioned MEK as a hot target. The first was a spectacular data set from a phase I of PLX4032, a drug developed by privately held Plexxikon and Roche for melanoma. Although PLX4032 does not target MEK , the fact that it hits a different target in the MAPK signaling pathway (BRAF) seems to further validate MEK as a target. Another reason for the great interest in MEK inhibitors was large amount of clinical data for AZD6244, the MEK inhibitor Array licensed out to AstraZeneca more than 6 years ago. Although, AZD6244 failed in three phase II trials and did not reach a registration trial, it still demonstrated interesting signs of efficacy, especially in tumors harboring certain mutations. This has led some to blame the AZD6244’s delays on a poorly designed clinical program rather than the compound itself. Therefore, ironically, the effort and money put into AZD6244 by AstraZeneca enabled Array to sign such a lucrative deal for its other MEK inhibitor with Novartis.
From Novartis’ stand point, it added another promising kinase inhibitor to the armamentarium. The company’s kinase inhibitor pipeline for cancer comprises of 9 compounds in development and three approved drugs, including Gleevec, the best selling kinase inhibitor to date. The company will probably have a fourth drug in the market next year following data from a registration study for INC424. This is the place to admit the size of the INC424 deal, which included an upfront payment of $210M for rights outside of the US, easily topped my predictions which turned out to be ridiculously low.
In contrast to INC424, ARRY-162 is still an early stage compound with no clear route for approval. As discussed above, its value will probably be in combination with other drugs, which is why large companies like Novartis aspire to have a diverse pipeline of targeted therapies that could be mixed and matched according to the clinical setting. Novartis, for example, has a strong presence in the PI3K arena, with one drug (Affinitor) already approved for kidney cancer and two others in clinical testing with what appear to be early signs of efficacy. On top of Array’s MEK inhibitor, Novartis also has a phase I compound which hits BRAF, another component of the MAPK pathway. Coincidentally or not, at last week’s AACR annual meeting, Array presented an extensive preclinical project on the combination of its ARRY-162 with Novartis’ Affinitor. This will probably be just one of many combinations Novartis will evaluate in the clinic.
In summary, the collaboration with Novartis is a great achievement for Array and a testament of excellent long term strategic planning. The company’s MEK inhibitors are now being developed in the hands of two of the top 5 pharmaceutical companies. The Novartis deal comes after a lucrative deal with Amgen in diabetes, which increase the number of clinical stage partnered compounds to 8. This is on top of four wholly owned compounds in the clinic.
Even after the recent climb, Array’s market cap is still under $200M, which is substantially lower than other companies with kinase inhibitor platforms such as Exelixis and Arqule. The only thing that separates Array from these companies is the fact that it does not have one drug with a clear path to market. Most of Array’s drugs are still in early clinical development, so investors cannot put a price tag on them. Once investors see the light at the end of the tunnel with at least one of Array’s drugs (partnered or unpartnered), the company’s valuation should increase dramatically. It is hard to predict the timing of such an event, but with the amount of shots on goal Array has, it is probably only a matter of time.
mfg ipollit
http://www.hammerstockblog.com/how-array-got-its-groove-back…
How Array Got its Groove Back
Array’s (ARRY) recent licensing deal with Novartis (NVS) is another evidence of pharma’s appetite for new oncology compounds, especially for targeted agents. Facing a patent cliff and dwindling internal pipelines, pharmaceutical companies are willing to pay a generous price for promising early stage compounds.
This is why companies with broad platform technologies that can feed the industry with new compounds represent an attractive investment opportunity. These companies include (in alphabetical order) Arqule (ARQL), Array, Exelixis (EXEL), Immunogen (IMGN), Micromet (MITI) and Seattle Genetics (SGEN). From that list, Array has been the worst performer in 2009 due to liquidity fears as well as lack of exciting clinical data for its proprietary compounds. The two recent deals with Amgen (AMGN) and Novartis helped Array strengthen its balance sheet, but more importantly, they prove that the company’s discovery and early stage development capabilities have been underestimated by the market.
Novartis in-licensed Array’s MEK inhibitors, including ARRY-162, currently in phase I in cancer patients. The deal included an upfront payment of $45M as well as double digit royalties on international sales and opt-in rights for the US. Although large licensing deals for early stage oncology compounds with limited efficacy data are not a rare event, Array’s deal with Novartis stands out as it has already out licensed a similar compound to AstraZeneca (AZN) several years ago. The AstraZeneca deal, which was announced in late 2003, covered AZD6244 (a preclinical stage compound nearing IND at the time) and backup compounds for an upfront payment of $10M.
The interesting part of the Astra agreement was that it permitted Array to develop other MEK inhibitors but only for non-oncology indications. This restriction was set to expire in April 2009. During that period, Array advanced two additional MEK inhibitors to clinical trials as part of its inflammation pipeline, one of which, ARRY-162, failed in a phase II trial in rheumatoid arthritis last year. Despite the failure, ARRY-162’s value kept going up, as the interest in MEK as a target for cancer therapy reached unprecedented levels in 2009.
MEK is part of the MAPK pathway, one of the two major signaling axes in cancer, on which multiple signals converge and lead to cell growth. The other axis is the PI3K pathway. The current approach with targeted therapies such as MEK inhibitors is to combine them with other drugs in order to block several cascades in parallel. The hottest trend right now is modulating both the MAPK and the PI3K pathways simultaneously, as cancer cells often activate both of these pathways. A good example for this trend is an ongoing phase I trial combining Astra’s MEK inhibitor (AZD6244) and Merck’s Akt inhibitor (MK-2206). This is probably the first time in history two pharmas decide to combine two drugs at such an early stage of development.
Array was not the first to strike a large MEK inhibitor deal. In April of last year, Ardea (RDEA) sold rights for a phase I MEK inhibitor to Bayer in a deal that included an upfront payment of $35M and low double-digit royalties. Array managed to get better terms, as its drug has a comprehensive safety and pharmacokinetics data package thanks to the inflammation program. In addition, ARRY-162 was the last unpartnered clinical stage MEK inhibitor in the market, which probably helped Array in the negotiation table.
On the clinical front there were mainly two factors that positioned MEK as a hot target. The first was a spectacular data set from a phase I of PLX4032, a drug developed by privately held Plexxikon and Roche for melanoma. Although PLX4032 does not target MEK , the fact that it hits a different target in the MAPK signaling pathway (BRAF) seems to further validate MEK as a target. Another reason for the great interest in MEK inhibitors was large amount of clinical data for AZD6244, the MEK inhibitor Array licensed out to AstraZeneca more than 6 years ago. Although, AZD6244 failed in three phase II trials and did not reach a registration trial, it still demonstrated interesting signs of efficacy, especially in tumors harboring certain mutations. This has led some to blame the AZD6244’s delays on a poorly designed clinical program rather than the compound itself. Therefore, ironically, the effort and money put into AZD6244 by AstraZeneca enabled Array to sign such a lucrative deal for its other MEK inhibitor with Novartis.
From Novartis’ stand point, it added another promising kinase inhibitor to the armamentarium. The company’s kinase inhibitor pipeline for cancer comprises of 9 compounds in development and three approved drugs, including Gleevec, the best selling kinase inhibitor to date. The company will probably have a fourth drug in the market next year following data from a registration study for INC424. This is the place to admit the size of the INC424 deal, which included an upfront payment of $210M for rights outside of the US, easily topped my predictions which turned out to be ridiculously low.
In contrast to INC424, ARRY-162 is still an early stage compound with no clear route for approval. As discussed above, its value will probably be in combination with other drugs, which is why large companies like Novartis aspire to have a diverse pipeline of targeted therapies that could be mixed and matched according to the clinical setting. Novartis, for example, has a strong presence in the PI3K arena, with one drug (Affinitor) already approved for kidney cancer and two others in clinical testing with what appear to be early signs of efficacy. On top of Array’s MEK inhibitor, Novartis also has a phase I compound which hits BRAF, another component of the MAPK pathway. Coincidentally or not, at last week’s AACR annual meeting, Array presented an extensive preclinical project on the combination of its ARRY-162 with Novartis’ Affinitor. This will probably be just one of many combinations Novartis will evaluate in the clinic.
In summary, the collaboration with Novartis is a great achievement for Array and a testament of excellent long term strategic planning. The company’s MEK inhibitors are now being developed in the hands of two of the top 5 pharmaceutical companies. The Novartis deal comes after a lucrative deal with Amgen in diabetes, which increase the number of clinical stage partnered compounds to 8. This is on top of four wholly owned compounds in the clinic.
Even after the recent climb, Array’s market cap is still under $200M, which is substantially lower than other companies with kinase inhibitor platforms such as Exelixis and Arqule. The only thing that separates Array from these companies is the fact that it does not have one drug with a clear path to market. Most of Array’s drugs are still in early clinical development, so investors cannot put a price tag on them. Once investors see the light at the end of the tunnel with at least one of Array’s drugs (partnered or unpartnered), the company’s valuation should increase dramatically. It is hard to predict the timing of such an event, but with the amount of shots on goal Array has, it is probably only a matter of time.
mfg ipollit
AVEO...
Morgan Stanley Initiated Coverage Of Aveo Pharmaceuticals With OW Rating, $15 PT (AVEO)
Written on Wed, 04/21/2010 - 09:09
By Amanda Smith
4/21/2010- Morgan Stanley analysts are initiating coverage of Aveo Pharmaceuticals (NASDAQ:AVEO) with an overweight rating and $15 price target.
Analysts Steven Harr and Sara Slifka said, "We expect Tivozanib, Aveo's lead drug candidate, to show superiority vs. Onyx' Nexavar on progression free survival (the primary endpoint) in its Phase III trial for advanced renal cell cancer (RCC). We see the potential for the drug to work in other indications with chemotherapy, and estimate WW peak sales of >$1.5 bn. We do not expect Aveo to raise capital, a key investor concern, before Phase III data in 2H11. We believe investors are underestimating tivozanib's strong data to date and its commercial potential. The Street appears to be pricing in $100-150 mn in peak sales vs. our estimate of >$1bn. If tivozanib proves superior to Nexavar, we see it gaining significant share from Nexavar and Pfizer's Sutent in the $1.6 bn and growing RCC market (as well as modest sales in other cancers)."
The bank sees a fiscal 2010 loss of $2.24 per share and a fiscal 2011 EPS loss of $1.12 per share.
mfg ipollit
Morgan Stanley Initiated Coverage Of Aveo Pharmaceuticals With OW Rating, $15 PT (AVEO)
Written on Wed, 04/21/2010 - 09:09
By Amanda Smith
4/21/2010- Morgan Stanley analysts are initiating coverage of Aveo Pharmaceuticals (NASDAQ:AVEO) with an overweight rating and $15 price target.
Analysts Steven Harr and Sara Slifka said, "We expect Tivozanib, Aveo's lead drug candidate, to show superiority vs. Onyx' Nexavar on progression free survival (the primary endpoint) in its Phase III trial for advanced renal cell cancer (RCC). We see the potential for the drug to work in other indications with chemotherapy, and estimate WW peak sales of >$1.5 bn. We do not expect Aveo to raise capital, a key investor concern, before Phase III data in 2H11. We believe investors are underestimating tivozanib's strong data to date and its commercial potential. The Street appears to be pricing in $100-150 mn in peak sales vs. our estimate of >$1bn. If tivozanib proves superior to Nexavar, we see it gaining significant share from Nexavar and Pfizer's Sutent in the $1.6 bn and growing RCC market (as well as modest sales in other cancers)."
The bank sees a fiscal 2010 loss of $2.24 per share and a fiscal 2011 EPS loss of $1.12 per share.
mfg ipollit
da ging ja gestern einiges schief... 
Neurosearch... sehr unerwartet: die Huntexil-PIII wurde neu ausgewertet bzw. wurde bei der ersten Auswertung offensichtlich eine nicht vorher definierte Anpassung vorgenommen. Nun ist der primäre Endpunkt laut Vorgaben nicht mehr signifikant!
http://www.evaluatepharma.com/Universal/View.aspx?type=Story…
NeuroSearch data blunder dents confidence
EP Vantage, April 28, 2010
NeuroSearch, the Danish biotech that dazzled earlier this year with impressive results from a new Huntington’s disease treatment, Huntexil, today lost some of its gloss. In an embarrassing disclosure, the company announced that a new analysis of the phase III data revealed the primary endpoint of the trial had actually been missed.
The disclosure cost NeuroSearch’s chief medical officer his job and the company a quarter of its market value, a value that had doubled in the wake of the initial announcement (NeuroSearch redeems itself with impressive Huntington’s phase III data, February 3, 2010). Although this certainly makes Huntexil look a riskier project than previously, this appears to be more of a statistical than a clinical glitch and stamping the trial a failure is probably a bit harsh. For faith to return, data due later this year from a phase IIb study being conducted in the US needs to be positive.
CAGn adjustment
The crux of the issue is an adjustment called CAGn, which refers to the differences in patients’ genetic disposition. The company argues that this is a clinically relevant adjustment to apply to the statistical analysis of the study, called MermaiHD.
Using this adjustment, the primary endpoint of the trial was met, establishing a significant improvement in motor function in patients, with a p value of 0.02. A value lower than 0.025 was required for the trial to be judged a success.
However, the problem has arisen because this adjustment was not pre-specified as part of the protocol for analysing the data for the primary endpoint. So when the data is analysed without the adjustment, a p value of 0.042 is reached, meaning on a strict reading, the trial failed.
Reputation dent
On the upside, analysis of the secondary endpoints is unaffected and on these measures, improvement of voluntary and involuntary motor symptoms, the trial remains a success. And at the end of the day the totality of the data still points to Huntexil as potentially an important new advance for this neurodegenerative condition, for which there are precious few really effective medicines available or in development (Therapeutic focus - Huntexil at spout of dry Huntington's disease pipeline, February 4, 2010).
Analysts believe that should it reach the market, the product could generate peak sales of close to $1bn, globally.
On the downside, this could raise some serious questions about the credibility of the study and NeuroSearch’s management. By firing chief medical officer Dr Dieter Meier the company is attempting to draw a line under the matter, but a dent to reputation might be unavoidable.
As to who knew what and when it is not clear, but being the last to know investors were understandably unimpressed. The drop in the company’s share price today, which in afternoon trade was 26% lower at DKr127, no doubt reflects fears of regulatory hold ups and concern about the outcome of the second large trial, called Hart.
From the Hart
Hart is actually a phase IIb study and much smaller than the phase III, recruiting only 220 patients to MermaiHD’s 437. It is also shorter, tracking patients over three months rather than six, and has more dosage arms.
The primary endpoint is the same, but some analysts have already expressed concern that due to the differences above, it is probably more likely to fail.
Coming after the re-evaluation of MermaiHD, this would not be taken well, but would not necessarily mean the end for Huntexil. In MermaiHD the drugs effects grew with time, so as long as Hart shows a trend for efficacy, it still might be enough for regulators.
Twelve month safety data from MermaiHD is also due this year, and the company plans to file the package with regulators early next year.
If Huntexil was not intended for such a niche patient population with an extreme medical need, this news would be a lot more damaging to the drug’s prospects, and the company. As it is, unless Hart completely fails and a further phase III trial is needed the drug still has a good chance of winning approval based on the studies already being conducted. However the remaining data due this year are now even more pivotal.
mfg ipollit

Neurosearch... sehr unerwartet: die Huntexil-PIII wurde neu ausgewertet bzw. wurde bei der ersten Auswertung offensichtlich eine nicht vorher definierte Anpassung vorgenommen. Nun ist der primäre Endpunkt laut Vorgaben nicht mehr signifikant!
http://www.evaluatepharma.com/Universal/View.aspx?type=Story…
NeuroSearch data blunder dents confidence
EP Vantage, April 28, 2010
NeuroSearch, the Danish biotech that dazzled earlier this year with impressive results from a new Huntington’s disease treatment, Huntexil, today lost some of its gloss. In an embarrassing disclosure, the company announced that a new analysis of the phase III data revealed the primary endpoint of the trial had actually been missed.
The disclosure cost NeuroSearch’s chief medical officer his job and the company a quarter of its market value, a value that had doubled in the wake of the initial announcement (NeuroSearch redeems itself with impressive Huntington’s phase III data, February 3, 2010). Although this certainly makes Huntexil look a riskier project than previously, this appears to be more of a statistical than a clinical glitch and stamping the trial a failure is probably a bit harsh. For faith to return, data due later this year from a phase IIb study being conducted in the US needs to be positive.
CAGn adjustment
The crux of the issue is an adjustment called CAGn, which refers to the differences in patients’ genetic disposition. The company argues that this is a clinically relevant adjustment to apply to the statistical analysis of the study, called MermaiHD.
Using this adjustment, the primary endpoint of the trial was met, establishing a significant improvement in motor function in patients, with a p value of 0.02. A value lower than 0.025 was required for the trial to be judged a success.
However, the problem has arisen because this adjustment was not pre-specified as part of the protocol for analysing the data for the primary endpoint. So when the data is analysed without the adjustment, a p value of 0.042 is reached, meaning on a strict reading, the trial failed.
Reputation dent
On the upside, analysis of the secondary endpoints is unaffected and on these measures, improvement of voluntary and involuntary motor symptoms, the trial remains a success. And at the end of the day the totality of the data still points to Huntexil as potentially an important new advance for this neurodegenerative condition, for which there are precious few really effective medicines available or in development (Therapeutic focus - Huntexil at spout of dry Huntington's disease pipeline, February 4, 2010).
Analysts believe that should it reach the market, the product could generate peak sales of close to $1bn, globally.
On the downside, this could raise some serious questions about the credibility of the study and NeuroSearch’s management. By firing chief medical officer Dr Dieter Meier the company is attempting to draw a line under the matter, but a dent to reputation might be unavoidable.
As to who knew what and when it is not clear, but being the last to know investors were understandably unimpressed. The drop in the company’s share price today, which in afternoon trade was 26% lower at DKr127, no doubt reflects fears of regulatory hold ups and concern about the outcome of the second large trial, called Hart.
From the Hart
Hart is actually a phase IIb study and much smaller than the phase III, recruiting only 220 patients to MermaiHD’s 437. It is also shorter, tracking patients over three months rather than six, and has more dosage arms.
The primary endpoint is the same, but some analysts have already expressed concern that due to the differences above, it is probably more likely to fail.
Coming after the re-evaluation of MermaiHD, this would not be taken well, but would not necessarily mean the end for Huntexil. In MermaiHD the drugs effects grew with time, so as long as Hart shows a trend for efficacy, it still might be enough for regulators.
Twelve month safety data from MermaiHD is also due this year, and the company plans to file the package with regulators early next year.
If Huntexil was not intended for such a niche patient population with an extreme medical need, this news would be a lot more damaging to the drug’s prospects, and the company. As it is, unless Hart completely fails and a further phase III trial is needed the drug still has a good chance of winning approval based on the studies already being conducted. However the remaining data due this year are now even more pivotal.
mfg ipollit
AMAG... Q1 ist zwar keine Katastrophe, liegt aber doch unter den Erwartungen nach den bereits schlechten Zahlen für Januar und Februar.
AMAG Pharma shares fall on weak 1Q results
On Wednesday April 28, 2010, 2:06 pm EDT
NEW YORK (AP) -- Shares of AMAG Pharmaceuticals Inc. fell Wednesday after the biotechnology company reported a wider first-quarter loss and weaker revenue than Wall Street expected.
The stock shed $3.42, or 9.3 percent, to $33.36 in afternoon trading. Shares earlier reached a 52-week low of $31.90. They have ranged from $33.11 to $58.23 over the past year.
On Tuesday, the company said it lost $23.1 million, or $1.15 per share, on revenue of $13.3 million during the quarter. Analysts polled by Thomson Reuters, on average, had forecast a loss of 85 cents per share on revenue of $16.8 million.
The bulk of the company's revenue comes from Feraheme, which treats iron deficiency anemia in chronic kidney disease patients.
Jefferies & Co. analyst Eun K. Yang reaffirmed a "Hold" rating on the stock with a $32 price target, citing the lower-than-expected Feraheme sales and wider-than-expected first-quarter loss.
"While its potential in non-dialysis remains, disappointing first-quarter sales suggests challenges it (Feraheme) faces in the competitive, pricing-sensitive intravenous iron market, with little advantages," Yang wrote in a note to investors.
mfg ipollit
AMAG Pharma shares fall on weak 1Q results
On Wednesday April 28, 2010, 2:06 pm EDT
NEW YORK (AP) -- Shares of AMAG Pharmaceuticals Inc. fell Wednesday after the biotechnology company reported a wider first-quarter loss and weaker revenue than Wall Street expected.
The stock shed $3.42, or 9.3 percent, to $33.36 in afternoon trading. Shares earlier reached a 52-week low of $31.90. They have ranged from $33.11 to $58.23 over the past year.
On Tuesday, the company said it lost $23.1 million, or $1.15 per share, on revenue of $13.3 million during the quarter. Analysts polled by Thomson Reuters, on average, had forecast a loss of 85 cents per share on revenue of $16.8 million.
The bulk of the company's revenue comes from Feraheme, which treats iron deficiency anemia in chronic kidney disease patients.
Jefferies & Co. analyst Eun K. Yang reaffirmed a "Hold" rating on the stock with a $32 price target, citing the lower-than-expected Feraheme sales and wider-than-expected first-quarter loss.
"While its potential in non-dialysis remains, disappointing first-quarter sales suggests challenges it (Feraheme) faces in the competitive, pricing-sensitive intravenous iron market, with little advantages," Yang wrote in a note to investors.
mfg ipollit
VPHM... ebenfalls Q1 unter den Erwartungen des Marktes
http://www.reuters.com/article/idCNSGE63R0EQ20100428?rpc=44
UPDATE 1-ViroPharma Q1 profit misses Street
* Q1 EPS $0.26 vs est $0.32
* Q1 rev $90.6 mln vs est $92.5 mln
* Raises lower end of 2010 Cinryze sales view
April 28 (Reuters) - Specialty pharmaceutical company ViroPharma Inc (VPHM.O) posted a lower-than-expected quarterly profit, hurt by higher expenses, but raised the bottom end of its 2010 sales outlook for key drug Cinryze.
For the first quarter, the company posted a net income of $21.2 million, or 26 cents a share, compared to a net loss of $59.2 million, or 77 cents a share, in the year-ago quarter.
Revenue for the quarter was up 50 percent to $90.6 million.
Analysts on average expected the company to earn 32 cents a share, on revenue of $92.5 million, according to Thomson Reuters I/B/E/S.
Net sales of Cinryze, used for the treatment of hereditary angioedema -- a potentially fatal genetic disease -- rose about 421 percent to $35 million, the company said.
ViroPharma said it now expects full-year Cinryze sales of $155 million to $175 million. It had earlier forecast sales of $150 million $175 million.
Shares of the company closed at $13.90 Tuesday on Nasdaq.
mfg ipollit
http://www.reuters.com/article/idCNSGE63R0EQ20100428?rpc=44
UPDATE 1-ViroPharma Q1 profit misses Street
* Q1 EPS $0.26 vs est $0.32
* Q1 rev $90.6 mln vs est $92.5 mln
* Raises lower end of 2010 Cinryze sales view
April 28 (Reuters) - Specialty pharmaceutical company ViroPharma Inc (VPHM.O) posted a lower-than-expected quarterly profit, hurt by higher expenses, but raised the bottom end of its 2010 sales outlook for key drug Cinryze.
For the first quarter, the company posted a net income of $21.2 million, or 26 cents a share, compared to a net loss of $59.2 million, or 77 cents a share, in the year-ago quarter.
Revenue for the quarter was up 50 percent to $90.6 million.
Analysts on average expected the company to earn 32 cents a share, on revenue of $92.5 million, according to Thomson Reuters I/B/E/S.
Net sales of Cinryze, used for the treatment of hereditary angioedema -- a potentially fatal genetic disease -- rose about 421 percent to $35 million, the company said.
ViroPharma said it now expects full-year Cinryze sales of $155 million to $175 million. It had earlier forecast sales of $150 million $175 million.
Shares of the company closed at $13.90 Tuesday on Nasdaq.
mfg ipollit
zu REGN:
Regeneron outlines a blockbuster future
By John Carroll
Created May 12 2010 - 11:40am
To hear Reuters tell it this morning, Regeneron Pharmaceuticals is on the cusp of reaching biotech's center ring. The developer has a research budget this year of more than $700 million--with Sanofi Aventis providing $160 million [1]--and will have data on seven late-stage trials.
If they're successful, Regeneron execs say they will have pivotal data on two potential blockbusters and a third drug that could earn $500 million a year.
"The cycle is getting to be in full swing," CEO Leonard Schleifer told Reuters. "We need a big hit on one of these drugs and then I think the world will start to pay a lot more attention to our pipeline" of new medicines.
In a separate release, Regeneron Pharmaceuticals (NASDAQ: REGN [2]) presented some encouraging news on two of its antibody programs partnered with Sanofi-Aventis--though one appeared to offer mixed results for pain.
In a Phase I trial of the protein-blocking REGN727 the developer said that researchers tracked "highly significant lowering of mean LDL (bad) cholesterol," with a maximum reduction of more than 60 percent at the highest dose. A Phase II for REGN475 involving 217 patients suffering from osteoarthritis of the knee, meanwhile, hit its primary endpoint for safety and demonstrated improvements in average walking pain scores over 8 weeks.
"Preliminary analysis of interim efficacy data from a Phase 2 trial of REGN 475 in the acute setting of nerve root compression induced pain (acute sciatica) suggests that REGN475 therapy will not be effective in this setting," said the company in a statement. The Tarrytown, NY-based biotech says that it will discuss the details of the data at an investors gathering July 15. Its shares tracked up about four percent this morning.
hier die PM von REGN zun den mabs:
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=46…
Regeneron outlines a blockbuster future
By John Carroll
Created May 12 2010 - 11:40am
To hear Reuters tell it this morning, Regeneron Pharmaceuticals is on the cusp of reaching biotech's center ring. The developer has a research budget this year of more than $700 million--with Sanofi Aventis providing $160 million [1]--and will have data on seven late-stage trials.
If they're successful, Regeneron execs say they will have pivotal data on two potential blockbusters and a third drug that could earn $500 million a year.
"The cycle is getting to be in full swing," CEO Leonard Schleifer told Reuters. "We need a big hit on one of these drugs and then I think the world will start to pay a lot more attention to our pipeline" of new medicines.
In a separate release, Regeneron Pharmaceuticals (NASDAQ: REGN [2]) presented some encouraging news on two of its antibody programs partnered with Sanofi-Aventis--though one appeared to offer mixed results for pain.
In a Phase I trial of the protein-blocking REGN727 the developer said that researchers tracked "highly significant lowering of mean LDL (bad) cholesterol," with a maximum reduction of more than 60 percent at the highest dose. A Phase II for REGN475 involving 217 patients suffering from osteoarthritis of the knee, meanwhile, hit its primary endpoint for safety and demonstrated improvements in average walking pain scores over 8 weeks.
"Preliminary analysis of interim efficacy data from a Phase 2 trial of REGN 475 in the acute setting of nerve root compression induced pain (acute sciatica) suggests that REGN475 therapy will not be effective in this setting," said the company in a statement. The Tarrytown, NY-based biotech says that it will discuss the details of the data at an investors gathering July 15. Its shares tracked up about four percent this morning.
hier die PM von REGN zun den mabs:
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=46…
COX.PA kriegt einen auf den Deckel ....
NicOx provides update on FDA Advisory Committee Meeting for naproxcinod
http://finance.yahoo.com/news/NicOx-provides-update-on-FDA-i…

NicOx provides update on FDA Advisory Committee Meeting for naproxcinod
http://finance.yahoo.com/news/NicOx-provides-update-on-FDA-i…
Antwort auf Beitrag Nr.: 39.512.665 von SLGramann am 13.05.10 09:26:23noch ein Artikel zu REGN...
http://www.reuters.com/article/idUSTRE64B37U20100512
Exclusive: Biotech Regeneron on verge of big leagues
(Reuters) - Regeneron Pharmaceuticals Inc, a relatively unknown biotechnology company based in sleepy Tarrytown, New York, is not exactly a household name. But that is likely to change fast.
The company plans to steal the limelight in the coming year with data from seven late-stage trials of its experimental drugs for gout, colon cancer and a leading cause of blindness. It is rare for a biotech of any size to have so much pivotal news coming out in such a short period of time.
"The cycle is getting to be in full swing," said Chief Executive Leonard Schleifer. "We need a big hit on one of these drugs and then I think the world will start to pay a lot more attention to our pipeline" of new medicines.
In an exclusive interview with Reuters at company headquarters, Schleifer and his research chief George Yancopoulos outlined their plan for leaping from obscurity to the big leagues.
They know from experience, however, that in biotech there is no sure thing. In March 2003, when the company's Axokine obesity treatment stumbled in clinical trials, Regeneron shares lost more than half their value.
But success in even one of its three treatments now in late-stage studies could propel Regeneron from a typical money-losing biotech into a profitable company with a bright future. Its products could eventually take on Roche Holding AG's $6 billion cancer drug, Avastin, and $3 billion Lucentis eye-disease medicine. The gout drug could attract annual sales of $500 million or more, analysts estimate.
Regeneron on Wednesday said two other drugs in earlier-stage trials significantly cut levels of "bad" LDL cholesterol and eased knee pain from osteoarthritis.
But Wall Street is more focused on its drugs in late-stage trials.
"We believe that Regeneron should represent a core holding in biotechnology portfolios, based on the depth of its pipeline and its partnerships," Roth Capital Partners analyst Joseph Pantginis said on Wednesday. He said he is encouraged by new data from the early-stage trials.
Regeneron has some big backers and one of the biggest research budgets of any biotech, at more than $700 million this year. That includes spending by collaborators and $160 million of support from research partner Sanofi-Aventis SA.
It currently sells only one product, called Arcalyst, that brings in less than $20 million a year and treats a rare inherited inflammatory condition.
But the three drugs now wrapping up late-stage studies, the final step before seeking regulatory approval, have far bigger sales potential.
NOT IMMUNE TO FAILURE
Most biotech companies are staking their futures on only one or two drugs and many have gone bankrupt trying. Regeneron, which is now testing eight drugs, plans to have as many as 40 more in trials by 2017 under its lucrative deal with Sanofi to test Regeneron antibodies against a wide range of diseases.
Regeneron claims to have perhaps the most effective drug-making technology -- mice with human immune-system genes that produce better antibodies than rival companies.
It cites another major weapon: the continual hands-on involvement of Merck & Co Inc's legendary former Chief Executive Roy Vagelos.
The stock, with big current stakes held by Sanofi, T. Rowe Price Group affiliates and Carl Icahn, has risen 57 percent over the past year, compared with a 70 percent gain for the NYSARC Biotech Index and a 27 percent advance for the Standard & Poor's 500. In morning trade, it rose a further $1.09 or 4.3 percent to $26.61 on Nasdaq.
Regeneron's market capitalization is about $2 billion, far below industry leaders such as Amgen Inc at $53 billion and $129 billion for Roche, owner of Genentech, a biotech Regeneron emulates.
The company is based in the hometown of author Washington Irving, whose famous tale "Rip Van Winkle" concerns a man who falls asleep for 20 years and awakens to find everything changed. But unlike Rip, who was habitually idle, Regeneron has worked at a feverish pace since 1988 to change everything about drug development.
By next month, Regeneron plans to release data from Phase III trials of its drug to prevent and treat pain from gout, a form of arthritis that often hits the big toe. It uses the same active ingredient as Arcalyst, rilonacept, and works by blocking an inflammation-causing protein called Interleukin-1.
In the second half of the year it plans to provide data from late-stage trials of its macular degeneration product, called VEGF Trap, and its aflibercept drug for colon cancer. Both drugs, like Avastin, block the VEGF protein which is involved in growth of blood vessels.
Schleifer said the company's top commercial opportunity is probably VEGF-Trap, being developed with Bayer AG.
BIG DREAMS, TINY OFFICE
The company was started in 1988 by Schleifer, an assistant professor of neurology at Cornell University Medical College. He planned to discover nerve-growth proteins that could cure some of the grimmest neurological scourges by regenerating neurons, which are nerve cells. Thus the name Regeneron.
"The original idea, which you can find in the original business plan, was that we would discover them and then spritz them on people who needed them. We would cure a couple of diseases such as Lou Gehrig's disease, maybe Parkinson's disease and Alzheimer's disease, and sail off into the sunset," Schleifer said.
Schleifer nurtured those lofty ambitions in a dilapidated studio apartment meant to house Cornell's medical staff, located on Manhattan's Upper East Side.
"The super was thrown out of the building because he was a drunk, so I convinced the hospital to let me sublet it for awhile. The rent was about $500. That was Regeneron's world headquarters. It was just me."
Schleifer, a native of New York City's borough of Queens with a gift of gab to match his scientific enthusiasm, attracted a scientific advisory board that included several Nobel laureates and obtained a $1 million grubstake from a venture capital unit of Merrill Lynch.
He set out to recruit a stable of scientists, most notably Yancopoulos, a 28-year-old molecular immunologist who had just won a kind of genius award for his research at Columbia Medical School and is now Regeneron's head of research.
The company spread its wings in 1989 by renting 15,000 square feet of an empty Union Carbide factory in Tarrytown, where laboratories were built.
Regeneron could afford the bigger digs because earlier that month it won $10 million in funding from Sumitomo Chemical Co Ltd in exchange for giving the company rights to sell up to three of its future drugs in Japan.
At the time, only one protein involved in the growth and survival of neurons -- or neurotrophic factor -- had been discovered: Nerve Growth Factor. But within weeks after Regeneron's lab opened, Yancopoulos discovered a second one that became known as Brain-Derived Neurotrophic Factor (BDNF).
"Literally within a week after that, we cloned another one called Neurotrophin 3 (NT3) and then a third one called Ciliary Neurotrophic Factor (CNTF)," Yancopoulos said.
Yancopoulos became one of the world's most-cited neurologists due to his rapid-fire discoveries. The attention led to a $100 million partnership in 1990 with Amgen, an up-and-coming new biotech, that involved trials of BDNF against Lou Gehrig's disease and of NT3 for nerve pain in diabetics.
Regeneron also began testing CNTF against Lou Gehrig's disease and signed a $135 million deal in 1997 with Procter & Gamble Co to develop a number of Regeneron drugs, including using CNTF to treat obesity.
But none of the drugs ultimately panned out in clinical trials during a 10-year period.
"In many ways you could look at that as a lost decade because the ultimate prize eluded us in terms of applying our first discoveries," Yancopoulos said. "These neurological diseases were really challenging and we recognized maybe we weren't as smart as we thought we were and didn't have the experience that maybe we needed."
A MODEL IN MERCK
Schleifer and Yancopoulos considered remodeling the company after Merck & Co, the U.S. drugmaker nicknamed "the house that research built" under the leadership of Vagelos. Merck spread its bets across many diseases and its many breakthroughs included the first "statin" cholesterol drug.
"Vagelos had always been a hero of mine because I'm also the son of a Greek immigrant," Yancopoulos said. "My father cut out a Greek-language newspaper article for me in the 1970s when Roy became head of research at Merck and said: 'If you're going to become a scientist instead of a doctor,' which was always his dream for me, 'at least become like this scientist.'"
Yancopoulos said he and Schleifer "always talked about Roy. And we thought just like he and Merck followed the science as a foundation, we could do that as well."
Schleifer startled Yancopoulos one day with a suggestion that they hire Vagelos instead of just emulating him.
"At that time, Roy had been a top CEO in America for a half dozen years in a row, so I told Len: 'Roy is not even going to answer your phone calls!'" Yancopoulos said.
"But Len called up Roy, and Roy answered. And he was planning to step down at Merck and wanted to do something different, to get into this biotech-type stuff. And Len convinced him to come visit Regeneron for a couple of days, for me to give him a total overview of the company."
Vagelos became chairman of Regeneron's board in 1995 and made a deep impression. He urged the company to study other diseases and to exploit its understanding of cell receptors -- proteins on the surface of cells that set into motion biological reactions within the cell.
Likewise, Vagelos stressed the company's other strong suit developed during its years of neuroscience research -- its expertise in cell signaling, communication among cells and how errors in that process can lead to disease.
Fifteen years later, Vagelos remains a totally involved company chairman and owns 1.8 percent of Regeneron's shares.
"He comes to every one of our scientific advisory meetings and talks on a daily basis to me and Len," Yancopoulos said. "He's in here at least one or two days a week and knows as much as I do on everything that's going on -- every single one of our programs. At the age of 80, he's in better physical shape than Len or I, and he's still my hero."
mfg ipollit
http://www.reuters.com/article/idUSTRE64B37U20100512
Exclusive: Biotech Regeneron on verge of big leagues
(Reuters) - Regeneron Pharmaceuticals Inc, a relatively unknown biotechnology company based in sleepy Tarrytown, New York, is not exactly a household name. But that is likely to change fast.
The company plans to steal the limelight in the coming year with data from seven late-stage trials of its experimental drugs for gout, colon cancer and a leading cause of blindness. It is rare for a biotech of any size to have so much pivotal news coming out in such a short period of time.
"The cycle is getting to be in full swing," said Chief Executive Leonard Schleifer. "We need a big hit on one of these drugs and then I think the world will start to pay a lot more attention to our pipeline" of new medicines.
In an exclusive interview with Reuters at company headquarters, Schleifer and his research chief George Yancopoulos outlined their plan for leaping from obscurity to the big leagues.
They know from experience, however, that in biotech there is no sure thing. In March 2003, when the company's Axokine obesity treatment stumbled in clinical trials, Regeneron shares lost more than half their value.
But success in even one of its three treatments now in late-stage studies could propel Regeneron from a typical money-losing biotech into a profitable company with a bright future. Its products could eventually take on Roche Holding AG's $6 billion cancer drug, Avastin, and $3 billion Lucentis eye-disease medicine. The gout drug could attract annual sales of $500 million or more, analysts estimate.
Regeneron on Wednesday said two other drugs in earlier-stage trials significantly cut levels of "bad" LDL cholesterol and eased knee pain from osteoarthritis.
But Wall Street is more focused on its drugs in late-stage trials.
"We believe that Regeneron should represent a core holding in biotechnology portfolios, based on the depth of its pipeline and its partnerships," Roth Capital Partners analyst Joseph Pantginis said on Wednesday. He said he is encouraged by new data from the early-stage trials.
Regeneron has some big backers and one of the biggest research budgets of any biotech, at more than $700 million this year. That includes spending by collaborators and $160 million of support from research partner Sanofi-Aventis SA.
It currently sells only one product, called Arcalyst, that brings in less than $20 million a year and treats a rare inherited inflammatory condition.
But the three drugs now wrapping up late-stage studies, the final step before seeking regulatory approval, have far bigger sales potential.
NOT IMMUNE TO FAILURE
Most biotech companies are staking their futures on only one or two drugs and many have gone bankrupt trying. Regeneron, which is now testing eight drugs, plans to have as many as 40 more in trials by 2017 under its lucrative deal with Sanofi to test Regeneron antibodies against a wide range of diseases.
Regeneron claims to have perhaps the most effective drug-making technology -- mice with human immune-system genes that produce better antibodies than rival companies.
It cites another major weapon: the continual hands-on involvement of Merck & Co Inc's legendary former Chief Executive Roy Vagelos.
The stock, with big current stakes held by Sanofi, T. Rowe Price Group affiliates and Carl Icahn, has risen 57 percent over the past year, compared with a 70 percent gain for the NYSARC Biotech Index and a 27 percent advance for the Standard & Poor's 500. In morning trade, it rose a further $1.09 or 4.3 percent to $26.61 on Nasdaq.
Regeneron's market capitalization is about $2 billion, far below industry leaders such as Amgen Inc at $53 billion and $129 billion for Roche, owner of Genentech, a biotech Regeneron emulates.
The company is based in the hometown of author Washington Irving, whose famous tale "Rip Van Winkle" concerns a man who falls asleep for 20 years and awakens to find everything changed. But unlike Rip, who was habitually idle, Regeneron has worked at a feverish pace since 1988 to change everything about drug development.
By next month, Regeneron plans to release data from Phase III trials of its drug to prevent and treat pain from gout, a form of arthritis that often hits the big toe. It uses the same active ingredient as Arcalyst, rilonacept, and works by blocking an inflammation-causing protein called Interleukin-1.
In the second half of the year it plans to provide data from late-stage trials of its macular degeneration product, called VEGF Trap, and its aflibercept drug for colon cancer. Both drugs, like Avastin, block the VEGF protein which is involved in growth of blood vessels.
Schleifer said the company's top commercial opportunity is probably VEGF-Trap, being developed with Bayer AG.
BIG DREAMS, TINY OFFICE
The company was started in 1988 by Schleifer, an assistant professor of neurology at Cornell University Medical College. He planned to discover nerve-growth proteins that could cure some of the grimmest neurological scourges by regenerating neurons, which are nerve cells. Thus the name Regeneron.
"The original idea, which you can find in the original business plan, was that we would discover them and then spritz them on people who needed them. We would cure a couple of diseases such as Lou Gehrig's disease, maybe Parkinson's disease and Alzheimer's disease, and sail off into the sunset," Schleifer said.
Schleifer nurtured those lofty ambitions in a dilapidated studio apartment meant to house Cornell's medical staff, located on Manhattan's Upper East Side.
"The super was thrown out of the building because he was a drunk, so I convinced the hospital to let me sublet it for awhile. The rent was about $500. That was Regeneron's world headquarters. It was just me."
Schleifer, a native of New York City's borough of Queens with a gift of gab to match his scientific enthusiasm, attracted a scientific advisory board that included several Nobel laureates and obtained a $1 million grubstake from a venture capital unit of Merrill Lynch.
He set out to recruit a stable of scientists, most notably Yancopoulos, a 28-year-old molecular immunologist who had just won a kind of genius award for his research at Columbia Medical School and is now Regeneron's head of research.
The company spread its wings in 1989 by renting 15,000 square feet of an empty Union Carbide factory in Tarrytown, where laboratories were built.
Regeneron could afford the bigger digs because earlier that month it won $10 million in funding from Sumitomo Chemical Co Ltd in exchange for giving the company rights to sell up to three of its future drugs in Japan.
At the time, only one protein involved in the growth and survival of neurons -- or neurotrophic factor -- had been discovered: Nerve Growth Factor. But within weeks after Regeneron's lab opened, Yancopoulos discovered a second one that became known as Brain-Derived Neurotrophic Factor (BDNF).
"Literally within a week after that, we cloned another one called Neurotrophin 3 (NT3) and then a third one called Ciliary Neurotrophic Factor (CNTF)," Yancopoulos said.
Yancopoulos became one of the world's most-cited neurologists due to his rapid-fire discoveries. The attention led to a $100 million partnership in 1990 with Amgen, an up-and-coming new biotech, that involved trials of BDNF against Lou Gehrig's disease and of NT3 for nerve pain in diabetics.
Regeneron also began testing CNTF against Lou Gehrig's disease and signed a $135 million deal in 1997 with Procter & Gamble Co to develop a number of Regeneron drugs, including using CNTF to treat obesity.
But none of the drugs ultimately panned out in clinical trials during a 10-year period.
"In many ways you could look at that as a lost decade because the ultimate prize eluded us in terms of applying our first discoveries," Yancopoulos said. "These neurological diseases were really challenging and we recognized maybe we weren't as smart as we thought we were and didn't have the experience that maybe we needed."
A MODEL IN MERCK
Schleifer and Yancopoulos considered remodeling the company after Merck & Co, the U.S. drugmaker nicknamed "the house that research built" under the leadership of Vagelos. Merck spread its bets across many diseases and its many breakthroughs included the first "statin" cholesterol drug.
"Vagelos had always been a hero of mine because I'm also the son of a Greek immigrant," Yancopoulos said. "My father cut out a Greek-language newspaper article for me in the 1970s when Roy became head of research at Merck and said: 'If you're going to become a scientist instead of a doctor,' which was always his dream for me, 'at least become like this scientist.'"
Yancopoulos said he and Schleifer "always talked about Roy. And we thought just like he and Merck followed the science as a foundation, we could do that as well."
Schleifer startled Yancopoulos one day with a suggestion that they hire Vagelos instead of just emulating him.
"At that time, Roy had been a top CEO in America for a half dozen years in a row, so I told Len: 'Roy is not even going to answer your phone calls!'" Yancopoulos said.
"But Len called up Roy, and Roy answered. And he was planning to step down at Merck and wanted to do something different, to get into this biotech-type stuff. And Len convinced him to come visit Regeneron for a couple of days, for me to give him a total overview of the company."
Vagelos became chairman of Regeneron's board in 1995 and made a deep impression. He urged the company to study other diseases and to exploit its understanding of cell receptors -- proteins on the surface of cells that set into motion biological reactions within the cell.
Likewise, Vagelos stressed the company's other strong suit developed during its years of neuroscience research -- its expertise in cell signaling, communication among cells and how errors in that process can lead to disease.
Fifteen years later, Vagelos remains a totally involved company chairman and owns 1.8 percent of Regeneron's shares.
"He comes to every one of our scientific advisory meetings and talks on a daily basis to me and Len," Yancopoulos said. "He's in here at least one or two days a week and knows as much as I do on everything that's going on -- every single one of our programs. At the age of 80, he's in better physical shape than Len or I, and he's still my hero."
mfg ipollit
Antwort auf Beitrag Nr.: 39.513.821 von BrauchGeld am 13.05.10 12:39:38ja leider, Naproxcinod wird ziemlich sicher nicht zugelassen... NicOx ist damit keine Investition mehr wert
http://www.businessweek.com/news/2010-05-13/nicox-falls-afte…
NicOx Falls After Failing to Win FDA Panel’s Backing (Update3)
May 13, 2010, 11:51 AM EDT
By Catherine Larkin and Michelle Fay Cortez
May 13 (Bloomberg) -- NicOx SA, a French drug developer, fell the most in seven years in Paris trading after failing to win a U.S. panel’s backing to introduce its first product.
NicOx plunged 2.33 euros, or 45 percent, to 2.90 euros. It was the stock’s biggest decline since Feb. 20, 2003.
An outside advisory panel to the Food and Drug Administration voted yesterday against approval of NicOx’s naproxcinod, a painkiller designed to pose fewer heart risks than current therapies, including Pfizer Inc.’s Celebrex. The panel said the drugmaker didn’t prove the medicine was less likely to raise blood pressure. While the FDA usually follows the advisory panel’s recommendations, it isn’t required to do so.
The vote, 16-1 with one abstention, may signal the end of the road for naproxcinod in the U.S., said Sam Fazeli, an analyst with Piper Jaffray in London. Panel members asked for definitive trials to show the drug reduces the risk of heart disease, is safer for the stomach and doesn’t cause kidney damage. Trials could take as long as five years and cost more than NicOx can afford, Fazeli said.
“These would need to be large multiyear studies, including high-risk and vulnerable populations,” Fazeli, who rates the stock “underweight,” wrote in a note to investors. “We expect the FDA to follow the panel’s opinion and, thus, have removed the U.S. opportunity from our model.”
Arthritis
About 20 million Americans have osteoarthritis, where cartilage that cushions the joints erodes and bones rub together painfully, according to the National Institutes of Health. Naproxcinod is a modified form of naproxen, a non-steroidal anti-inflammatory drug, or NSAID, in the same family of medicines as Celebrex and Merck & Co.’s recalled Vioxx painkiller.
The FDA is scheduled to decide on NicOx’s application by July 24. The drugmaker has also asked for marketing permission from European regulators.
“This is just advice,” Elizabeth Robinson, president of NicOx’s research institute, said in an interview after the vote. “Our next step would be to continue talking with the FDA.”
The FDA said NicOx’s data showed inconsistent blood-pressure benefits for naproxcinod compared with naproxen. Naproxcinod was primarily associated with lower blood pressure one to four hours after dosing, the agency said.
“Typically, drugs with meaningful effects on cardiac outcomes have an effect on systolic and diastolic blood pressure that persists through the entire dosing interval,” Suchitra Balakrishnan, a medical officer in the FDA’s Division of Cardiovascular and Renal Products, told the panel. “In this case, the potential impact on cardiac outcomes is unclear.”
mfg ipollit
http://www.businessweek.com/news/2010-05-13/nicox-falls-afte…
NicOx Falls After Failing to Win FDA Panel’s Backing (Update3)
May 13, 2010, 11:51 AM EDT
By Catherine Larkin and Michelle Fay Cortez
May 13 (Bloomberg) -- NicOx SA, a French drug developer, fell the most in seven years in Paris trading after failing to win a U.S. panel’s backing to introduce its first product.
NicOx plunged 2.33 euros, or 45 percent, to 2.90 euros. It was the stock’s biggest decline since Feb. 20, 2003.
An outside advisory panel to the Food and Drug Administration voted yesterday against approval of NicOx’s naproxcinod, a painkiller designed to pose fewer heart risks than current therapies, including Pfizer Inc.’s Celebrex. The panel said the drugmaker didn’t prove the medicine was less likely to raise blood pressure. While the FDA usually follows the advisory panel’s recommendations, it isn’t required to do so.
The vote, 16-1 with one abstention, may signal the end of the road for naproxcinod in the U.S., said Sam Fazeli, an analyst with Piper Jaffray in London. Panel members asked for definitive trials to show the drug reduces the risk of heart disease, is safer for the stomach and doesn’t cause kidney damage. Trials could take as long as five years and cost more than NicOx can afford, Fazeli said.
“These would need to be large multiyear studies, including high-risk and vulnerable populations,” Fazeli, who rates the stock “underweight,” wrote in a note to investors. “We expect the FDA to follow the panel’s opinion and, thus, have removed the U.S. opportunity from our model.”
Arthritis
About 20 million Americans have osteoarthritis, where cartilage that cushions the joints erodes and bones rub together painfully, according to the National Institutes of Health. Naproxcinod is a modified form of naproxen, a non-steroidal anti-inflammatory drug, or NSAID, in the same family of medicines as Celebrex and Merck & Co.’s recalled Vioxx painkiller.
The FDA is scheduled to decide on NicOx’s application by July 24. The drugmaker has also asked for marketing permission from European regulators.
“This is just advice,” Elizabeth Robinson, president of NicOx’s research institute, said in an interview after the vote. “Our next step would be to continue talking with the FDA.”
The FDA said NicOx’s data showed inconsistent blood-pressure benefits for naproxcinod compared with naproxen. Naproxcinod was primarily associated with lower blood pressure one to four hours after dosing, the agency said.
“Typically, drugs with meaningful effects on cardiac outcomes have an effect on systolic and diastolic blood pressure that persists through the entire dosing interval,” Suchitra Balakrishnan, a medical officer in the FDA’s Division of Cardiovascular and Renal Products, told the panel. “In this case, the potential impact on cardiac outcomes is unclear.”
mfg ipollit
VPHM... zwar schon ein wenig älter, aber noch ein paar Analystenmeinungen zu Viropharma zusammengetragen im Blog notablecalls...
http://notablecalls.blogspot.com/2010/03/viropharma-nasdaqvp…
Monday, March 22, 2010
Viropharma (NASDAQ:VPHM): Analysts positive after investor day; Target raised to $19 at Oppenheimer
Viropharma (NASDAQ:VPHM) is getting a lot of positive commentary this morning after the co held an investor event on Friday, March 19.
- Oppenheimer is raising their price target on VPHM to a new Street high of $19 (prev. $15) noting the investor meeting focused on Cinryze and the opportunity in hereditary angioedema (HAE), as well as future expansion opportunities. Importantly, 1) with only ~400 patients on Cinryze at YE09, out of a total of ~2,800 patients, the firm believes a robust US Cinryze launch should continue. 2) With ~12,500 HAE patients, they believe the EU represents a significant expansion opportunity. 3) Opco sees potential Cinryze expansion opportunities in renal transplantation, where a POC study will begin in late-2010/early-2011. 4) They raise their price target to $19 from $15 on higher peak US Cinryze sales ($400M) and addition of European sales. Reits Outperform.
- Jefferies ups their target to $18 (prev. $16) and reits Buy as Cinryze sales in Europe, as well as all other new geographies discussed at VPHM's analyst meeting, represent upside to firm's estimates. They continue to believe that the Street has underappreciated the Cinryze market opportunity.
For the first time, VPHM also outlined its plans for obtaining Cinryze approval in other countries outside of Europe. There are 28 countries targeted for Cinryze regulatory filing in 2010-2012 and another five countries where filings will occur in 2013-2014. The company believes that this could expand the target patient population by over 90,000 patients, assuming a consistent worldwide HAE prevalence of 1:40,000 (compared to 23,000-24,000 patients in the U.S. and Europe).
In addition, Jefferies calls VPHM one of the beneficiaries of the Health Care Reform. The new health care reform act is a major positive for companies with biologics, as it creates a 12-year exclusivity period prior to biosimilar competition. Orphan drug exclusivity in the U.S. for seven years is the primary protection for both VPHM"s Cinryze (exclusivity expires in Oct 2015) and AUXL's Xiaflex (February 2017). Both drugs would benefit from an additional five years of exclusivity under the new health care reform act. As they had modeled the impact of biosimilars differently in each case, the firm estimates that a 12-year exclusivity period would add $0-1 per share to their VPHM price target of $18 and $10 per share to their AUXL price target of $44.
- Thomas Weisel is raising their price target to $17 (prev. $14) to reflect the impact of an aggressive and comprehensive ex-U.S. commercialization strategy and believe the notion of potential label expansion into the transplant setting will support continued multiple expansion.
The role of complement inhibition is gaining traction in the transplant setting following the recent presentation of promising investigator-sponsored, proof-of-concept data with Alexion’s Soliris.
VPHM provided a compelling review of the biology behind C1 inhibition and believes Cinryze can be used to treat antibody mediated rejection (AMR) and delayed graft function (DGF) in kidney transplant patients and plans to initiate two proof-of-concept Phase II trials in each setting later this year.
TWP believes the HAE market opportunity remains underappreciated and believe the stock should continue to find decent support at these levels as Consensus numbers and excitement about the transplant opportunity increase after Friday’s event.
Notablecalls: Well, what's there not to like? VPHM used to be a crummy biopharma play with Vancocin, their best-selling drug going generic in a year or two. That's until Cinryze showed up. It's priced around $400,000/year putting VPHM in the same league with the other rare-disease plays like GENZ or BMRN.
Now it seems it's all about Cinryze and the analyst community has fallen in love with the drug and its potential.
mfg ipollit
http://notablecalls.blogspot.com/2010/03/viropharma-nasdaqvp…
Monday, March 22, 2010
Viropharma (NASDAQ:VPHM): Analysts positive after investor day; Target raised to $19 at Oppenheimer
Viropharma (NASDAQ:VPHM) is getting a lot of positive commentary this morning after the co held an investor event on Friday, March 19.
- Oppenheimer is raising their price target on VPHM to a new Street high of $19 (prev. $15) noting the investor meeting focused on Cinryze and the opportunity in hereditary angioedema (HAE), as well as future expansion opportunities. Importantly, 1) with only ~400 patients on Cinryze at YE09, out of a total of ~2,800 patients, the firm believes a robust US Cinryze launch should continue. 2) With ~12,500 HAE patients, they believe the EU represents a significant expansion opportunity. 3) Opco sees potential Cinryze expansion opportunities in renal transplantation, where a POC study will begin in late-2010/early-2011. 4) They raise their price target to $19 from $15 on higher peak US Cinryze sales ($400M) and addition of European sales. Reits Outperform.
- Jefferies ups their target to $18 (prev. $16) and reits Buy as Cinryze sales in Europe, as well as all other new geographies discussed at VPHM's analyst meeting, represent upside to firm's estimates. They continue to believe that the Street has underappreciated the Cinryze market opportunity.
For the first time, VPHM also outlined its plans for obtaining Cinryze approval in other countries outside of Europe. There are 28 countries targeted for Cinryze regulatory filing in 2010-2012 and another five countries where filings will occur in 2013-2014. The company believes that this could expand the target patient population by over 90,000 patients, assuming a consistent worldwide HAE prevalence of 1:40,000 (compared to 23,000-24,000 patients in the U.S. and Europe).
In addition, Jefferies calls VPHM one of the beneficiaries of the Health Care Reform. The new health care reform act is a major positive for companies with biologics, as it creates a 12-year exclusivity period prior to biosimilar competition. Orphan drug exclusivity in the U.S. for seven years is the primary protection for both VPHM"s Cinryze (exclusivity expires in Oct 2015) and AUXL's Xiaflex (February 2017). Both drugs would benefit from an additional five years of exclusivity under the new health care reform act. As they had modeled the impact of biosimilars differently in each case, the firm estimates that a 12-year exclusivity period would add $0-1 per share to their VPHM price target of $18 and $10 per share to their AUXL price target of $44.
- Thomas Weisel is raising their price target to $17 (prev. $14) to reflect the impact of an aggressive and comprehensive ex-U.S. commercialization strategy and believe the notion of potential label expansion into the transplant setting will support continued multiple expansion.
The role of complement inhibition is gaining traction in the transplant setting following the recent presentation of promising investigator-sponsored, proof-of-concept data with Alexion’s Soliris.
VPHM provided a compelling review of the biology behind C1 inhibition and believes Cinryze can be used to treat antibody mediated rejection (AMR) and delayed graft function (DGF) in kidney transplant patients and plans to initiate two proof-of-concept Phase II trials in each setting later this year.
TWP believes the HAE market opportunity remains underappreciated and believe the stock should continue to find decent support at these levels as Consensus numbers and excitement about the transplant opportunity increase after Friday’s event.
Notablecalls: Well, what's there not to like? VPHM used to be a crummy biopharma play with Vancocin, their best-selling drug going generic in a year or two. That's until Cinryze showed up. It's priced around $400,000/year putting VPHM in the same league with the other rare-disease plays like GENZ or BMRN.
Now it seems it's all about Cinryze and the analyst community has fallen in love with the drug and its potential.
mfg ipollit
Regeneron Schedules June 9, 2010 Teleconference and Webcast to Discuss Phase 3 Trial Results in Gout
TARRYTOWN, N.Y., June 8 /PRNewswire-FirstCall/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) today announced that it will hold a teleconference and webcast at 8:30 a.m. Eastern Time tomorrow, Wednesday, June 9, to discuss Phase 3 trial results for ARCALYST® (rilonacept) in the prevention of drug-induced gout flares and in the treatment of acute gout attacks. A press release will be issued tomorrow prior to the call.
----------
Ich bin extrem gespannt!
TARRYTOWN, N.Y., June 8 /PRNewswire-FirstCall/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) today announced that it will hold a teleconference and webcast at 8:30 a.m. Eastern Time tomorrow, Wednesday, June 9, to discuss Phase 3 trial results for ARCALYST® (rilonacept) in the prevention of drug-induced gout flares and in the treatment of acute gout attacks. A press release will be issued tomorrow prior to the call.
----------
Ich bin extrem gespannt!
Antwort auf Beitrag Nr.: 39.515.966 von ipollit am 13.05.10 17:56:46
In Ergänzung zu dem von ipollit geposteten Artikel über REGN:
Regeneron’s $2 Billion Research Surge Mimics Genentech Tactics
June 09, 2010, 12:20 AM EDT
June 9 (Bloomberg) -- Regeneron Pharmaceuticals Inc. will report study results as early as today on the first of three new drugs that may generate at least $2 billion a year in revenue.
The medicines -- for gout, eye disease and cancer -- are in the final phase of testing needed for U.S. marketing approval. The first finding, on the gout drug Arcalyst, may help generate $500 million a year in additional revenue, said Joseph Pantginis, an analyst with Roth Capital Partners in New York.
Having three drugs in late-stage trials is “pretty uncommon for a stand-alone biotech today,” said Ted Tenthoff, a Piper Jaffray & Co. analyst in New York. The treatments are emerging from Regeneron’s development of a drug discovery technology that may help the 22-year-old company with just one $20 million-a-year product rival Roche Holding AG’s Genentech Inc. unit, with its 10 products and $9.5 billion in sales, said Chief Executive Officer Len Schleifer in an interview.
“We don’t want the company to sink or swim on the back of any one thing, which is why we’re trying to move an army of ideas forward,” said Schleifer, in an interview at Regeneron’s headquarters in Tarrytown, New York. “We’ve modeled ourselves on the companies like Genentech that had a cadre of people and technologies.
Regeneron fell almost 2 percent, or 53 cents, to $26.50 yesterday in Nasdaq Stock Market composite trading after rising 66 percent in the 12 months before today. Shares could increase to $31.22, according to the average price target of nine analysts surveyed by Bloomberg.
‘Better Than 50-50 Chance’
Results from seven late-stage trials on the drugs are expected to be reported within 12 months, Schleifer said. Pantginis, of Roth Capital Partners, gives the treatments a “better than 50-50 chance” of success. If all the studies prove positive, the three products may generate at least $2 billion a year, Pantginis said.
That will represent a turnaround for a company that has had three research failures since it began in 1988.
In March 1994, the company’s shares tumbled 33 percent in a single day after weight loss and flulike symptoms were linked to its experimental drug for amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease. A second Lou Gehrig’s disease drug failed in January 1997, and shares lost about half their value.
In March 2003, an obesity treatment failed and, once again, the company’s value was cut in half.
‘Three Major Failures’
“The difference between a small company and a large company is how you fail,” Schleifer said. “In a large company, you bury the program in the middle of the night and no one comes to the funeral. We do it in the middle of the day, and it is front-page news.”
Following the failure of the two Lou Gehrig’s disease drugs, “it was quite clear we needed to do something different,” said P. Roy Vagelos, the chairman of Regeneron’s board. Vagelos, a physician, led research at Whitehouse, New Jersey-based Merck & Co. from 1976 to 1985, and was the drugmaker’s CEO for 10 years after that.
After the research failures, management met and decided to focus on development of a technology their scientists had been working on, rather than a single drug, Vagelos said. “We came up with the approach of traps to neutralize molecules that might be involved with the disease processes, an approach I liked,” he said.
Chemical “traps” work by binding to certain proteins, stopping them from activating cell receptors that spur a reaction. Arcalyst works by binding to interleukin-1, a protein that can trigger inflammation. Gout, a form of arthritis, occurs when uric acid builds up in the bloodstream, causing a painful swelling of joints in the toes and foot.
Eye Disease, Cancer
Regeneron scientists used the same concept to create an as- yet unnamed drug that works against the eye disease age-related macular degeneration and the cancer therapy aflibercept. In those cases, the binding process prevents blood vessel growth. Aflibercept is being tested as a first medication for prostate tumors and as a treatment for patients who fail initial therapy for colorectal and lung malignancies.
Approval for aflibercept in all three cancer indications would give it “blockbuster potential,” meaning it may sell $1 billion a year or more, according to Roth Capital’s Pantginis. The eye treatment may generate $500 million after regulatory clearance, he said.
Michael Yee, an analyst for RBC Capital Partners in San Francisco, said the company gains from having “strong partners” in Paris-based Sanofi-Aventis SA and Bayer AG, of Leverkusen, Germany, to help support drug development.
‘Big Pharma Partners’
The clinical trial load “is possible only because they have big pharma partners that are funding 50 percent of the studies,” RBC’s Yee, one of three analysts with a hold rating on the company surveyed by Bloomberg, said in a telephone interview. Eight analysts rate the stock a buy.
Bayer is collaborating on the eye drug. Regeneron will get all the U.S. sales, and Regeneron and Bayer will share profits outside the U.S., according to an October 2006 agreement. Bayer made an upfront payment of $75 million, and Regeneron may earn up to $245 million in sales milestones.
Sanofi owns 18.6 percent of Regeneron’s shares and pays the company $160 million a year to help with its research. Regeneron stands to receive as much as $250 million in payments if the products top $1 billion in revenue outside the U.S.
Under the agreement, Sanofi has the option to co-develop each new antibody Regeneron discovers. The profits in the U.S. will be shared equally, and outside the U.S. will be split on a sliding scale, with Sanofi’s share ranging from 65 percent to 55 percent. The partnership began November 2007 and was expanded in November 2009.
‘Excited About Alliance’
“We’re very excited about that alliance,” said Paul Chew, the U.S. chief medical officer for Sanofi. “Sanofi has the resources, and Regeneron has the technology and the know-how. We’ve preserved the strengths of each.”
Genentech too gained from its relationship with a partner. Swiss drugmaker Roche owned 56 percent of the South San Francisco, California-based biotechnology company for more than 18 years, until acquiring Genentech last year. Sanofi’s Chew declined to say whether Sanofi might acquire Regeneron in the future.
Schleifer said his drive to be like Genentech stops at the point when that company was acquired by its partner. He’s not isn’t interested in being bought, he said.
“We’re not building a company to sell a company,” Schleifer said. “We’re building a company to deliver drugs that make a difference and that will deliver value to shareholders. If you really want to capture the innovativeness of a small company, you leave them alone.”
Not ‘Sanofized’
Having Sanofi as a partner will help with that goal, Schleifer said. The French drugmaker hasn’t “Sanofized” Regeneron, he said, and he doesn’t believe they’ll try.
“The diversified strategy is something we like to see in biotech,” said Mark Monane, a New York-based analyst for Needham & Co., in a telephone interview. “It’s an important year for the company, as we’ll get to open the envelope on the late-stage products.”
--Editors: Reg Gale, Andrew Pollack
To contact the reporter on this story: Elizabeth Lopatto in New York at elopatto@bloomberg.net.
To contact the editor responsible for this story: Reg Gale at rgale5@bloomberg.net.
----------------
In etwas mehr als einer Stunde sind wir schlauer...
In Ergänzung zu dem von ipollit geposteten Artikel über REGN:
Regeneron’s $2 Billion Research Surge Mimics Genentech Tactics
June 09, 2010, 12:20 AM EDT
June 9 (Bloomberg) -- Regeneron Pharmaceuticals Inc. will report study results as early as today on the first of three new drugs that may generate at least $2 billion a year in revenue.
The medicines -- for gout, eye disease and cancer -- are in the final phase of testing needed for U.S. marketing approval. The first finding, on the gout drug Arcalyst, may help generate $500 million a year in additional revenue, said Joseph Pantginis, an analyst with Roth Capital Partners in New York.
Having three drugs in late-stage trials is “pretty uncommon for a stand-alone biotech today,” said Ted Tenthoff, a Piper Jaffray & Co. analyst in New York. The treatments are emerging from Regeneron’s development of a drug discovery technology that may help the 22-year-old company with just one $20 million-a-year product rival Roche Holding AG’s Genentech Inc. unit, with its 10 products and $9.5 billion in sales, said Chief Executive Officer Len Schleifer in an interview.
“We don’t want the company to sink or swim on the back of any one thing, which is why we’re trying to move an army of ideas forward,” said Schleifer, in an interview at Regeneron’s headquarters in Tarrytown, New York. “We’ve modeled ourselves on the companies like Genentech that had a cadre of people and technologies.
Regeneron fell almost 2 percent, or 53 cents, to $26.50 yesterday in Nasdaq Stock Market composite trading after rising 66 percent in the 12 months before today. Shares could increase to $31.22, according to the average price target of nine analysts surveyed by Bloomberg.
‘Better Than 50-50 Chance’
Results from seven late-stage trials on the drugs are expected to be reported within 12 months, Schleifer said. Pantginis, of Roth Capital Partners, gives the treatments a “better than 50-50 chance” of success. If all the studies prove positive, the three products may generate at least $2 billion a year, Pantginis said.
That will represent a turnaround for a company that has had three research failures since it began in 1988.
In March 1994, the company’s shares tumbled 33 percent in a single day after weight loss and flulike symptoms were linked to its experimental drug for amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease. A second Lou Gehrig’s disease drug failed in January 1997, and shares lost about half their value.
In March 2003, an obesity treatment failed and, once again, the company’s value was cut in half.
‘Three Major Failures’
“The difference between a small company and a large company is how you fail,” Schleifer said. “In a large company, you bury the program in the middle of the night and no one comes to the funeral. We do it in the middle of the day, and it is front-page news.”
Following the failure of the two Lou Gehrig’s disease drugs, “it was quite clear we needed to do something different,” said P. Roy Vagelos, the chairman of Regeneron’s board. Vagelos, a physician, led research at Whitehouse, New Jersey-based Merck & Co. from 1976 to 1985, and was the drugmaker’s CEO for 10 years after that.
After the research failures, management met and decided to focus on development of a technology their scientists had been working on, rather than a single drug, Vagelos said. “We came up with the approach of traps to neutralize molecules that might be involved with the disease processes, an approach I liked,” he said.
Chemical “traps” work by binding to certain proteins, stopping them from activating cell receptors that spur a reaction. Arcalyst works by binding to interleukin-1, a protein that can trigger inflammation. Gout, a form of arthritis, occurs when uric acid builds up in the bloodstream, causing a painful swelling of joints in the toes and foot.
Eye Disease, Cancer
Regeneron scientists used the same concept to create an as- yet unnamed drug that works against the eye disease age-related macular degeneration and the cancer therapy aflibercept. In those cases, the binding process prevents blood vessel growth. Aflibercept is being tested as a first medication for prostate tumors and as a treatment for patients who fail initial therapy for colorectal and lung malignancies.
Approval for aflibercept in all three cancer indications would give it “blockbuster potential,” meaning it may sell $1 billion a year or more, according to Roth Capital’s Pantginis. The eye treatment may generate $500 million after regulatory clearance, he said.
Michael Yee, an analyst for RBC Capital Partners in San Francisco, said the company gains from having “strong partners” in Paris-based Sanofi-Aventis SA and Bayer AG, of Leverkusen, Germany, to help support drug development.
‘Big Pharma Partners’
The clinical trial load “is possible only because they have big pharma partners that are funding 50 percent of the studies,” RBC’s Yee, one of three analysts with a hold rating on the company surveyed by Bloomberg, said in a telephone interview. Eight analysts rate the stock a buy.
Bayer is collaborating on the eye drug. Regeneron will get all the U.S. sales, and Regeneron and Bayer will share profits outside the U.S., according to an October 2006 agreement. Bayer made an upfront payment of $75 million, and Regeneron may earn up to $245 million in sales milestones.
Sanofi owns 18.6 percent of Regeneron’s shares and pays the company $160 million a year to help with its research. Regeneron stands to receive as much as $250 million in payments if the products top $1 billion in revenue outside the U.S.
Under the agreement, Sanofi has the option to co-develop each new antibody Regeneron discovers. The profits in the U.S. will be shared equally, and outside the U.S. will be split on a sliding scale, with Sanofi’s share ranging from 65 percent to 55 percent. The partnership began November 2007 and was expanded in November 2009.
‘Excited About Alliance’
“We’re very excited about that alliance,” said Paul Chew, the U.S. chief medical officer for Sanofi. “Sanofi has the resources, and Regeneron has the technology and the know-how. We’ve preserved the strengths of each.”
Genentech too gained from its relationship with a partner. Swiss drugmaker Roche owned 56 percent of the South San Francisco, California-based biotechnology company for more than 18 years, until acquiring Genentech last year. Sanofi’s Chew declined to say whether Sanofi might acquire Regeneron in the future.
Schleifer said his drive to be like Genentech stops at the point when that company was acquired by its partner. He’s not isn’t interested in being bought, he said.
“We’re not building a company to sell a company,” Schleifer said. “We’re building a company to deliver drugs that make a difference and that will deliver value to shareholders. If you really want to capture the innovativeness of a small company, you leave them alone.”
Not ‘Sanofized’
Having Sanofi as a partner will help with that goal, Schleifer said. The French drugmaker hasn’t “Sanofized” Regeneron, he said, and he doesn’t believe they’ll try.
“The diversified strategy is something we like to see in biotech,” said Mark Monane, a New York-based analyst for Needham & Co., in a telephone interview. “It’s an important year for the company, as we’ll get to open the envelope on the late-stage products.”
--Editors: Reg Gale, Andrew Pollack
To contact the reporter on this story: Elizabeth Lopatto in New York at elopatto@bloomberg.net.
To contact the editor responsible for this story: Reg Gale at rgale5@bloomberg.net.
----------------
In etwas mehr als einer Stunde sind wir schlauer...
Regeneron Pharmaceuticals, Inc. Announces ARCALYST (rilonacept) Meets Primary And All Secondary Endpoints In Phase 3 Trial Of Prevention Of Gout Flares In Patients Initiating Allopurinol Therapy
6:55am EDT
Regeneron Pharmaceuticals, Inc. announced that a Phase 3 study in gout patients initiating allopurinol therapy to lower their uric acid levels showed that ARCALYST (rilonacept), also known as IL-1 Trap, prevented gout attacks, as measured by the primary endpoint of the number of gout flares per patient over the 16 week treatment period. Primary Endpoint; Patients who received ARCALYST at a weekly, self-administered, subcutaneous dose of 160 milligrams (mg) had an 80% decrease in mean number of gout flares compared to the placebo group over the 16 week treatment period (0.21 flares vs. 1.06 flares, p<0.0001). Patients who received ARCALYST at a weekly dose of 80 mg had a 73% decrease compared to the placebo group (0.29 flares vs. 1.06 flares, p<0.0001). Key Secondary Endpoints; All secondary endpoints of the study were highly positive (p<0.001 vs. placebo). These include: Treatment with ARCALYST reduced the proportion of patients who experienced two or more flares during the study period by up to 88% (3.7% with ARCALYST 160 mg, 5.0% with ARCALYST 80 mg, and 31.6% with placebo, p<0.0001). Treatment with ARCALYST reduced the proportion of patients who experienced at least one gout flare during the study period by up to 65% (16.3% with ARCALYST 160 mg, 18.8% with ARCALYST 80 mg, and 46.8% with placebo, p<0.001). ARCALYST was generally well tolerated with no reported drug-related serious adverse events. - - - - - (repeated)
-----------
Klingt für mich wie ein Volltreffer. Bin aber kein Experte und warte mal auf fachmännische Kommentare bzw. auf die Reaktion des Marktes.
6:55am EDT
Regeneron Pharmaceuticals, Inc. announced that a Phase 3 study in gout patients initiating allopurinol therapy to lower their uric acid levels showed that ARCALYST (rilonacept), also known as IL-1 Trap, prevented gout attacks, as measured by the primary endpoint of the number of gout flares per patient over the 16 week treatment period. Primary Endpoint; Patients who received ARCALYST at a weekly, self-administered, subcutaneous dose of 160 milligrams (mg) had an 80% decrease in mean number of gout flares compared to the placebo group over the 16 week treatment period (0.21 flares vs. 1.06 flares, p<0.0001). Patients who received ARCALYST at a weekly dose of 80 mg had a 73% decrease compared to the placebo group (0.29 flares vs. 1.06 flares, p<0.0001). Key Secondary Endpoints; All secondary endpoints of the study were highly positive (p<0.001 vs. placebo). These include: Treatment with ARCALYST reduced the proportion of patients who experienced two or more flares during the study period by up to 88% (3.7% with ARCALYST 160 mg, 5.0% with ARCALYST 80 mg, and 31.6% with placebo, p<0.0001). Treatment with ARCALYST reduced the proportion of patients who experienced at least one gout flare during the study period by up to 65% (16.3% with ARCALYST 160 mg, 18.8% with ARCALYST 80 mg, and 46.8% with placebo, p<0.001). ARCALYST was generally well tolerated with no reported drug-related serious adverse events. - - - - - (repeated)
-----------
Klingt für mich wie ein Volltreffer. Bin aber kein Experte und warte mal auf fachmännische Kommentare bzw. auf die Reaktion des Marktes.
Antwort auf Beitrag Nr.: 39.655.540 von SLGramann am 09.06.10 14:02:29
Ambitious Regeneron unveils mixed PhIII results for gout drug
June 9, 2010 — 9:09am ET | By John Carroll
Inspired by a string of development failures, Tarrytown, NY-based Regeneron Pharmaceuticals is endeavoring to find out if it can duplicate the ambitious R&D strategy that made Genentech a star in the biotech world. And if the developer can bring in approvals on all three of its drug programs now in late-stage development, analysts estimate the potential reward at $2 billion in annual revenue.
This morning, Regeneron Pharmaceuticals announced that its gout drug Arcalyst hit its goals in a late-stage study examining its ability to prevent new attacks of the disease but flunked a separate Phase III designed to demonstrate its effectiveness at reducing gout-related pain.
Patients enrolled in the gout attack trial demonstrated an 80 percent reduction in flare-ups compared to placebo when given a 160 mg dose; there was a 73 percent reduction for subjects taking an 80 mg dose. In the separate trial, Arcalyst failed to demonstrate an ability to reduce pain once the condition was triggered. And Regeneron says it will now wait for two more rounds of late-stage data on the drug before it pursues an FDA approval in mid-2011.
Regeneron's gout trials are being pursued at the same time the biotech company has several other Phase IIIs underway for a new cancer medication as well as an eye disease therapy--a broad range of disease targets and studies for a relatively small biotech company to undertake at one time. Regeneron has data from five more late-stage trials due over the next 12 months.
"We don't want the company to sink or swim on the back of any one thing, which is why we're trying to move an army of ideas forward," said CEO Len Schleifer, in an interview with Bloomberg. "We've modeled ourselves on the companies like Genentech that had a cadre of people and technologies."
---------
Aktie ca. 7% im Minus.
Ambitious Regeneron unveils mixed PhIII results for gout drug
June 9, 2010 — 9:09am ET | By John Carroll
Inspired by a string of development failures, Tarrytown, NY-based Regeneron Pharmaceuticals is endeavoring to find out if it can duplicate the ambitious R&D strategy that made Genentech a star in the biotech world. And if the developer can bring in approvals on all three of its drug programs now in late-stage development, analysts estimate the potential reward at $2 billion in annual revenue.
This morning, Regeneron Pharmaceuticals announced that its gout drug Arcalyst hit its goals in a late-stage study examining its ability to prevent new attacks of the disease but flunked a separate Phase III designed to demonstrate its effectiveness at reducing gout-related pain.
Patients enrolled in the gout attack trial demonstrated an 80 percent reduction in flare-ups compared to placebo when given a 160 mg dose; there was a 73 percent reduction for subjects taking an 80 mg dose. In the separate trial, Arcalyst failed to demonstrate an ability to reduce pain once the condition was triggered. And Regeneron says it will now wait for two more rounds of late-stage data on the drug before it pursues an FDA approval in mid-2011.
Regeneron's gout trials are being pursued at the same time the biotech company has several other Phase IIIs underway for a new cancer medication as well as an eye disease therapy--a broad range of disease targets and studies for a relatively small biotech company to undertake at one time. Regeneron has data from five more late-stage trials due over the next 12 months.
"We don't want the company to sink or swim on the back of any one thing, which is why we're trying to move an army of ideas forward," said CEO Len Schleifer, in an interview with Bloomberg. "We've modeled ourselves on the companies like Genentech that had a cadre of people and technologies."
---------
Aktie ca. 7% im Minus.
hallo sl gramann
wie ist denn deine meinung (einschätzung) zu onyx ?????der doch herbe rückgang gerechtfertigt oder durch zocker runter geprügelt ??
bin gestern bei 21,90$ eingestiegen.
danke
asics
wie ist denn deine meinung (einschätzung) zu onyx ?????der doch herbe rückgang gerechtfertigt oder durch zocker runter geprügelt ??
bin gestern bei 21,90$ eingestiegen.
danke
asics
UPDATE 3-Regeneron gout drug works in one trial, fails other
Wed Jun 9, 2010 12:51pm EDT
Stocks
Regeneron Pharmaceuticals, Inc.
REGN.O
* Gout drug works as preventive therapy, fails as treatment
* Co testing the drug in two other preventive trials
* Co sees filing for approval mid-2011
* Shares fall as much as 8 percent (Adds details, more analyst comments; updates stock movement)
By Esha Dey
BANGALORE, June 9 (Reuters) - Regeneron Pharmaceuticals Inc (REGN.O) said a late-stage study of its experimental gout drug met the main trial goal of preventing gout attacks, while a second late-stage study failed to improve gout-related pain, sending its shares down 8 percent.
However, analysts viewed Tuesday's news as mostly positive and do not expect the failure of the pain trial to affect the chances of the drug Arcalyst getting approved as a preventive therapy.
Roth Capital Partners analyst Joseph Pantginis, who raised his price target on the stock to $38 from $35, said he now sees a 60 percent chance of the drug geting approval in gout prevention, compared to his prior estimate of a 50 percent chance of success.
Pantginis maintained his "buy" rating on the stock.
Gout, a painful type of arthritis in which a build up of uric acid causes swollen joints, affects about 5 million Americans.
The company is developing Arcalyst for preventing or treating gout flares in patients who received uric acid-lowering therapy. The therapy often triggers a rise in frequency of gout attacks in the first several months of treatment.
The drug is already approved by U.S. regulators for the treatment of Cryopyrin-Associated Periodic Syndromes -- a group of rare, inherited auto-inflammatory conditions.
The late-stage study that tested the drug, generically known as rilonacept, as a preventive therapy in 241 patients showed 160 milligrams dose of the drug decreased gout flares by 80 percent as compared to a dummy drug, while the 80 mg dose showed a 73 percent reduction.
The second late-stage trial, which evaluated the drug as a treatment for acute gout flares in 225 patients, did not meet the main goal of significantly reducing the average intensity of gout pain, as compared to a common, but potent, painkiller.
"The pain study was higher risk, as less data is available there previously," RBC Capital Markets analyst Michael Yee said, adding that he sees the prevention data to be "robust."
The company said pending successful results from two other ongoing studies on the drug as a preventive therapy, it plans to file for regulatory approval by mid-2011.
RBC analyst Yee said flare prevention is a few hundred million dollars opportunity, though the challenge for the company will be to get doctors to use the drug upfront even before a flare has happened.
Treatment with Arcalyst, in both the studies, was generally well tolerated with no reported drug-related serious adverse events, Regeneron said in a statement.
The most commonly reported adverse event in the preventive study was injection site reaction, and that in the pain treatment study was headache.
Other U.S. companies developing gout treatments are Savient Pharmaceuticals Inc (SVNT.O), Ardea Biosciences Inc (RDEA.O) and BioCryst Pharmaceuticals Inc (BCRX.O). Swiss drugmaker Novartis AG (NOVN.VX) is also developing a treatment for gout.
Shares of Regeneron were down 3 percent at $25.83 in afternoon trade on Nasdaq. They touched a low of $24.32 earlier in the day. (Reporting by Esha Dey in Bangalore; Editing by Aradhana Aravindan, Roshni Menon)
------------------------
@asics, sorry, da habe ich keine Einschätzung. Vielleicht möchte sich ipollit dazu äußern?
Wed Jun 9, 2010 12:51pm EDT
Stocks
Regeneron Pharmaceuticals, Inc.
REGN.O
* Gout drug works as preventive therapy, fails as treatment
* Co testing the drug in two other preventive trials
* Co sees filing for approval mid-2011
* Shares fall as much as 8 percent (Adds details, more analyst comments; updates stock movement)
By Esha Dey
BANGALORE, June 9 (Reuters) - Regeneron Pharmaceuticals Inc (REGN.O) said a late-stage study of its experimental gout drug met the main trial goal of preventing gout attacks, while a second late-stage study failed to improve gout-related pain, sending its shares down 8 percent.
However, analysts viewed Tuesday's news as mostly positive and do not expect the failure of the pain trial to affect the chances of the drug Arcalyst getting approved as a preventive therapy.
Roth Capital Partners analyst Joseph Pantginis, who raised his price target on the stock to $38 from $35, said he now sees a 60 percent chance of the drug geting approval in gout prevention, compared to his prior estimate of a 50 percent chance of success.
Pantginis maintained his "buy" rating on the stock.
Gout, a painful type of arthritis in which a build up of uric acid causes swollen joints, affects about 5 million Americans.
The company is developing Arcalyst for preventing or treating gout flares in patients who received uric acid-lowering therapy. The therapy often triggers a rise in frequency of gout attacks in the first several months of treatment.
The drug is already approved by U.S. regulators for the treatment of Cryopyrin-Associated Periodic Syndromes -- a group of rare, inherited auto-inflammatory conditions.
The late-stage study that tested the drug, generically known as rilonacept, as a preventive therapy in 241 patients showed 160 milligrams dose of the drug decreased gout flares by 80 percent as compared to a dummy drug, while the 80 mg dose showed a 73 percent reduction.
The second late-stage trial, which evaluated the drug as a treatment for acute gout flares in 225 patients, did not meet the main goal of significantly reducing the average intensity of gout pain, as compared to a common, but potent, painkiller.
"The pain study was higher risk, as less data is available there previously," RBC Capital Markets analyst Michael Yee said, adding that he sees the prevention data to be "robust."
The company said pending successful results from two other ongoing studies on the drug as a preventive therapy, it plans to file for regulatory approval by mid-2011.
RBC analyst Yee said flare prevention is a few hundred million dollars opportunity, though the challenge for the company will be to get doctors to use the drug upfront even before a flare has happened.
Treatment with Arcalyst, in both the studies, was generally well tolerated with no reported drug-related serious adverse events, Regeneron said in a statement.
The most commonly reported adverse event in the preventive study was injection site reaction, and that in the pain treatment study was headache.
Other U.S. companies developing gout treatments are Savient Pharmaceuticals Inc (SVNT.O), Ardea Biosciences Inc (RDEA.O) and BioCryst Pharmaceuticals Inc (BCRX.O). Swiss drugmaker Novartis AG (NOVN.VX) is also developing a treatment for gout.
Shares of Regeneron were down 3 percent at $25.83 in afternoon trade on Nasdaq. They touched a low of $24.32 earlier in the day. (Reporting by Esha Dey in Bangalore; Editing by Aradhana Aravindan, Roshni Menon)
------------------------
@asics, sorry, da habe ich keine Einschätzung. Vielleicht möchte sich ipollit dazu äußern?
So, ein letzter Artikel von mir zum Thema Arcalyst bei Gicht. Diesmal von Prohost (bin mir aber nicht sicher, was von denen zu halten ist...):
Regeneron’s (REGN) Phase 3 study in gout patients initiating allopurinol therapy to lower their uric acid levels showed that the company’s drug Arcalyst (rilonacept), known as IL-1 Trap, prevented gout attacks, as measured by the primary endpoint of the number of gout flares per patient over the 16 week treatment period.
Is this s good, or bad news?
Judging from investors’ reaction to the news, one would believe the clinical trial results are a failure. However, judging based on understanding the disease, its causes, complications, symptoms and what current conventional treatments are accomplishing, or lacking would prove that Arcalyst might be the missing link in the treatment of gout. The drug neutralizes a major mediator of the severe attacks of inflammatory symptom flare up the patients suffer from after taking the uric acid lowering drugs, which is the fundamental treatment of gout. That’s what makes the bulk of patients who abandon the treatments that deal with the cause of disease, i.e., elevated uric acid. of the cause of disease. No conventional drug taken at a safe dose has been effective in preventing the flare up of the symptoms, which take their toll on patients and probably lead to worsening of the disease. It is easier for gossipers to perpetrate skepticism towards Regeneron’s drug Arcalyst if the listeners do not understand what gout is and what treatments are required for its successful management. The fact is that, gout requires more than two classes of drugs to stop the main cause of this disease, control its disabling symptoms, and prevent the attacks, thus, the consequent complications if the disease is not well treated.
Gout occurs in patients who have elevated levels of uric acid. The uric acid is deposited in the joints and other body tissues. In the joints uric acid causes joint inflammation, joint pain, stiffness swelling, redness and heat. Deposited in soft tissues, uric acid causes the same it does the joints. Generally, patients experience flare up of symptoms that no drug is yet capable of preventing. The current management of gout aims first at lowering uric acid by either decreasing its formation or increasing its elimination from the body with uric acid-lowering drugs. Drugs exist that can tackle one or the other of the two actions. For example, the drug Allopurinol, decreases uric acid formation and Probenecid helps the body eliminate excess uric acid through the kidneys and into the urine. The problem is that controlling the level of uric acid does not stop all the symptoms right away but the pain and suffering might increase and persist for long. The reason is that the breakup of uric acid crystals boosts inflammatory mediators, especially interleukin-1 (IL-1), which causes the flare. IL-1 is targeted for inhibition by Regeneron’s drug Arcalyst. The flare the patients experience after the treatment makes it difficult for them to adhere to uric acid-lowering therapies and discontinue the treatment within the first few months of therapy. That’s why Arcalyst is badly needed.
Currently, physicians are using the anti-inflammatory drug colchicine to calm down inflammation. In many cases, however, the dose required for the control of severe inflammatory symptoms is high and must be taken for a long time, which makes its side effects prohibiting. Patients would suffer severe diarrhea, abdominal cramps, nausea, and vomiting. Non-steroid anti-inflammatory drugs are used also, but in many cases they do not control the severe inflammatory symptoms during the flare, unless very high doses are given for a long time. Long-term use of high dose can provoke grave side effects, including peptic ulcer, kidney failure and liver failure. Steroids are resorted to, yet, again, their long-term high dose use could result in detrimental side effects, which go from immune system suppression, to osteoporosis, bone fracture, endocrine system deregulation, difficulty of wound healing, diabetes and the list goes on and on.
So, the most important results from Arcalyst phase 3 study is the one, which confirms that the concomitant use of the drug with uric acid-lowering therapy for the first several months may help avoid gout flares. The use of Arcalyst is expected to improve patients’ adherence to uric acid lowering drugs, sustain their wellbeing and improve the outcome of their disease. Is this bad news?
Regeneron’s (REGN) Phase 3 study in gout patients initiating allopurinol therapy to lower their uric acid levels showed that the company’s drug Arcalyst (rilonacept), known as IL-1 Trap, prevented gout attacks, as measured by the primary endpoint of the number of gout flares per patient over the 16 week treatment period.
Is this s good, or bad news?
Judging from investors’ reaction to the news, one would believe the clinical trial results are a failure. However, judging based on understanding the disease, its causes, complications, symptoms and what current conventional treatments are accomplishing, or lacking would prove that Arcalyst might be the missing link in the treatment of gout. The drug neutralizes a major mediator of the severe attacks of inflammatory symptom flare up the patients suffer from after taking the uric acid lowering drugs, which is the fundamental treatment of gout. That’s what makes the bulk of patients who abandon the treatments that deal with the cause of disease, i.e., elevated uric acid. of the cause of disease. No conventional drug taken at a safe dose has been effective in preventing the flare up of the symptoms, which take their toll on patients and probably lead to worsening of the disease. It is easier for gossipers to perpetrate skepticism towards Regeneron’s drug Arcalyst if the listeners do not understand what gout is and what treatments are required for its successful management. The fact is that, gout requires more than two classes of drugs to stop the main cause of this disease, control its disabling symptoms, and prevent the attacks, thus, the consequent complications if the disease is not well treated.
Gout occurs in patients who have elevated levels of uric acid. The uric acid is deposited in the joints and other body tissues. In the joints uric acid causes joint inflammation, joint pain, stiffness swelling, redness and heat. Deposited in soft tissues, uric acid causes the same it does the joints. Generally, patients experience flare up of symptoms that no drug is yet capable of preventing. The current management of gout aims first at lowering uric acid by either decreasing its formation or increasing its elimination from the body with uric acid-lowering drugs. Drugs exist that can tackle one or the other of the two actions. For example, the drug Allopurinol, decreases uric acid formation and Probenecid helps the body eliminate excess uric acid through the kidneys and into the urine. The problem is that controlling the level of uric acid does not stop all the symptoms right away but the pain and suffering might increase and persist for long. The reason is that the breakup of uric acid crystals boosts inflammatory mediators, especially interleukin-1 (IL-1), which causes the flare. IL-1 is targeted for inhibition by Regeneron’s drug Arcalyst. The flare the patients experience after the treatment makes it difficult for them to adhere to uric acid-lowering therapies and discontinue the treatment within the first few months of therapy. That’s why Arcalyst is badly needed.
Currently, physicians are using the anti-inflammatory drug colchicine to calm down inflammation. In many cases, however, the dose required for the control of severe inflammatory symptoms is high and must be taken for a long time, which makes its side effects prohibiting. Patients would suffer severe diarrhea, abdominal cramps, nausea, and vomiting. Non-steroid anti-inflammatory drugs are used also, but in many cases they do not control the severe inflammatory symptoms during the flare, unless very high doses are given for a long time. Long-term use of high dose can provoke grave side effects, including peptic ulcer, kidney failure and liver failure. Steroids are resorted to, yet, again, their long-term high dose use could result in detrimental side effects, which go from immune system suppression, to osteoporosis, bone fracture, endocrine system deregulation, difficulty of wound healing, diabetes and the list goes on and on.
So, the most important results from Arcalyst phase 3 study is the one, which confirms that the concomitant use of the drug with uric acid-lowering therapy for the first several months may help avoid gout flares. The use of Arcalyst is expected to improve patients’ adherence to uric acid lowering drugs, sustain their wellbeing and improve the outcome of their disease. Is this bad news?
Exelixis Announces June 21 Webcast of Conference Call Update on Development Collaboration with Bristol-Myers Squibb Company for XL184
SOUTH SAN FRANCISCO, Calif.--(BUSINESS WIRE)--Exelixis, Inc. (Nasdaq:EXEL) will provide an update on its collaboration with Bristol-Myers Squibb Company for XL184 on Monday, June 21, 2010 before the markets open. The announcement will be followed by a live webcast at 5:00 a.m. PT/ 8:00 a.m. ET. The webcast may be accessed in the Event Calendar page under Investors at http://www.exelixis.com.
An audio replay of the webcast will be available until 11:59 p.m ET/ 8:59 p.m. PT on July 5, 2010. Access numbers for the replay are: 888-286-8010 (domestic) and 617-801-6888 (international), and the passcode is 21140740.
----------------
Was kommt denn da auf uns zu?
SOUTH SAN FRANCISCO, Calif.--(BUSINESS WIRE)--Exelixis, Inc. (Nasdaq:EXEL) will provide an update on its collaboration with Bristol-Myers Squibb Company for XL184 on Monday, June 21, 2010 before the markets open. The announcement will be followed by a live webcast at 5:00 a.m. PT/ 8:00 a.m. ET. The webcast may be accessed in the Event Calendar page under Investors at http://www.exelixis.com.
An audio replay of the webcast will be available until 11:59 p.m ET/ 8:59 p.m. PT on July 5, 2010. Access numbers for the replay are: 888-286-8010 (domestic) and 617-801-6888 (international), and the passcode is 21140740.
----------------
Was kommt denn da auf uns zu?
Antwort auf Beitrag Nr.: 39.708.316 von SLGramann am 19.06.10 12:00:15
Jemand im Yahoo-Board verweist auf diesen Trial und spekuliert etwas herum:
http://clinicaltrials.gov/ct2/show/NCT00704730
Note that Primary Outcome measure for MTC is due in June according to the above link. The way it is going to be announced (Monday pre-market), it has to be really good news. Otherwise they would have announced it after Hrs Friday! ALso, note that they said nothing about this trial in ASCO!! I never got that.
But it looks like this will be the turning point for EXEL. If data looks good, this may also be the basis for BMY to make an offer. In fact, I am sure BMY already tried it, but EXEL instead raised money otherwise, rather than take the cheap offers, that BMY always does (remember MEDX?). With positive result of MTC, and final validation for exels pipeline, 20-25 pop by next week is not far-fetched!!
-------
Es ist wahrscheinlich, dass das mit dem Trial zusammenhängt. Ob es gute News sein werden, steht auf einem ganz anderen Blatt. Wir müssens halt abwarten.
Jemand im Yahoo-Board verweist auf diesen Trial und spekuliert etwas herum:
http://clinicaltrials.gov/ct2/show/NCT00704730
Note that Primary Outcome measure for MTC is due in June according to the above link. The way it is going to be announced (Monday pre-market), it has to be really good news. Otherwise they would have announced it after Hrs Friday! ALso, note that they said nothing about this trial in ASCO!! I never got that.
But it looks like this will be the turning point for EXEL. If data looks good, this may also be the basis for BMY to make an offer. In fact, I am sure BMY already tried it, but EXEL instead raised money otherwise, rather than take the cheap offers, that BMY always does (remember MEDX?). With positive result of MTC, and final validation for exels pipeline, 20-25 pop by next week is not far-fetched!!
-------
Es ist wahrscheinlich, dass das mit dem Trial zusammenhängt. Ob es gute News sein werden, steht auf einem ganz anderen Blatt. Wir müssens halt abwarten.
Antwort auf Beitrag Nr.: 39.708.356 von SLGramann am 19.06.10 12:11:16so eine Ankündigkung hört sich schon etwas merkwürdig an... würden Daten kommen, würde dies dann nicht deutlicher angekündigt wie z.B. zuletzt bei REGN? Bei Daten wäre ein "Development Update" eher etwas negatives, denke ich, da damit ein Fehlschlag verschleiert werden kann. Aber es ist alles reine Spekulation... im yahoo-Board gab es ja auch den Hinweis, dass EXEL die Option besitzt, sich nicht wie bisher an den Entwicklungskosten zur Hälfte zu beteiligen, sondern dass BMS die Entwicklung voll übernimmt und EXEL dafür von 50% der US-Gewinne auf die normalen zweistelligen Royalties zurückgestuft wird:
The companies have agreed to co-develop XL184. Exelixis will have the option to co-promote XL184 in the United States. The companies will share worldwide development costs and commercial profits on XL184 in the United States. Exelixis will be eligible to receive sales performance milestones of up to $150 million and double-digit royalties on sales outside the United States. The clinical development of XL184 will be directed by a joint committee. It is anticipated that Exelixis will conduct a significant portion of clinical development activities through 2010. Exelixis may opt out of the co-development of XL184, in which case Exelixis would instead be eligible to receive development and regulatory milestones of up to $295 million, double-digit royalties on XL184 product sales worldwide, and sales performance milestones.
wäre auch eine Möglichkeit um Ausgaben weiter zu reduzieren
mfg ipollit
The companies have agreed to co-develop XL184. Exelixis will have the option to co-promote XL184 in the United States. The companies will share worldwide development costs and commercial profits on XL184 in the United States. Exelixis will be eligible to receive sales performance milestones of up to $150 million and double-digit royalties on sales outside the United States. The clinical development of XL184 will be directed by a joint committee. It is anticipated that Exelixis will conduct a significant portion of clinical development activities through 2010. Exelixis may opt out of the co-development of XL184, in which case Exelixis would instead be eligible to receive development and regulatory milestones of up to $295 million, double-digit royalties on XL184 product sales worldwide, and sales performance milestones.
wäre auch eine Möglichkeit um Ausgaben weiter zu reduzieren
mfg ipollit
Antwort auf Beitrag Nr.: 39.365.254 von SLGramann am 20.04.10 09:07:45habe es es jetzt auch mal mit einer kleinen Position AVEO probiert... MK nur ca. 215 USD
http://www.xconomy.com/boston/2009/06/29/aveo-pieces-togethe…
Aveo Pieces Together a Plan to Rival Big Boys of Cancer Drug World
Luke Timmerman 6/29/09
Great biotechnology stories have three essential ingredients—science, medicine, and business. Aveo Pharmaceuticals CEO Tuan Ha-Ngoc told me last week that he thinks about these same elements in his quest to build a sustainable company. Few companies ever put together all the pieces, though, and it’s too early to say if Cambridge, MA-based Aveo is one of them.
Until a few weeks ago, Aveo was probably best known for the first piece of the puzzle, the science. It has what it calls a more accurate method for mimicking cancer in mouse experimental models, compared with the traditional “xenograft” approach.
Then, in late May, Aveo made headlines on the medical side, when researchers presented promising results for its lead experimental drug for kidney cancer. The first drug to come from Aveo’s experimental platform, tivozanib (or AV-951) is an oral pill designed to block three specific types of molecules that allow the formation of new blood vessels; tumors rely on new vessels for nourishment as they grow. The study showed the drug could slow the spread of malignancy with minimal side effects. If the data can be confirmed in a larger trial to start later this year, Aveo’s drug could be in a position to compete with Pfizer’s sunitinib (Sutent) and Bayer and Onyx Pharmaceuticals’ sorafenib (Nexavar), which combined pulled in $1.5 billion last year.
But even if things go right, Aveo won’t have its moneymaking drug on the market until 2012 at the earliest. So if you’re Ha-Ngoc, how do you keep the business moving forward for years when venture capital is hard to come by? You keep 100 percent ownership of your crown jewels in North America, find partners to commercialize them elsewhere, all structure the deals so they pay for your R&D engine. Then plow the profits back into R&D and do it again.
“To build a sustainable company, you can’t be a single-product company or a single-indication company,” Ha-Ngoc says. “We want to be a full-fledged company.”
The Aveo story began in 2002. The company spun out of the lab of Ronald DePinho and Lynda Chin at the Dana-Farber Cancer Institute in Boston. They started with the premise that since cancer has been cured many times in mice, and never in humans, maybe the existing mouse models for the disease could use some improvement. The conventional
“xenograft” approach involves taking human cancer cells, growing them in the foreign environment of a lab dish, then injecting them into a mouse with a suppressed immune system—which all adds variables and creates an artificial environment, Ha-Ngoc says.
The Aveo model, on the other hand, involves splicing cancer-causing genes into a mouse embryo. The DNA encoding those genes is rigged so they’re turned off when the mouse is born and can be switched on later in life by simply feeding the mouse a specific molecule, such as an antibiotic. This allows the mouse to develop normally, and can avoid the kinds of mutations that can arise in the lab dish, skewing traditional models, Ha-Ngoc says.
The Aveo approach was compelling enough to Merck that the pharma giant signed a partnership with Aveo to identify new drug targets in 2003. Two years later, Merck expanded the partnership to encompass testing potential drug compounds against those targets, Ha-Ngoc says. By 2007, Eli Lilly, OSI Pharmaceuticals, and Schering-Plough had also formed deals to use the Aveo technology. This year, Biogen Idec joined the pack. All these deals have added up to provide $140 million in financing for Aveo, on top of the $90 million the company has raised in venture capital.
Aveo is now in “active discussions” to find a partner on its most valuable asset—tivozanib for kidney cancer. Aveo’s goal is to hold onto 100 percent of the rights to this in North America, something few biotech companies ever get to do with cancer drugs.
What surprised me more is how ambitious Aveo is in its clinical development plan for this drug, even before it has found a partner. Aveo is working on a trial design that will enroll “several hundred” patients with kidney cancer, randomly assigned to get its drug or an “active comparator,” which means a drug that might work, instead the more traditional route of putting the new drug up against a straw man like a placebo or a drug like interferon, which doesn’t work. The main goal will be to provide a clear answer on whether the Aveo drug can slow down the spread of kidney tumors, with a secondary goal will be to see whether it can help people live longer.
Besides that big trial, which is expected to start enrolling patients before the end of this year, Aveo is already lining up ways to differentiate its drug from Pfizer and Bayer’s competitors. Those drugs block the same molecule that Aveo’s drug targets, a receptor called VEGF, but they also hit other receptors involved in rapid cell growth and proliferation. By hitting multiple targets, these drugs cause “overlapping toxicities” and can’t be given in combination with drugs like Wyeth’s temsirolimus (Torisel), Ha-Ngoc says. By being very specific (Aveo’s drug blocks three forms of VEGF but no other targets), and because of its mild side effect profile, the Aveo drug should be useful in more combinations than the Pfizer and Bayer drugs, he says. Aveo expects to publish the company’s first data to support this idea in the second half of the year, Ha-Ngoc says.
Usually venture-backed biotechs don’t have more than one serious late-stage drug candidate to talk about, but like Ha-Ngoc says, he’s not looking to build a one-drug company. Aveo’s second candidate, AV-299, is an antibody drug that’s designed to block hepatocyte growth factor, a cell-growth protein that’s involved in malignancies. Aveo, which is developing this with Schering-Plough, has completed an early-stage safety study of the drug and is now gearing up to start two mid-stage trials of 200 patients each.
On my way out the door, Ha-Ngoc showed me Aveo’s “Wall of Fame,” which bears individual color photos of all 136 of the company’s employees, in order of which year they joined the firm. There were a lot of photos pasted on 2008, and not as many in 2009, but he says he wants that list to keep growing.
“We want to create an organization that discovers and develops drugs in a different way, that takes out the guesswork,” Ha-Ngoc says. “Hopefully five years from now we’ll be recognized as a company that has commercialized an important new drug, and that has a pipeline that can reproduce it in a sustained way.”
mfg ipollit
http://www.xconomy.com/boston/2009/06/29/aveo-pieces-togethe…
Aveo Pieces Together a Plan to Rival Big Boys of Cancer Drug World
Luke Timmerman 6/29/09
Great biotechnology stories have three essential ingredients—science, medicine, and business. Aveo Pharmaceuticals CEO Tuan Ha-Ngoc told me last week that he thinks about these same elements in his quest to build a sustainable company. Few companies ever put together all the pieces, though, and it’s too early to say if Cambridge, MA-based Aveo is one of them.
Until a few weeks ago, Aveo was probably best known for the first piece of the puzzle, the science. It has what it calls a more accurate method for mimicking cancer in mouse experimental models, compared with the traditional “xenograft” approach.
Then, in late May, Aveo made headlines on the medical side, when researchers presented promising results for its lead experimental drug for kidney cancer. The first drug to come from Aveo’s experimental platform, tivozanib (or AV-951) is an oral pill designed to block three specific types of molecules that allow the formation of new blood vessels; tumors rely on new vessels for nourishment as they grow. The study showed the drug could slow the spread of malignancy with minimal side effects. If the data can be confirmed in a larger trial to start later this year, Aveo’s drug could be in a position to compete with Pfizer’s sunitinib (Sutent) and Bayer and Onyx Pharmaceuticals’ sorafenib (Nexavar), which combined pulled in $1.5 billion last year.
But even if things go right, Aveo won’t have its moneymaking drug on the market until 2012 at the earliest. So if you’re Ha-Ngoc, how do you keep the business moving forward for years when venture capital is hard to come by? You keep 100 percent ownership of your crown jewels in North America, find partners to commercialize them elsewhere, all structure the deals so they pay for your R&D engine. Then plow the profits back into R&D and do it again.
“To build a sustainable company, you can’t be a single-product company or a single-indication company,” Ha-Ngoc says. “We want to be a full-fledged company.”
The Aveo story began in 2002. The company spun out of the lab of Ronald DePinho and Lynda Chin at the Dana-Farber Cancer Institute in Boston. They started with the premise that since cancer has been cured many times in mice, and never in humans, maybe the existing mouse models for the disease could use some improvement. The conventional
“xenograft” approach involves taking human cancer cells, growing them in the foreign environment of a lab dish, then injecting them into a mouse with a suppressed immune system—which all adds variables and creates an artificial environment, Ha-Ngoc says.
The Aveo model, on the other hand, involves splicing cancer-causing genes into a mouse embryo. The DNA encoding those genes is rigged so they’re turned off when the mouse is born and can be switched on later in life by simply feeding the mouse a specific molecule, such as an antibiotic. This allows the mouse to develop normally, and can avoid the kinds of mutations that can arise in the lab dish, skewing traditional models, Ha-Ngoc says.
The Aveo approach was compelling enough to Merck that the pharma giant signed a partnership with Aveo to identify new drug targets in 2003. Two years later, Merck expanded the partnership to encompass testing potential drug compounds against those targets, Ha-Ngoc says. By 2007, Eli Lilly, OSI Pharmaceuticals, and Schering-Plough had also formed deals to use the Aveo technology. This year, Biogen Idec joined the pack. All these deals have added up to provide $140 million in financing for Aveo, on top of the $90 million the company has raised in venture capital.
Aveo is now in “active discussions” to find a partner on its most valuable asset—tivozanib for kidney cancer. Aveo’s goal is to hold onto 100 percent of the rights to this in North America, something few biotech companies ever get to do with cancer drugs.
What surprised me more is how ambitious Aveo is in its clinical development plan for this drug, even before it has found a partner. Aveo is working on a trial design that will enroll “several hundred” patients with kidney cancer, randomly assigned to get its drug or an “active comparator,” which means a drug that might work, instead the more traditional route of putting the new drug up against a straw man like a placebo or a drug like interferon, which doesn’t work. The main goal will be to provide a clear answer on whether the Aveo drug can slow down the spread of kidney tumors, with a secondary goal will be to see whether it can help people live longer.
Besides that big trial, which is expected to start enrolling patients before the end of this year, Aveo is already lining up ways to differentiate its drug from Pfizer and Bayer’s competitors. Those drugs block the same molecule that Aveo’s drug targets, a receptor called VEGF, but they also hit other receptors involved in rapid cell growth and proliferation. By hitting multiple targets, these drugs cause “overlapping toxicities” and can’t be given in combination with drugs like Wyeth’s temsirolimus (Torisel), Ha-Ngoc says. By being very specific (Aveo’s drug blocks three forms of VEGF but no other targets), and because of its mild side effect profile, the Aveo drug should be useful in more combinations than the Pfizer and Bayer drugs, he says. Aveo expects to publish the company’s first data to support this idea in the second half of the year, Ha-Ngoc says.
Usually venture-backed biotechs don’t have more than one serious late-stage drug candidate to talk about, but like Ha-Ngoc says, he’s not looking to build a one-drug company. Aveo’s second candidate, AV-299, is an antibody drug that’s designed to block hepatocyte growth factor, a cell-growth protein that’s involved in malignancies. Aveo, which is developing this with Schering-Plough, has completed an early-stage safety study of the drug and is now gearing up to start two mid-stage trials of 200 patients each.
On my way out the door, Ha-Ngoc showed me Aveo’s “Wall of Fame,” which bears individual color photos of all 136 of the company’s employees, in order of which year they joined the firm. There were a lot of photos pasted on 2008, and not as many in 2009, but he says he wants that list to keep growing.
“We want to create an organization that discovers and develops drugs in a different way, that takes out the guesswork,” Ha-Ngoc says. “Hopefully five years from now we’ll be recognized as a company that has commercialized an important new drug, and that has a pipeline that can reproduce it in a sustained way.”
mfg ipollit
Antwort auf Beitrag Nr.: 39.708.691 von ipollit am 19.06.10 14:33:31interessant sind vielleicht auch ZIOP (MK 200 Mio USD) und Wilex (MK 140 Mio USD)? ... ARRY ist mit einer MK von 175 Mio USD auch nicht sehr teuer. Mal schauen...
mfg ipollit
mfg ipollit
okay, das so ziemlich Schlechteste ist nun Tatsache:
Bristol returns cancer drug XL184 to Exelixis
Deena Beasley
LOS ANGELES
Sun Jun 20, 2010 11:57pm EDTLOS ANGELES (Reuters) - Bristol-Myers Squibb Co will pay another $17 million to end its development collaboration for Exelixis Inc's experimental cancer drug XL184.
Health
Exelixis said on Sunday it will continue to develop the drug, which is being studied as a treatment for thyroid cancer, glioblastoma and other solid tumors.
"We have the resources to take this forward for some time," Exelixis Chief Executive George Scangos told Reuters, noting that the company may seek another partnership.
Under a deal signed in late 2008, Bristol-Myers had paid Exelixis $240 million for the rights to XL184 and another drug, called XL281 -- a $195 million upfront cash payment, plus additional license payments of $45 million in 2009.
Bristol's decision to return the drug to Exelixis "really does reflect a need for both companies to prioritize their portfolios," Scangos said. "We were unable to reach an agreement with BMS on what the appropriate clinical program would be."
He said Exelixis plans to move forward later this year with a Phase 3 trial of the drug in patients with recurrent glioblastoma, the most common and deadly form of brain cancer.
The company first announced plans for the pivotal trial earlier this month, disappointing some investors who had speculated that the developers might be able to use results from an ongoing mid-stage trial for a regulatory filing.
Results from a Phase 3 trial of XL184 in patients with medullary thyroid cancer are expected in the first half of next year.
Scangos said Exelixis also expects to report at a November medical meeting in Berlin early-stage data on XL184 in patients with five other types of solid tumors.
XL184 is an oral drug designed to block the vascular endothelial growth factor, the same target as drugs like Roche Holding AG's Avastin, as well as MET and RET, two other drivers of tumor formation,
Scangos said Exelixis, which recently entered into two debt financing deals for a combined $160 million -- $80 million of which will be used to extend the maturity of an existing obligation -- will update investors on the company's cash position when second-quarter results are reported.
The CEO said the "incremental cost" for the XL184 program is less than $20 million this year.
(Editing by Marguerita Choy)
-----------
Jetzt wissen wir auch, warum Exelixis diesen Personalabbau angekündigt hat. XL184 allein zu stemmen, wird schwer. Zumal das Zutrauen zu diesem Projekt jetzt in den Keller gehen dürfte.
Wird heute ein schönes Minus geben. Mal sehen, ob wir mit 30% hinkommen.
Bristol returns cancer drug XL184 to Exelixis
Deena Beasley
LOS ANGELES
Sun Jun 20, 2010 11:57pm EDTLOS ANGELES (Reuters) - Bristol-Myers Squibb Co will pay another $17 million to end its development collaboration for Exelixis Inc's experimental cancer drug XL184.
Health
Exelixis said on Sunday it will continue to develop the drug, which is being studied as a treatment for thyroid cancer, glioblastoma and other solid tumors.
"We have the resources to take this forward for some time," Exelixis Chief Executive George Scangos told Reuters, noting that the company may seek another partnership.
Under a deal signed in late 2008, Bristol-Myers had paid Exelixis $240 million for the rights to XL184 and another drug, called XL281 -- a $195 million upfront cash payment, plus additional license payments of $45 million in 2009.
Bristol's decision to return the drug to Exelixis "really does reflect a need for both companies to prioritize their portfolios," Scangos said. "We were unable to reach an agreement with BMS on what the appropriate clinical program would be."
He said Exelixis plans to move forward later this year with a Phase 3 trial of the drug in patients with recurrent glioblastoma, the most common and deadly form of brain cancer.
The company first announced plans for the pivotal trial earlier this month, disappointing some investors who had speculated that the developers might be able to use results from an ongoing mid-stage trial for a regulatory filing.
Results from a Phase 3 trial of XL184 in patients with medullary thyroid cancer are expected in the first half of next year.
Scangos said Exelixis also expects to report at a November medical meeting in Berlin early-stage data on XL184 in patients with five other types of solid tumors.
XL184 is an oral drug designed to block the vascular endothelial growth factor, the same target as drugs like Roche Holding AG's Avastin, as well as MET and RET, two other drivers of tumor formation,
Scangos said Exelixis, which recently entered into two debt financing deals for a combined $160 million -- $80 million of which will be used to extend the maturity of an existing obligation -- will update investors on the company's cash position when second-quarter results are reported.
The CEO said the "incremental cost" for the XL184 program is less than $20 million this year.
(Editing by Marguerita Choy)
-----------
Jetzt wissen wir auch, warum Exelixis diesen Personalabbau angekündigt hat. XL184 allein zu stemmen, wird schwer. Zumal das Zutrauen zu diesem Projekt jetzt in den Keller gehen dürfte.
Wird heute ein schönes Minus geben. Mal sehen, ob wir mit 30% hinkommen.
Antwort auf Beitrag Nr.: 39.711.770 von SLGramann am 21.06.10 07:19:19
Na ja, meine Fähigkeiten als Kursprognostiker habe ich mal wieder "glänzend" unter Beweis gestellt. Statt befürchteter -30% sind wir jetzt gerade mal bei -6%.
Indes hat der Tag auch gerade erst begonnen.
Und in jedem Fall finde ich die Nachricht einfach nur grottenschlecht und glaube bis zum Beweis des Gegenteils absolut nicht an das Gerede von Portfoliooptimierung bei BMY.
@ipollit, AVEO und ARRY habe ich mir auch gegönnt, auch eine sehr kleine Position Celldex. Gerade AVEO wird aber nur mit wenigen Stücken gehandelt und der Kurs fällt langsam aber kontinuierlich. Hast Du eine explizite Meinung zu Tivozanib? Wie siehst Du die Chancen gegen Nexavar?
Na ja, meine Fähigkeiten als Kursprognostiker habe ich mal wieder "glänzend" unter Beweis gestellt. Statt befürchteter -30% sind wir jetzt gerade mal bei -6%.
Indes hat der Tag auch gerade erst begonnen.
Und in jedem Fall finde ich die Nachricht einfach nur grottenschlecht und glaube bis zum Beweis des Gegenteils absolut nicht an das Gerede von Portfoliooptimierung bei BMY.
@ipollit, AVEO und ARRY habe ich mir auch gegönnt, auch eine sehr kleine Position Celldex. Gerade AVEO wird aber nur mit wenigen Stücken gehandelt und der Kurs fällt langsam aber kontinuierlich. Hast Du eine explizite Meinung zu Tivozanib? Wie siehst Du die Chancen gegen Nexavar?
Antwort auf Beitrag Nr.: 39.711.770 von SLGramann am 21.06.10 07:19:19
Meinung:
Exelixis and BMS part ways over XL184
It came as no big surprise this morning to hear that Exelixis and BMS have announced they are terminating their agreement over XL184. The compound is being tested in medullary thyroid cancer, glioblastoma multiforme (GBM) and non-small cell lung cancer (NSCLC). This is a small molecule that inhibits several targets, namely MET, RET and VEGFR2.
According to Exelixis, the CEO stated in their press release:
"We certainly understand BMS' need to make pipeline and prioritization decisions."
It looks as if they couldn't agree on the priorities for the clinical development, which would be a little odd given the $240M invested in XL184 and XL281 in 2008, with the same indications planned.
It could also be a question of risk management for several reasons:
1. Thyroid cancer is slow growing and thus development times will be relatively long, lung cancer is notoriously difficult to crack, as is GBM.
2. BMS also have another VEGF inhibitor in late stage development called brivanib (BMS582664), which is in phase III and inhibits both VEGFR2 and FGFR. This compound is being tested in a number of indications, including liver and colon cancers.
Recent BMS analyst meetings from have focused on brivanib as one of the promising new oncology agents in the pipeline, so my suspicion is that they probably decided they only needed one VEGF inhibitor and killed the Exelixis agent rather than their own homegrown one. These things happen all the time. Sometimes you develop several molecules in the hope that one looks more promising in trials.
A few years ago, I remember reading about an incredibly brave and strong patient in one of the early brivanib trials, for advanced cancer. In this case, the feisty young lady had a non-differentiated spindle cell sarcoma and blogged about the encouraging impact of her new treatment:
"My Scans came back with wonderful results. The Brivanib pills are working! My tumors are stable and haven't grown since my last scan! One tumor in my lymph node has actually died! There is no blood flow to the tumor! This is the best news I could get. My doctor is so happy with these results.
I will be on the pills for 12 weeks. After that I will be given either a Placebo or continue on the pills. Because it is a trial it's a 50, 50 shot that I could get the Placebo. Booooo! I will know right away by how I am feeling. The reaction happens within 15 minutes after I take the pills. I am a walking zombie. If I do get the Placebo, I can then go back on the trial."
You can follow her incredible journey back to health here; it's an inspiration to us all.
Thankfully, she's still blogging 2 years later, a testament to her resolve and ability to fight the disease. Long may she continue! Not everyone who gets cancer is elderly, often many people are diagnosed in their teens, twenties and thirties too.
I don't know about you, but I love happy stories and hope to be following her blog for a very long time.
dazu Kommentare:
craig schrieb...
Is Exelexis even a viable company at this point in terms of their ability to continue?
This is the second time someone has passed over XL184 (Glaxo was the first).
Is XL184 just a weak, dead compound?
I am beginning to get rather sick and tired of hearing EXEL's CEO brag about "best in class" when he can't even get a seat in the class room.
MaverickNY schrieb...
Craig,
XL184 was always going to struggle given the indications they selected and 2 rejections from suitors will make people shy the 3rd time round.
That said, they still have a portfolio of interesting compounds, but the odds of several of them making it in highly competitive markets is for anyone to guess. Certainly, the news makes it harder to generate enthusiasm going forward.
I do agree with you about the 'best in class' fanfare. It is a marketing tactic to be used when you have great data and are not first to market (from that old adage, be first or be best), not at every opportunity or the ploy becomes tired very quickly.
Sometimes it is better to under promise and over deliver, not the other way round.
http://www.pharmastrategyblog.com/2010/06/exelixis-and-bms-p…
Meinung:
Exelixis and BMS part ways over XL184
It came as no big surprise this morning to hear that Exelixis and BMS have announced they are terminating their agreement over XL184. The compound is being tested in medullary thyroid cancer, glioblastoma multiforme (GBM) and non-small cell lung cancer (NSCLC). This is a small molecule that inhibits several targets, namely MET, RET and VEGFR2.
According to Exelixis, the CEO stated in their press release:
"We certainly understand BMS' need to make pipeline and prioritization decisions."
It looks as if they couldn't agree on the priorities for the clinical development, which would be a little odd given the $240M invested in XL184 and XL281 in 2008, with the same indications planned.
It could also be a question of risk management for several reasons:
1. Thyroid cancer is slow growing and thus development times will be relatively long, lung cancer is notoriously difficult to crack, as is GBM.
2. BMS also have another VEGF inhibitor in late stage development called brivanib (BMS582664), which is in phase III and inhibits both VEGFR2 and FGFR. This compound is being tested in a number of indications, including liver and colon cancers.
Recent BMS analyst meetings from have focused on brivanib as one of the promising new oncology agents in the pipeline, so my suspicion is that they probably decided they only needed one VEGF inhibitor and killed the Exelixis agent rather than their own homegrown one. These things happen all the time. Sometimes you develop several molecules in the hope that one looks more promising in trials.
A few years ago, I remember reading about an incredibly brave and strong patient in one of the early brivanib trials, for advanced cancer. In this case, the feisty young lady had a non-differentiated spindle cell sarcoma and blogged about the encouraging impact of her new treatment:
"My Scans came back with wonderful results. The Brivanib pills are working! My tumors are stable and haven't grown since my last scan! One tumor in my lymph node has actually died! There is no blood flow to the tumor! This is the best news I could get. My doctor is so happy with these results.
I will be on the pills for 12 weeks. After that I will be given either a Placebo or continue on the pills. Because it is a trial it's a 50, 50 shot that I could get the Placebo. Booooo! I will know right away by how I am feeling. The reaction happens within 15 minutes after I take the pills. I am a walking zombie. If I do get the Placebo, I can then go back on the trial."
You can follow her incredible journey back to health here; it's an inspiration to us all.
Thankfully, she's still blogging 2 years later, a testament to her resolve and ability to fight the disease. Long may she continue! Not everyone who gets cancer is elderly, often many people are diagnosed in their teens, twenties and thirties too.
I don't know about you, but I love happy stories and hope to be following her blog for a very long time.
dazu Kommentare:
craig schrieb...
Is Exelexis even a viable company at this point in terms of their ability to continue?
This is the second time someone has passed over XL184 (Glaxo was the first).
Is XL184 just a weak, dead compound?
I am beginning to get rather sick and tired of hearing EXEL's CEO brag about "best in class" when he can't even get a seat in the class room.
MaverickNY schrieb...
Craig,
XL184 was always going to struggle given the indications they selected and 2 rejections from suitors will make people shy the 3rd time round.
That said, they still have a portfolio of interesting compounds, but the odds of several of them making it in highly competitive markets is for anyone to guess. Certainly, the news makes it harder to generate enthusiasm going forward.
I do agree with you about the 'best in class' fanfare. It is a marketing tactic to be used when you have great data and are not first to market (from that old adage, be first or be best), not at every opportunity or the ploy becomes tired very quickly.
Sometimes it is better to under promise and over deliver, not the other way round.
http://www.pharmastrategyblog.com/2010/06/exelixis-and-bms-p…
Antwort auf Beitrag Nr.: 39.714.407 von SLGramann am 21.06.10 15:44:09zuletzt hat EXEL sich ja bereits recht schlecht entwickelt... vielleicht ist dadurch auch schon ein wenig im Kurs enthalten gewesen, zumindest dass die Reaktion sich jetzt in Grenzen hält. Eigentlich ist es nicht sonderlich günstig, seine Programme mit den Pharmas zu verpartnern und dann sie zurückzubekommen, wenn es teuer wird. Andererseits hat BMS ja bereits einiges dafür bezahlt... unter dem Strich dürfte es sich für EXEL doch gelohnt haben, auch wenn es jetzt schwieriger wird. XL184 wurde zuvor ja bereits von GSK an EXEL abgegeben... würden die das auch machen, wenn dieses Programm ein großes Potential besitzen würde, zumal es ja bereits in der PIII ist?

AVEO... mehr Infos als die, die du bereits gepostet hast, habe ich auch nicht. Bezüglich Tivozanib hört es sich ganz gut an, dass AVEO recht große PIII-Studien und direkt gegen den Standard Nexavar testet... sie scheinen sich also recht sicher zu sein, dass Tivozanib besser ist als Nexavar. Der Ansatz, selektiver bei dem einen validierten Target anzusetzen, macht für mich auch Sinn, um damit die Nebenwirkungen zu reduzieren und die Kombinationsmöglichkeiten mit anderen Mitteln zu erhöhen. Bei Nexavar weiß man oft nicht genau, warum es genau wirkt, da es an vielen Stellen ansetzt... hier und da kann es ein Vorteil sein, wenn es etwas positives macht, was man nicht vorhergesehen hat, oft kann sich das aber auch negativ auswirken. Vielleicht ist das auch ein Problem bei XL184. Bei der noch geringen MK riskiere ich nun mal diese Position... am Ende weiß man nie sicher, ob es funktioniert, selbst wenn man eine Experte wäre. Daher versuche ich möglichst breit zu streuen, damit selbst ein Totalausfall nur geringe Auswirkungen hat... Chancen bietet AVEO jedenfalls denke ich.

****
habe mir heute ein paar ZIOP zugelegt... kleine Verzögerungen und fehlendes SPA sind für mich keine Argumente (zumal ein SPA nach dem GPC-Desaster für mich keinen Wert besitzt) für den heutigen Kurssturz. Vielleicht bekommen sie mit dem PFS-Endpunkt Probleme... naja, mal schauen.

JMP "Buy weakness. Reiterate $10 target & Outperform"
Ziopharm (3) (ZIOP - $4.85, $202M market cap) announced disappointing news with regard to progress towards securing an SPA for the palifosfamide pivotal trial; reiterate Market Outperform rating and $10 price target.
We believe the FDA has likely made a policy change regarding the acceptability of PFS for regular approval but still sees strong PFS advantages as perhaps approvable under its accelerated approval program.
As a result of minor protocol changes and completing discussions with the FDA, the palifosfamide Phase III trial will now start in 3Q10, not 2Q10, and will not have an SPA.
Our $10 price target continues to be derived from a sum-of-the-parts analysis of the palifosfamide revenue opportunity in sarcoma, including both U.S. sales and ex-U.S. royalties.
We view this as a minor setback but expect shares to trade down this morning.
However, we maintain a positive outlook for the long term based on our confidence in the efficacy and safety of palifosfamide in sarcoma.
We believe that the company has been diligent in seeking advise from key experts and the FDA and that the trial design will be consistent with our previous expectations.
While we acknowledge that an SPA would have been seen to lower the risk to the program, the eventual approval decision should still be driven by a compelling demonstration of efficacy.
News highlights risks of over-transparency.
We believe the primary driver of the stock's weakness will be that management had set very clear expectations for the SPA as a near-certainty to be finalized in the very near term.
In our view, the news highlights three points: 1) Nothing is certain with the FDA until it is final; 2) Management's credibility will be challenged by investors, especially in light of the recent equity financing; 3) However, we believe this is consistent with recent FDA views on PFS and that the agency appears committed to enabling the development of palifosfamide.
************
http://messages.finance.yahoo.com/Stocks_%28A_to_Z%29/Stocks…
Needham highlights
-Palifosfamide phase III will be only front line patients. Phase II was front and second line and the second line patients didn't show as significant a difference. So they are stacking the deck in their favor.
-The rationale for the last money raise was to expand and go after more indications, such as pediatric cancers and small cell lung cancer.
-Small cell lung cancer trial being run by Dr. Larry Einhorn, the guy behind approval of ifosfamide. Phase II trial in planning stages.
-Palifosfamide will be priced less than generic
-Oral phase I data soon
-Extensive partnering discussions with the biggest pharma company in the world and many other smaller ones on all 3 programs.
-6-9 months ago they were very eager to partner, but now they're not...
-They are trying to be "as strategic as possible," retaining rights as much as possible for palifosfamide.
-Sounds like they are only going to partner dariniparsin (ongoing discussions on Asian partnership) and indibulin.
-Partnering discussions in early, mid, and late stages - no idea as to timing.
**************
JMP SECURITIES
Biotechnology – Update
ZIOPHARM Oncology, Inc. (1,3)
Data Presentation at ASCO Well Received by Physician Community
MARKET OUTPERFORM ZIOP
Target Price $10.00
INVESTMENT HIGHLIGHTS
o PICASSO presentation at ASCO well received, boding well for pivotal accrual; reiterate Market Outperform rating and $10 price target on Ziopharm. Results from the PICASSO trial were presented in an oral session on Monday afternoon and, in our view, the data were well received by the attending physician community. While the key data from the presentation had already been previously released, we believe that the most important aim of this presentation was to attract the attention of the broader sarcoma physician community, and it is our view that this aim was achieved. Our diligence with potential study investigators for the pivotal trial increases our confidence for timely enrollment following the initiation of this trial in mid-year. We anticipate that the further presentations on the “Best of ASCO” tour will generate additional physician interest in the pivotal trial in the US and also in Europe. Furthermore, a breakfast meeting we held with the presenter of the data provided several key insights on the pivotal trial, market need, and future adoption of palifosfamide. Our $10 price target is derived from a sum-of-the-parts analysis of the palifosfamide revenue opportunity in sarcoma, including both US sales and ex-US royalties.
o Breakfast with primary investigator provides several incremental positives. During the conference we hosted a breakfast meeting with Dr. Claire Verschraegen, the first author on the PICASSO abstract. Dr. Verschraegen’s comments yielded or confirmed several positives both for the pivotal trial and commercial potential of palifosfamide. Her overall impression of the data was overwhelmingly positive, specifically with respect to the hazard ratio and favorable safety profile of the combination. Dr. Verschraegen noted that typically combination therapy is expected to have increased the toxicity profile of doxorubicin. She also highlighted the cost savings with palifosfamide over ifosfamide as mesna co-therapy is not required, noting that ifosfamide treatment requires hospitalization for one week for rehydration. Dr. Verschraegen confirmed that the primary endpoint for the pivotal trial will be PFS and she was positive on the prospects for patient enrollment. Her belief was that the only other competing agent for trial patients is Yondelis and the strict enrollment criteria rules out 2 of 3 patients in this trial. Finally, in the non-clinical trial setting, she believes that palifosfamide will be dosed through progression due to lack of treatment options.
o PICASSO data does not disappoint. The PICASSO trial was a randomized Phase II trial assessing the efficacy and safety of palifosfamide in combination with doxorubicin compared to doxorubicin alone. The trial was halted early due to efficacy following a planned review by the independent Data Safety Monitoring Committee (DSMB). At data cutoff there were 66 patients who had been treated in the trial and 62 were eligible for evaluation. A response was reported for 7 of 30 patients (23%) in the palifosfamide combination arm and for 3 of 32 patients (9%) in the doxorubicin alone arm. There were 18 progression events in the doxorubicin alone arm compared to 10 in the palifosfamide arm, representing a statistically significant hazard ratio of 0.43 (p=0.019).
The median PFS in the palifosfamide arm was 7.8 months, 3.4 months greater than in the doxorubicin alone arm. An analysis of patients receiving treatment in either arm of the trial for 6 cycles or less (the standard treatment period for doxorubicin) demonstrated a hazard ratio of 0.39 (p= 0.023) in favor of palifosfamide. Overall survival data are not yet mature and unlikely to yield a divergence between the groups due to patient crossover following the DSMB review; however, trend is currently in favor of palifosfamide. We view this survival data as positive, as a positive survival trend will likely be viewed as supportive of approval in the pivotal trial, in addition to the primary endpoint of PFS.
***
o Key takeaways include rigor in Pali protocol design, possible timelines, IP and additional indications, with capital efficiency remaining “Job #1.” Some of the key discussion points on which we gleaned incremental insights for the upcoming palifosfamide pivotal program in sarcoma include endpoints (clearly PFS but powered for OS), various ways to deal with discordant data (primary or site read vs. independent reader), and possible DSMB futility/safety reviews, as well as timelines for enrollment (start in 3Q10) and early data (PFS from roughly 2/3 events in 1H12). All said, this incremental info and our discussions on rigorous trial design, endpoints, and statistical plan provide us confidence in the Pali sarcoma program. Beyond the important start of this lead program, for additional value creation prior to Pali pivotal data we now look to increasing visibility of possible Pali utility from soon-to-start trials in small cell lung/breast cancer, and early (Phase I and preclinical) data for Daraniparsin in PTCL (1H11) and an undisclosed large market indication (oral version, 2H10). Finally, we are pleased with the IP on Pali and Dara, with composition-of-matter claims (on Pali) extending beyond 2025. Although we believe the clinical data from other indications for Pali and perhaps those for Daraniparsin and Indibulin (breast cancer) will result in many monetizable assets while also diversifying risk, we hesitate to value these until data is in hand. Therefore, our valuation for ZIOP shares continues to be driven by the sarcoma potential of Pali.
o Updating model to include financing. Last week, Ziopharm completed an equity financing, raising net proceeds of approximately $33MM, excluding overallotment allocations, through the sale of 7 million shares of common stock at $5 per share. This cash adds to the cash balance of $45MM at the end of 1Q10, giving a pro forma cash position of $78MM. We view this cash balance as more than sufficient to complete the clinical development of palifosfamide in advanced sarcoma, as well as limited clinical development for other candidates. Following the completion of this transaction, Ziopharm has basic shares outstanding of 48.8 million. We have updated our model to increase the weighted average share count in 2Q10 and beyond. In addition, the fully diluted share count includes stock options outstanding of 3.5 million, stock options available for issue of 3.1 million, and warrants of 15.9 million (exercisable at $4.11 per share). The net impact of the changes to our model does not have any impact on our valuation or price target on ZIOP shares.
mfg ipollit
AVEO... mehr Infos als die, die du bereits gepostet hast, habe ich auch nicht. Bezüglich Tivozanib hört es sich ganz gut an, dass AVEO recht große PIII-Studien und direkt gegen den Standard Nexavar testet... sie scheinen sich also recht sicher zu sein, dass Tivozanib besser ist als Nexavar. Der Ansatz, selektiver bei dem einen validierten Target anzusetzen, macht für mich auch Sinn, um damit die Nebenwirkungen zu reduzieren und die Kombinationsmöglichkeiten mit anderen Mitteln zu erhöhen. Bei Nexavar weiß man oft nicht genau, warum es genau wirkt, da es an vielen Stellen ansetzt... hier und da kann es ein Vorteil sein, wenn es etwas positives macht, was man nicht vorhergesehen hat, oft kann sich das aber auch negativ auswirken. Vielleicht ist das auch ein Problem bei XL184. Bei der noch geringen MK riskiere ich nun mal diese Position... am Ende weiß man nie sicher, ob es funktioniert, selbst wenn man eine Experte wäre. Daher versuche ich möglichst breit zu streuen, damit selbst ein Totalausfall nur geringe Auswirkungen hat... Chancen bietet AVEO jedenfalls denke ich.
****
habe mir heute ein paar ZIOP zugelegt... kleine Verzögerungen und fehlendes SPA sind für mich keine Argumente (zumal ein SPA nach dem GPC-Desaster für mich keinen Wert besitzt) für den heutigen Kurssturz. Vielleicht bekommen sie mit dem PFS-Endpunkt Probleme... naja, mal schauen.
JMP "Buy weakness. Reiterate $10 target & Outperform"
Ziopharm (3) (ZIOP - $4.85, $202M market cap) announced disappointing news with regard to progress towards securing an SPA for the palifosfamide pivotal trial; reiterate Market Outperform rating and $10 price target.
We believe the FDA has likely made a policy change regarding the acceptability of PFS for regular approval but still sees strong PFS advantages as perhaps approvable under its accelerated approval program.
As a result of minor protocol changes and completing discussions with the FDA, the palifosfamide Phase III trial will now start in 3Q10, not 2Q10, and will not have an SPA.
Our $10 price target continues to be derived from a sum-of-the-parts analysis of the palifosfamide revenue opportunity in sarcoma, including both U.S. sales and ex-U.S. royalties.
We view this as a minor setback but expect shares to trade down this morning.
However, we maintain a positive outlook for the long term based on our confidence in the efficacy and safety of palifosfamide in sarcoma.
We believe that the company has been diligent in seeking advise from key experts and the FDA and that the trial design will be consistent with our previous expectations.
While we acknowledge that an SPA would have been seen to lower the risk to the program, the eventual approval decision should still be driven by a compelling demonstration of efficacy.
News highlights risks of over-transparency.
We believe the primary driver of the stock's weakness will be that management had set very clear expectations for the SPA as a near-certainty to be finalized in the very near term.
In our view, the news highlights three points: 1) Nothing is certain with the FDA until it is final; 2) Management's credibility will be challenged by investors, especially in light of the recent equity financing; 3) However, we believe this is consistent with recent FDA views on PFS and that the agency appears committed to enabling the development of palifosfamide.
************
http://messages.finance.yahoo.com/Stocks_%28A_to_Z%29/Stocks…
Needham highlights
-Palifosfamide phase III will be only front line patients. Phase II was front and second line and the second line patients didn't show as significant a difference. So they are stacking the deck in their favor.
-The rationale for the last money raise was to expand and go after more indications, such as pediatric cancers and small cell lung cancer.
-Small cell lung cancer trial being run by Dr. Larry Einhorn, the guy behind approval of ifosfamide. Phase II trial in planning stages.
-Palifosfamide will be priced less than generic
-Oral phase I data soon
-Extensive partnering discussions with the biggest pharma company in the world and many other smaller ones on all 3 programs.
-6-9 months ago they were very eager to partner, but now they're not...
-They are trying to be "as strategic as possible," retaining rights as much as possible for palifosfamide.
-Sounds like they are only going to partner dariniparsin (ongoing discussions on Asian partnership) and indibulin.
-Partnering discussions in early, mid, and late stages - no idea as to timing.
**************
JMP SECURITIES
Biotechnology – Update
ZIOPHARM Oncology, Inc. (1,3)
Data Presentation at ASCO Well Received by Physician Community
MARKET OUTPERFORM ZIOP
Target Price $10.00
INVESTMENT HIGHLIGHTS
o PICASSO presentation at ASCO well received, boding well for pivotal accrual; reiterate Market Outperform rating and $10 price target on Ziopharm. Results from the PICASSO trial were presented in an oral session on Monday afternoon and, in our view, the data were well received by the attending physician community. While the key data from the presentation had already been previously released, we believe that the most important aim of this presentation was to attract the attention of the broader sarcoma physician community, and it is our view that this aim was achieved. Our diligence with potential study investigators for the pivotal trial increases our confidence for timely enrollment following the initiation of this trial in mid-year. We anticipate that the further presentations on the “Best of ASCO” tour will generate additional physician interest in the pivotal trial in the US and also in Europe. Furthermore, a breakfast meeting we held with the presenter of the data provided several key insights on the pivotal trial, market need, and future adoption of palifosfamide. Our $10 price target is derived from a sum-of-the-parts analysis of the palifosfamide revenue opportunity in sarcoma, including both US sales and ex-US royalties.
o Breakfast with primary investigator provides several incremental positives. During the conference we hosted a breakfast meeting with Dr. Claire Verschraegen, the first author on the PICASSO abstract. Dr. Verschraegen’s comments yielded or confirmed several positives both for the pivotal trial and commercial potential of palifosfamide. Her overall impression of the data was overwhelmingly positive, specifically with respect to the hazard ratio and favorable safety profile of the combination. Dr. Verschraegen noted that typically combination therapy is expected to have increased the toxicity profile of doxorubicin. She also highlighted the cost savings with palifosfamide over ifosfamide as mesna co-therapy is not required, noting that ifosfamide treatment requires hospitalization for one week for rehydration. Dr. Verschraegen confirmed that the primary endpoint for the pivotal trial will be PFS and she was positive on the prospects for patient enrollment. Her belief was that the only other competing agent for trial patients is Yondelis and the strict enrollment criteria rules out 2 of 3 patients in this trial. Finally, in the non-clinical trial setting, she believes that palifosfamide will be dosed through progression due to lack of treatment options.
o PICASSO data does not disappoint. The PICASSO trial was a randomized Phase II trial assessing the efficacy and safety of palifosfamide in combination with doxorubicin compared to doxorubicin alone. The trial was halted early due to efficacy following a planned review by the independent Data Safety Monitoring Committee (DSMB). At data cutoff there were 66 patients who had been treated in the trial and 62 were eligible for evaluation. A response was reported for 7 of 30 patients (23%) in the palifosfamide combination arm and for 3 of 32 patients (9%) in the doxorubicin alone arm. There were 18 progression events in the doxorubicin alone arm compared to 10 in the palifosfamide arm, representing a statistically significant hazard ratio of 0.43 (p=0.019).
The median PFS in the palifosfamide arm was 7.8 months, 3.4 months greater than in the doxorubicin alone arm. An analysis of patients receiving treatment in either arm of the trial for 6 cycles or less (the standard treatment period for doxorubicin) demonstrated a hazard ratio of 0.39 (p= 0.023) in favor of palifosfamide. Overall survival data are not yet mature and unlikely to yield a divergence between the groups due to patient crossover following the DSMB review; however, trend is currently in favor of palifosfamide. We view this survival data as positive, as a positive survival trend will likely be viewed as supportive of approval in the pivotal trial, in addition to the primary endpoint of PFS.
***
o Key takeaways include rigor in Pali protocol design, possible timelines, IP and additional indications, with capital efficiency remaining “Job #1.” Some of the key discussion points on which we gleaned incremental insights for the upcoming palifosfamide pivotal program in sarcoma include endpoints (clearly PFS but powered for OS), various ways to deal with discordant data (primary or site read vs. independent reader), and possible DSMB futility/safety reviews, as well as timelines for enrollment (start in 3Q10) and early data (PFS from roughly 2/3 events in 1H12). All said, this incremental info and our discussions on rigorous trial design, endpoints, and statistical plan provide us confidence in the Pali sarcoma program. Beyond the important start of this lead program, for additional value creation prior to Pali pivotal data we now look to increasing visibility of possible Pali utility from soon-to-start trials in small cell lung/breast cancer, and early (Phase I and preclinical) data for Daraniparsin in PTCL (1H11) and an undisclosed large market indication (oral version, 2H10). Finally, we are pleased with the IP on Pali and Dara, with composition-of-matter claims (on Pali) extending beyond 2025. Although we believe the clinical data from other indications for Pali and perhaps those for Daraniparsin and Indibulin (breast cancer) will result in many monetizable assets while also diversifying risk, we hesitate to value these until data is in hand. Therefore, our valuation for ZIOP shares continues to be driven by the sarcoma potential of Pali.
o Updating model to include financing. Last week, Ziopharm completed an equity financing, raising net proceeds of approximately $33MM, excluding overallotment allocations, through the sale of 7 million shares of common stock at $5 per share. This cash adds to the cash balance of $45MM at the end of 1Q10, giving a pro forma cash position of $78MM. We view this cash balance as more than sufficient to complete the clinical development of palifosfamide in advanced sarcoma, as well as limited clinical development for other candidates. Following the completion of this transaction, Ziopharm has basic shares outstanding of 48.8 million. We have updated our model to increase the weighted average share count in 2Q10 and beyond. In addition, the fully diluted share count includes stock options outstanding of 3.5 million, stock options available for issue of 3.1 million, and warrants of 15.9 million (exercisable at $4.11 per share). The net impact of the changes to our model does not have any impact on our valuation or price target on ZIOP shares.
mfg ipollit
ein weiterer Erfolg für ISIS:
Sanofi signs up to $750 mln drug deal with Regulus
Tue Jun 22, 2010 2:30am EDTStocks
* Regulus gets $25 mln upfront plus $10 equity investment * Regulus is jointly owned by Alnylam and Isis
(Adds details on deal value, background)
PARIS, June 22 (Reuters) - French drugmaker Sanofi-Aventis (SASY.PA) has struck a strategic alliance potentially worth up to $750 million with Regulus Therapeutics, taking it into the emerging field of microRNA.
The alliance will initially focus on developing new treatments for fibrosis, an excessive growth of fibrous tissue that can cause various disorders, Sanofi said on Tuesday.
MicroRNAs are small scraps of genetic material which are being explored as a new class of therapies for a range of diseases, given their ability to regulate gene expression.
Sanofi and Regulus will collaborate on microRNA drug discovery and preclinical development for an initial four targets, including the lead fibrosis programme. Sanofi also has an option to develop and commercialise other compounds, beyond the first four targets.
Regulus, which is jointly owned by Alnylam Pharmaceuticals (ALNY.O) and Isis Pharmaceuticals (ISIS.O), will receive a $25 million upfront fee and a future $10 million equity investment.
In total, the alliance could be valued at over $750 million when taking into account upfront payments, equity investment, research funding, and potential preclinical, clinical and commercial milestone payments for multiple products. (Reporting by Ben Hirschler; Editing by Hans Peters)
Sanofi signs up to $750 mln drug deal with Regulus
Tue Jun 22, 2010 2:30am EDTStocks
* Regulus gets $25 mln upfront plus $10 equity investment * Regulus is jointly owned by Alnylam and Isis
(Adds details on deal value, background)
PARIS, June 22 (Reuters) - French drugmaker Sanofi-Aventis (SASY.PA) has struck a strategic alliance potentially worth up to $750 million with Regulus Therapeutics, taking it into the emerging field of microRNA.
The alliance will initially focus on developing new treatments for fibrosis, an excessive growth of fibrous tissue that can cause various disorders, Sanofi said on Tuesday.
MicroRNAs are small scraps of genetic material which are being explored as a new class of therapies for a range of diseases, given their ability to regulate gene expression.
Sanofi and Regulus will collaborate on microRNA drug discovery and preclinical development for an initial four targets, including the lead fibrosis programme. Sanofi also has an option to develop and commercialise other compounds, beyond the first four targets.
Regulus, which is jointly owned by Alnylam Pharmaceuticals (ALNY.O) and Isis Pharmaceuticals (ISIS.O), will receive a $25 million upfront fee and a future $10 million equity investment.
In total, the alliance could be valued at over $750 million when taking into account upfront payments, equity investment, research funding, and potential preclinical, clinical and commercial milestone payments for multiple products. (Reporting by Ben Hirschler; Editing by Hans Peters)
Antwort auf Beitrag Nr.: 39.716.094 von ipollit am 21.06.10 20:11:13
Hi ipollit,
hier einige kritische Anmerkungen zu ZIOP:
http://www.pharmastrategyblog.com/2010/06/pharma-rd-is-a-rol…
Hi ipollit,
hier einige kritische Anmerkungen zu ZIOP:
http://www.pharmastrategyblog.com/2010/06/pharma-rd-is-a-rol…
zu SGEN in Bezug auf Lintuzumab:
http://www.xconomy.com/seattle/2010/06/22/seattle-genetics-d…
Artikel ist umfassend und m.E. lesenswert.
Die Daten sollten bis August vorliegen. Vielleicht gibts ja mal ne positive Überraschung?
http://www.xconomy.com/seattle/2010/06/22/seattle-genetics-d…
Artikel ist umfassend und m.E. lesenswert.
Die Daten sollten bis August vorliegen. Vielleicht gibts ja mal ne positive Überraschung?
zu AVEO:
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced it has received orphan medicinal product designation for tivozanib (N-{2-chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl}-N'-(5-methyl-3-isoxazolyl) urea hydrochloride monohydrate), its oral, triple VEGF receptor inhibitor, for the treatment of renal cell carcinoma by the European Medicines Agency (EMA).
http://www.businesswire.com/portal/site/home/permalink/?ndmV…
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced it has received orphan medicinal product designation for tivozanib (N-{2-chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl}-N'-(5-methyl-3-isoxazolyl) urea hydrochloride monohydrate),
its oral, triple VEGF receptor inhibitor, for the treatment of renal cell carcinoma by the European Medicines Agency (EMA).http://www.businesswire.com/portal/site/home/permalink/?ndmV…
Antwort auf Beitrag Nr.: 39.720.950 von SLGramann am 22.06.10 16:53:07diese kritischen Anmerkungen zu ZIOP kann ich teilweise nicht so ganz nachvollziehen...
z.B. "Secondly, the big sticking point for the FDA was the proposed primary endpoint, ie PFS rather than overall survival (OS). One does not always follow the other, but seriously, accepting OS as the goal may well have been an easy win-win for Ziopharm."
das ist einfach gesagt... ein einfacher win-win, wenn man OS als primären Endpunkt nimmt. Finde ich schon merkwürdig, dass das nicht differenzierter hinterfragt wird. In den meisten Kommentaren wird angeführt, dass OS ein Risiko wäre, da hierfür keine PII-Daten vorliegen, anderes als für PFS. Das halte ich für Blödsinn, da wohl Palifosfamide wohl nur dann einen wirklichen Nutzen bringen wird, wenn es sich auch positiv auf das Überleben auswirkt. Der Punkt ist für mich aber der, dass eine OS-PIII u.U. extrem viel länger braucht als die Bestimmung von PFS... OS heißt ja, dass man warten muss, bis entsprechend viele Patienten gestorben sind. Sind sie sehr sehr krank, dann dauert es nicht so lang, obwohl es dann auch schwierig sein kann, überhaupt eine Wirkung von Palifosfamide zu erkennen. Bei der Krebsart scheint es aber doch (zum Glück) recht lange zu dauern, bis die Patienten daran sterben. Das macht die Studie teuer, weil sie lange dauert, unkalkulierbarer, weil sich in der langen Zeit viel ändern kann, was dann selbst positive Ergebnisse unbrauchbar macht. U.U. lässt sich eine solche Studie garnicht sinnvoll durchführen. Einfach auf OS zu wechseln, um ein SPA zu bekommen, ist sehr kurz gedacht. Z.B. hier steht auch so etwas... http://investorshub.advfn.com/boards/read_msg.aspx?message_i… "But all sarcoma trials that I am aware of use PFS as their primary endpoint... Note that OS sounds nice and straightforward, but in a condition like sarcoma where the majority of newly diagnosed patients survive 5 years and more, it's actually a very murky endpoint indeed. When people progress, they drop out of the trial and move on to other therapies or are lost to follow-up. So really it's not feasible as a primary endpoint here."
Außerdem was bringt schon ein SPA? Die Aussage "If changing the endpoint from PFS to OS is all that is needed for garnering an SPA, I think I would grab it and go. It's much easier to gain approval if you meet the criteria for a pre-negotiated SPA, as Roche/Genentech have shown with Tarceva several times, even with marginal, yet significant improvements, based on well powered studies." ist doch auch ein ziemlicher Blödsinn! Wie ist das denn mit GPC und Satraplatin gelaufen, womit ich ziemlich auf die Nase gefallen bin?... die hatten ein SPA und gemäß des SPA war die PIII ganz klar bezüglich PFS erfolgreich. Und was hat die FDA gemacht? Der war das SPA plötzlich völlig egal und sie haben die Endpunkte einfach nicht mehr akzeptiert... das SPA war da ein absoluter Witz und garnichts wert!
ZIOP hat den Orphan Drug Status... damit können sie eine beschleunigte Zulassung aufgrund der PFS-Daten beantragen, was nicht ungewöhnlich ist... so haben zuletzt z.B. auch Genmabs Arzerra und Allos Folotyn die Zulassung erhalten. Die endgültige Zulassung erfolgt dann später wenn positive OS-Daten vorliegen.
mfg ipollit
z.B. "Secondly, the big sticking point for the FDA was the proposed primary endpoint, ie PFS rather than overall survival (OS). One does not always follow the other, but seriously, accepting OS as the goal may well have been an easy win-win for Ziopharm."
das ist einfach gesagt... ein einfacher win-win, wenn man OS als primären Endpunkt nimmt. Finde ich schon merkwürdig, dass das nicht differenzierter hinterfragt wird. In den meisten Kommentaren wird angeführt, dass OS ein Risiko wäre, da hierfür keine PII-Daten vorliegen, anderes als für PFS. Das halte ich für Blödsinn, da wohl Palifosfamide wohl nur dann einen wirklichen Nutzen bringen wird, wenn es sich auch positiv auf das Überleben auswirkt. Der Punkt ist für mich aber der, dass eine OS-PIII u.U. extrem viel länger braucht als die Bestimmung von PFS... OS heißt ja, dass man warten muss, bis entsprechend viele Patienten gestorben sind. Sind sie sehr sehr krank, dann dauert es nicht so lang, obwohl es dann auch schwierig sein kann, überhaupt eine Wirkung von Palifosfamide zu erkennen. Bei der Krebsart scheint es aber doch (zum Glück) recht lange zu dauern, bis die Patienten daran sterben. Das macht die Studie teuer, weil sie lange dauert, unkalkulierbarer, weil sich in der langen Zeit viel ändern kann, was dann selbst positive Ergebnisse unbrauchbar macht. U.U. lässt sich eine solche Studie garnicht sinnvoll durchführen. Einfach auf OS zu wechseln, um ein SPA zu bekommen, ist sehr kurz gedacht. Z.B. hier steht auch so etwas... http://investorshub.advfn.com/boards/read_msg.aspx?message_i… "But all sarcoma trials that I am aware of use PFS as their primary endpoint... Note that OS sounds nice and straightforward, but in a condition like sarcoma where the majority of newly diagnosed patients survive 5 years and more, it's actually a very murky endpoint indeed. When people progress, they drop out of the trial and move on to other therapies or are lost to follow-up. So really it's not feasible as a primary endpoint here."
Außerdem was bringt schon ein SPA? Die Aussage "If changing the endpoint from PFS to OS is all that is needed for garnering an SPA, I think I would grab it and go. It's much easier to gain approval if you meet the criteria for a pre-negotiated SPA, as Roche/Genentech have shown with Tarceva several times, even with marginal, yet significant improvements, based on well powered studies." ist doch auch ein ziemlicher Blödsinn! Wie ist das denn mit GPC und Satraplatin gelaufen, womit ich ziemlich auf die Nase gefallen bin?... die hatten ein SPA und gemäß des SPA war die PIII ganz klar bezüglich PFS erfolgreich. Und was hat die FDA gemacht? Der war das SPA plötzlich völlig egal und sie haben die Endpunkte einfach nicht mehr akzeptiert... das SPA war da ein absoluter Witz und garnichts wert!
ZIOP hat den Orphan Drug Status... damit können sie eine beschleunigte Zulassung aufgrund der PFS-Daten beantragen, was nicht ungewöhnlich ist... so haben zuletzt z.B. auch Genmabs Arzerra und Allos Folotyn die Zulassung erhalten. Die endgültige Zulassung erfolgt dann später wenn positive OS-Daten vorliegen.
mfg ipollit
REGN...
Pfizer hat sein Tanezumab-Programm wegen Nebenwirkungen gestoppt... unter Umständen hat dies Auswirkungen auf REGN475, das in PII ist... beide AKs richten sich gegen NGF. Wenn das Problem beim Target liegt, wird auch REGN475 keinen Erfolg haben.
http://www.fiercebiotech.com/story/safety-concerns-force-pfi…
Safety concerns force Pfizer to suspend pain trials
June 24, 2010 — 9:15am ET
Pfizer can add one more promising experimental drug that has been hobbled by unexpected safety issues. The pharma giant says it has been forced to suspend an ambitious slate of trials of its experimental pain drug tanezumab after patients taking the therapy for osteoarthritis saw their condition worsen to the point they required joint replacement surgery. Pfizer, which only days ago touted "extraordinary" late-stage data for the drug, says that at the request of the FDA its researchers have stopped enrolling patients or providing the drug in all of its osteoarthritis trials.
"Patient safety is a top priority for Pfizer," a Pfizer spokesperson told Bloomberg in an e-mail. "We are coordinating closely with regulatory authorities and investigators to implement the clinical hold in the osteoarthritis studies and for patients with co-morbid osteoarthritis in the chronic pain program."
Tanezumab, an infused biologic which is designed to block pain by inhibiting nerve growth factor, moved into late-stage testing a year ago. And Pfizer execs had high hopes that the therapy could go on to become the first biotech therapy approved for pain. A Bloomberg analyst survey concluded that the drug had the potential to earn $260 million a year by 2013. Instead, the drug company faces a fresh round of criticism for its plague of development problems despite an annual R&D budget of $7 billion.
In addition to osteoarthritis Pfizer was studying tanezumab for cancer pain, lower back pain and other types of pain. "I don't think it's definitely over for this product, but this is a potential big red flag for some of those other indications," Morningstar analyst Damien Conover told Reuters.
mfg ipollit
Pfizer hat sein Tanezumab-Programm wegen Nebenwirkungen gestoppt... unter Umständen hat dies Auswirkungen auf REGN475, das in PII ist... beide AKs richten sich gegen NGF. Wenn das Problem beim Target liegt, wird auch REGN475 keinen Erfolg haben.
http://www.fiercebiotech.com/story/safety-concerns-force-pfi…
Safety concerns force Pfizer to suspend pain trials
June 24, 2010 — 9:15am ET
Pfizer can add one more promising experimental drug that has been hobbled by unexpected safety issues. The pharma giant says it has been forced to suspend an ambitious slate of trials of its experimental pain drug tanezumab after patients taking the therapy for osteoarthritis saw their condition worsen to the point they required joint replacement surgery. Pfizer, which only days ago touted "extraordinary" late-stage data for the drug, says that at the request of the FDA its researchers have stopped enrolling patients or providing the drug in all of its osteoarthritis trials.
"Patient safety is a top priority for Pfizer," a Pfizer spokesperson told Bloomberg in an e-mail. "We are coordinating closely with regulatory authorities and investigators to implement the clinical hold in the osteoarthritis studies and for patients with co-morbid osteoarthritis in the chronic pain program."
Tanezumab, an infused biologic which is designed to block pain by inhibiting nerve growth factor, moved into late-stage testing a year ago. And Pfizer execs had high hopes that the therapy could go on to become the first biotech therapy approved for pain. A Bloomberg analyst survey concluded that the drug had the potential to earn $260 million a year by 2013. Instead, the drug company faces a fresh round of criticism for its plague of development problems despite an annual R&D budget of $7 billion.
In addition to osteoarthritis Pfizer was studying tanezumab for cancer pain, lower back pain and other types of pain. "I don't think it's definitely over for this product, but this is a potential big red flag for some of those other indications," Morningstar analyst Damien Conover told Reuters.
mfg ipollit
Antwort auf Beitrag Nr.: 39.734.578 von ipollit am 25.06.10 00:11:57
ipollit, danke für das Statement, das sehr aufschlussreich ist. Was Du über GPC schreibst, ist schon erstaunlich. Ich habe das damals alles noch überhaupt nicht mitbekommen. Nach meinem Verständnis ist ein SPA schon eine Vereinbarung, die starke Bindungswirkung hat? Die FDA muss damals doch auch rationale Gründe gehabt haben? Oder ist GPC da einem generellen Politikwechsel zum Opfer gefallen? Aber vielleicht führt es zu weit, so "olle Kamellen" jetzt zu diskutieren.
Gruß
ipollit, danke für das Statement, das sehr aufschlussreich ist. Was Du über GPC schreibst, ist schon erstaunlich. Ich habe das damals alles noch überhaupt nicht mitbekommen. Nach meinem Verständnis ist ein SPA schon eine Vereinbarung, die starke Bindungswirkung hat? Die FDA muss damals doch auch rationale Gründe gehabt haben? Oder ist GPC da einem generellen Politikwechsel zum Opfer gefallen? Aber vielleicht führt es zu weit, so "olle Kamellen" jetzt zu diskutieren.
Gruß
Antwort auf Beitrag Nr.: 39.734.766 von SLGramann am 25.06.10 06:39:30"Nach meinem Verständnis ist ein SPA schon eine Vereinbarung, die starke Bindungswirkung hat"
Das scheint nach meiner Erfahrung mit GPC aber nicht immer so zu sein. Letztlich ist es eigentlich egal, weil Satraplatin sich in der PIII am Ende nicht auf OS positiv ausgewirkt hat, so dass aus Satraplatin mit diesen Daten nichts geworden wäre. Aber zu diesem Zeitpunkt, als GPC wie geplant eine beschleunigte Zulassung aufgrund von positiven PFS-Daten in einer PIII mit SPA beantragt hat, hätte ich nicht damit gerechnet, dass sie nicht etwa die Ergebnisse in Frage stellen, sondern trotz SPA z.B. die Aussagekraft des Endpunkts, die Messmethoden usw... ich dachte, dass dies im SPA vorher bereits eindeutig geklärt worden wäre. z.B. hier... http://www.fda.gov/ohrms/dockets/AC/07/transcripts/2007-4309…
"The first issue is the definition of one of the two co-primary endpoints, PFS. PFS is defined as a composite endpoint, consisting of radiographic progression, symptomatic progression, including pain, analgesic consumption, ECOG performance status, weight loss and other clinical events related to prostate cancer, and also skeletal-related events.
The FDA has no experience with this composite endpoint. This concern was clearly communicated to the commercial sponsor during the development phase and planning for the trial. The FDA has recommended that the trial's primary endpoint be overall survival in several meetings and correspondence with the sponsor. Although a special protocol assessment was submitted, the agency did not agree with the definition of PFS and stated that the acceptability of the sponsor-defined PFS epidemiologic would be a review issue. The acceptability of this endpoint would be subject to the evaluation of the magnitude of effect on the endpoint's component, the reliability and objectivity and the measurement of the endpoint, and the clinical significance of the claimed effect.
The FDA will seek ODAC advice on the acceptability and reliability of this composite endpoint as the basis for marketing approval. ... Satraplatin was better than placebo on the composite endpoint with a median PFS of 11.1 weeks versus 9.7 weeks. ...
vielleicht hat die FDA im SPA auch vorher eindeutig gesagt, dass sie PFS so nicht akzeptieren... dann verstehe ich aber nicht, warum GPC trotzdem die Studie so durchgeführt hat. Oder die FDA hat es einfach offen gelassen, ob sie trotz SPA dieses PFS akzeptieren. Wie dem auch sei... ein SPA ist demnach keine Garantie, dass die FDA nicht hinterher die Messmethoden und Studiendurchführung kritisiert. Letzteres sollte das SPA ja eigentlich verhindern.
mfg ipollit
Das scheint nach meiner Erfahrung mit GPC aber nicht immer so zu sein. Letztlich ist es eigentlich egal, weil Satraplatin sich in der PIII am Ende nicht auf OS positiv ausgewirkt hat, so dass aus Satraplatin mit diesen Daten nichts geworden wäre. Aber zu diesem Zeitpunkt, als GPC wie geplant eine beschleunigte Zulassung aufgrund von positiven PFS-Daten in einer PIII mit SPA beantragt hat, hätte ich nicht damit gerechnet, dass sie nicht etwa die Ergebnisse in Frage stellen, sondern trotz SPA z.B. die Aussagekraft des Endpunkts, die Messmethoden usw... ich dachte, dass dies im SPA vorher bereits eindeutig geklärt worden wäre. z.B. hier... http://www.fda.gov/ohrms/dockets/AC/07/transcripts/2007-4309…
"The first issue is the definition of one of the two co-primary endpoints, PFS. PFS is defined as a composite endpoint, consisting of radiographic progression, symptomatic progression, including pain, analgesic consumption, ECOG performance status, weight loss and other clinical events related to prostate cancer, and also skeletal-related events.
The FDA has no experience with this composite endpoint. This concern was clearly communicated to the commercial sponsor during the development phase and planning for the trial. The FDA has recommended that the trial's primary endpoint be overall survival in several meetings and correspondence with the sponsor. Although a special protocol assessment was submitted, the agency did not agree with the definition of PFS and stated that the acceptability of the sponsor-defined PFS epidemiologic would be a review issue. The acceptability of this endpoint would be subject to the evaluation of the magnitude of effect on the endpoint's component, the reliability and objectivity and the measurement of the endpoint, and the clinical significance of the claimed effect.
The FDA will seek ODAC advice on the acceptability and reliability of this composite endpoint as the basis for marketing approval. ... Satraplatin was better than placebo on the composite endpoint with a median PFS of 11.1 weeks versus 9.7 weeks. ...
vielleicht hat die FDA im SPA auch vorher eindeutig gesagt, dass sie PFS so nicht akzeptieren... dann verstehe ich aber nicht, warum GPC trotzdem die Studie so durchgeführt hat. Oder die FDA hat es einfach offen gelassen, ob sie trotz SPA dieses PFS akzeptieren. Wie dem auch sei... ein SPA ist demnach keine Garantie, dass die FDA nicht hinterher die Messmethoden und Studiendurchführung kritisiert. Letzteres sollte das SPA ja eigentlich verhindern.
mfg ipollit
Antwort auf Beitrag Nr.: 39.741.842 von ipollit am 26.06.10 19:45:03bezügl. SPA und dieser GPC-Geschichte... hab hier noch einen aktuellen Artikel von David Miller gefunden, in dem dieser Fall erwähnt wird.
Hier geht es um BMS(Medarex) Ipilimumab... sind Therapien, die in das Immunsystem eingreifen, doch nicht so ohne, wie viele vielleicht annehmen? Hierbei wird ja das Immunsystem (also etwas natürliches) dazu gebracht, den Krebs zu bekämpfen... ist das sicher beherrschbar? Im Falle von Ipilimumab scheint es ja über das Immunsystem auch zu tötlichen Nebenwirkungen zu kommen. Könnte das nicht auch bei anderen Ansätzen wie z.B. MITI's Bite-AKs ein mögliches Problem sein: dass das Immunsystem zu sensibel gegen körpereigene Zellen wird, was sich dann nicht mehr abstellen lässt und zu einer chronischen vielleicht sogar tötlichen Autoimmunreaktion führen könnte?
http://www.minyanville.com/businessmarkets/articles/david-mi…
Bristol's Melanoma Drug Could Make the Stock a Buy
By David Miller Jun 07, 2010 10:00 am
...
The loud one is ipilimumab, or “ipi” for short. This drug was developed by Medarex, which was acquired by Bristol-Myers Squibb (BMY). Bristol is claiming ipi demonstrated the first survival advantage in Stage IV metastatic melanoma ever seen. It's clearly the most hyped drug at this conference.
So does it live up to the hype?
My firm used to cover Medarex, so I’m plenty familiar with the drug. Ipi is something called a CTLA-4 inhibitor. You have an immune system that is capable of attacking any cell in your body. Yet your immune system generally only attacks foreign invaders. CTLA-4 is the “check point regulator” preventing your own immune cells from turning you into a puddle of goo. By inhibiting CTLA-4, ipi hopes to take the brakes off the immune system a little so your immune system finally recognizes and destroys cancer cells.
This removal of the brakes comes at a cost. Patients who get ipi not only have their cancer cells attacked, but also healthy cells. Some patients get the brakes taken off too much and die from the drug, typically due to perforations or other issues with the gastrointestinal tract.
Single-digit percentages of patients died of ipi in the clinical trial trumpeted by Bristol at ASCO. However, ipi increased survival and tumor response rates over the control arm -- which was a cancer peptide vaccine called gp100 that has failed more trials than I have fingers and toes.
So does it mean anything if ipi beat a known-failed drug? That’s a question I'm certain an FDA advisory panel will have to sort out when Bristol applies for approval. And that’s the point I want to make here amidst all the investor-side hype and all the understandable hope from melanoma patients -- approval of ipilimumab by this FDA is not a sure thing.
I believe ipi works. I'm surprised it worked in melanoma because no drugs work in melanoma. But I’m not one of the ipi doubters.
The problem with the ipi melanoma trial is not efficacy. It is potentially the trial design. Ameliorating this is the original trial design was subject to a Special Protocol Agreement (shortened to SPA) with the FDA. Having a SPA means the FDA has blessed the statistical and structural design of your trial. If you follow the agreed statistical and structural design precisely, the drug proves safe and effective, and some other advancement doesn’t happen in your field while you are running the trial -- you’re likely good to go for approval. Deviations or safety issues can derail the SPA, however. Just ask those who bet on satraplatin approval.
Bristol changed the trial from the original design. One would hope it did so with FDA approval, but the three presentations we’ve seen here at ASCO never mentioned the SPA was amended. One amendment was to use overall survival as the primary endpoint instead of objective response. We doubt the FDA had any problem with that because the original endpoint was also statistically significant. The other amendment was to shift the primary analysis of the trial from ipi+gp100 as the primary study arm to “any arm containing ipilimumab.” This is subtle biostatistical geek stuff, but could be important.
This trial showed gp100 was inert at best and harmful at worst. Ipi monotherapy was the winner. The trial wasn’t originally designed to show that, and the biostatisticians at the FDA will have a problem with that unless the SPA was amended. I agree that makes no sense. But those who have read most everything I've written here at Minyanville know I consistently counsel how FDA biostatistical reality and common sense rarely meet.
So that’s one issue. Anyone interested in buying Bristol-Myers Squibb on this news needs to confirm they formally amended the SPA with the FDA before pressing that buy button.
The second issue is a good issue to have, and that has to do with product labeling after approval. Because gp100 only works in a certain patient population (HLA-A2 positive patients), enrollment in the trial is restricted to HAL-A2+ patients. While there's no indication ipi activity is restricted in this way, that’s the patient population studied.
If ipi is approved, will the label restrict use to HLA-A2+ patients? This is a non-trivial issue because only 50% of the intended “customer” base for the drug is HLA-A2+. I’ve seen a couple of revenue projections for ipi from other analysts and they don't adjust for this 50% haircut.
Here’s what the FDA’s Office of Oncologic Drugs (OOD) had to say in a similar situation in a recent drug approval:
“Indication statements… are supported by trials which have shown an effect, such as an improvement in overall survival, on the patient’s underlying disease. [T]he indication for [__________] should specify the study patient population in which the key trial supporting this [drug application] was conducted.”
To translate, OOD is suggesting if the trial didn’t study a population then that population should not be allowed on the label. If OOD is going to be consistent in the application of this rule, then the initial label for ipilimumab will be limited to the 50% of patients who are HLA-A2+.
I would expect many of the other 50% will get the drug off label. How widespread off-label use is depends on pricing. I also believe some insurance companies will be convinced to cover the cost for non-HLA-A2+ patients given the clinical history. If you're doing financial modeling on this drug, the safe bet is the FDA will have a restrictive label and anything else is gravy.
The final issue here is safety. Melanoma docs are neither trained nor prepared to deal with the kinds of serious adverse events that pop up in ipi trials. I believe the FDA will require an above-average surveillance program (called a REMS for you FDA geeks) to go along with any ipi approval. This program will require more regular and rapid reporting of side effects. It will also likely require long-term following of patients to understand the long-term side effects of CTLA-4 inhibition.
For a company like Bristol, these requests wouldn't be showstoppers. They will increase the cost of ipilimumab and, because of the paperwork and training involved for prescribing doctors, will tend to slow the initial sales ramp.
Bottom line, though, it looks like late-stage, metastatic melanoma patients finally have a new drug. Ipi isn't a picnic to take, but it's better than the alternatives. Depending on the SPA amendment issue, I think the FDA is likely to approve the drug. The market will know more when the briefing documents appear for what I believe will be an inevitable advisory panel.
mfg ipollit
Hier geht es um BMS(Medarex) Ipilimumab... sind Therapien, die in das Immunsystem eingreifen, doch nicht so ohne, wie viele vielleicht annehmen? Hierbei wird ja das Immunsystem (also etwas natürliches) dazu gebracht, den Krebs zu bekämpfen... ist das sicher beherrschbar? Im Falle von Ipilimumab scheint es ja über das Immunsystem auch zu tötlichen Nebenwirkungen zu kommen. Könnte das nicht auch bei anderen Ansätzen wie z.B. MITI's Bite-AKs ein mögliches Problem sein: dass das Immunsystem zu sensibel gegen körpereigene Zellen wird, was sich dann nicht mehr abstellen lässt und zu einer chronischen vielleicht sogar tötlichen Autoimmunreaktion führen könnte?
http://www.minyanville.com/businessmarkets/articles/david-mi…
Bristol's Melanoma Drug Could Make the Stock a Buy
By David Miller Jun 07, 2010 10:00 am
...
The loud one is ipilimumab, or “ipi” for short. This drug was developed by Medarex, which was acquired by Bristol-Myers Squibb (BMY). Bristol is claiming ipi demonstrated the first survival advantage in Stage IV metastatic melanoma ever seen. It's clearly the most hyped drug at this conference.
So does it live up to the hype?
My firm used to cover Medarex, so I’m plenty familiar with the drug. Ipi is something called a CTLA-4 inhibitor. You have an immune system that is capable of attacking any cell in your body. Yet your immune system generally only attacks foreign invaders. CTLA-4 is the “check point regulator” preventing your own immune cells from turning you into a puddle of goo. By inhibiting CTLA-4, ipi hopes to take the brakes off the immune system a little so your immune system finally recognizes and destroys cancer cells.
This removal of the brakes comes at a cost. Patients who get ipi not only have their cancer cells attacked, but also healthy cells. Some patients get the brakes taken off too much and die from the drug, typically due to perforations or other issues with the gastrointestinal tract.
Single-digit percentages of patients died of ipi in the clinical trial trumpeted by Bristol at ASCO. However, ipi increased survival and tumor response rates over the control arm -- which was a cancer peptide vaccine called gp100 that has failed more trials than I have fingers and toes.
So does it mean anything if ipi beat a known-failed drug? That’s a question I'm certain an FDA advisory panel will have to sort out when Bristol applies for approval. And that’s the point I want to make here amidst all the investor-side hype and all the understandable hope from melanoma patients -- approval of ipilimumab by this FDA is not a sure thing.
I believe ipi works. I'm surprised it worked in melanoma because no drugs work in melanoma. But I’m not one of the ipi doubters.
The problem with the ipi melanoma trial is not efficacy. It is potentially the trial design. Ameliorating this is the original trial design was subject to a Special Protocol Agreement (shortened to SPA) with the FDA. Having a SPA means the FDA has blessed the statistical and structural design of your trial. If you follow the agreed statistical and structural design precisely, the drug proves safe and effective, and some other advancement doesn’t happen in your field while you are running the trial -- you’re likely good to go for approval. Deviations or safety issues can derail the SPA, however. Just ask those who bet on satraplatin approval.
Bristol changed the trial from the original design. One would hope it did so with FDA approval, but the three presentations we’ve seen here at ASCO never mentioned the SPA was amended. One amendment was to use overall survival as the primary endpoint instead of objective response. We doubt the FDA had any problem with that because the original endpoint was also statistically significant. The other amendment was to shift the primary analysis of the trial from ipi+gp100 as the primary study arm to “any arm containing ipilimumab.” This is subtle biostatistical geek stuff, but could be important.
This trial showed gp100 was inert at best and harmful at worst. Ipi monotherapy was the winner. The trial wasn’t originally designed to show that, and the biostatisticians at the FDA will have a problem with that unless the SPA was amended. I agree that makes no sense. But those who have read most everything I've written here at Minyanville know I consistently counsel how FDA biostatistical reality and common sense rarely meet.
So that’s one issue. Anyone interested in buying Bristol-Myers Squibb on this news needs to confirm they formally amended the SPA with the FDA before pressing that buy button.
The second issue is a good issue to have, and that has to do with product labeling after approval. Because gp100 only works in a certain patient population (HLA-A2 positive patients), enrollment in the trial is restricted to HAL-A2+ patients. While there's no indication ipi activity is restricted in this way, that’s the patient population studied.
If ipi is approved, will the label restrict use to HLA-A2+ patients? This is a non-trivial issue because only 50% of the intended “customer” base for the drug is HLA-A2+. I’ve seen a couple of revenue projections for ipi from other analysts and they don't adjust for this 50% haircut.
Here’s what the FDA’s Office of Oncologic Drugs (OOD) had to say in a similar situation in a recent drug approval:
“Indication statements… are supported by trials which have shown an effect, such as an improvement in overall survival, on the patient’s underlying disease. [T]he indication for [__________] should specify the study patient population in which the key trial supporting this [drug application] was conducted.”
To translate, OOD is suggesting if the trial didn’t study a population then that population should not be allowed on the label. If OOD is going to be consistent in the application of this rule, then the initial label for ipilimumab will be limited to the 50% of patients who are HLA-A2+.
I would expect many of the other 50% will get the drug off label. How widespread off-label use is depends on pricing. I also believe some insurance companies will be convinced to cover the cost for non-HLA-A2+ patients given the clinical history. If you're doing financial modeling on this drug, the safe bet is the FDA will have a restrictive label and anything else is gravy.
The final issue here is safety. Melanoma docs are neither trained nor prepared to deal with the kinds of serious adverse events that pop up in ipi trials. I believe the FDA will require an above-average surveillance program (called a REMS for you FDA geeks) to go along with any ipi approval. This program will require more regular and rapid reporting of side effects. It will also likely require long-term following of patients to understand the long-term side effects of CTLA-4 inhibition.
For a company like Bristol, these requests wouldn't be showstoppers. They will increase the cost of ipilimumab and, because of the paperwork and training involved for prescribing doctors, will tend to slow the initial sales ramp.
Bottom line, though, it looks like late-stage, metastatic melanoma patients finally have a new drug. Ipi isn't a picnic to take, but it's better than the alternatives. Depending on the SPA amendment issue, I think the FDA is likely to approve the drug. The market will know more when the briefing documents appear for what I believe will be an inevitable advisory panel.
mfg ipollit
Antwort auf Beitrag Nr.: 39.745.936 von ipollit am 28.06.10 14:53:38hier auch noch etwas zum SPA... also selbst ein SPA ist keine Garantie... aber immerhin wohl etwas mehr als kein SPA zu haben
http://www.minyanville.com/articles/print.php?a=12868
While I might get surprised in July by a negative ODAC panel review of Satraplatin...
If I’m wrong, the biotech sector will be a tough place. Satraplatin’s trials were operated under a Special Protocol Assessment (SPA). By all accounts, the sponsors of the satraplatin trial held up their end of that agreement. For Pazdur to turn down a drug operated under a successful SPA and that hit its primary endpoint with a high degree of statistical significance would be a big deal in biotech – a big negative deal.
*******
The FDA requires a statistical analysis plan be filed with every pivotal trial along with the details about how
the trial is run. The stat plan and the trial details are often discussed at length by the FDA and the sponsor
before the sponsor makes a formal submission of the design.
The FDA must accept the trial design, which includes the stat plan, prior to the trial enrolling its first patient.
However, the FDA does not have to agree with the trial design or the stat plan when it accepts the design.
In fact, there are often material disagreements with both the trial design and the stat plan between the FDA
and the sponsor.
This subtle distinction is perhaps the single most costly mistake about regulatory issues that biotech
investors make. This is partly because it makes no sense (why would the FDA accept a trial design it knows
it won’t approve?!?) and partly because many biotech management teams don’t realize the difference
themselves.
There's only one way a biotech company's management and its investors can be certain the FDA agrees with
the stat plan and the trial design: When the sponsor and the FDA enter into something called a Special
Protocol Assessment. A SPA is a negotiated agreement between the sponsor and the FDA where both parties
agree on every important aspect of the trial design and stat plan.
At my firm, management teams have a great deal of explaining to do if they enter a pivotal trial without an
SPA in hand. Post the satraplatin debacle, where those management teams played fast and loose with the
facts surrounding what was and wasn’t in their SPA, we ask some very specific questions about designs and
stat plans. That said, we consider SPAs absolutely critical - particularly in the oncology space.
mfg ipollit
http://www.minyanville.com/articles/print.php?a=12868
While I might get surprised in July by a negative ODAC panel review of Satraplatin...
If I’m wrong, the biotech sector will be a tough place. Satraplatin’s trials were operated under a Special Protocol Assessment (SPA). By all accounts, the sponsors of the satraplatin trial held up their end of that agreement. For Pazdur to turn down a drug operated under a successful SPA and that hit its primary endpoint with a high degree of statistical significance would be a big deal in biotech – a big negative deal.
*******
The FDA requires a statistical analysis plan be filed with every pivotal trial along with the details about how
the trial is run. The stat plan and the trial details are often discussed at length by the FDA and the sponsor
before the sponsor makes a formal submission of the design.
The FDA must accept the trial design, which includes the stat plan, prior to the trial enrolling its first patient.
However, the FDA does not have to agree with the trial design or the stat plan when it accepts the design.
In fact, there are often material disagreements with both the trial design and the stat plan between the FDA
and the sponsor.
This subtle distinction is perhaps the single most costly mistake about regulatory issues that biotech
investors make. This is partly because it makes no sense (why would the FDA accept a trial design it knows
it won’t approve?!?) and partly because many biotech management teams don’t realize the difference
themselves.
There's only one way a biotech company's management and its investors can be certain the FDA agrees with
the stat plan and the trial design: When the sponsor and the FDA enter into something called a Special
Protocol Assessment. A SPA is a negotiated agreement between the sponsor and the FDA where both parties
agree on every important aspect of the trial design and stat plan.
At my firm, management teams have a great deal of explaining to do if they enter a pivotal trial without an
SPA in hand. Post the satraplatin debacle, where those management teams played fast and loose with the
facts surrounding what was and wasn’t in their SPA, we ask some very specific questions about designs and
stat plans. That said, we consider SPAs absolutely critical - particularly in the oncology space.
mfg ipollit
Antwort auf Beitrag Nr.: 39.746.001 von ipollit am 28.06.10 15:04:21
Alles sehr interessant! Scheint mir aber durchaus so zu sein, dass die letzte Verantwortung für ein vernünftiges Design auch bei einem SPA beim Unternehmen bleibt.
"The bearish counter argument is that while satraplatin demonstrated statistical significance on their designated primary endpoint, the benefit is not clinically significant at less than two weeks median benefit."
Tja, SPA scheint mir sehr wichtig und sehr positiv zu sein - vorausgesetzt, man kann dem Entwickler vertrauen, dass er wirklich die Zulassung im Blick hat und das Design dementsprechend abstimmt. Insofern war der Endpunkt im Fall Satraplatin wahrscheinlich nicht hinreichend sicher ausgewählt.
Ist aber für mich leider trotzdem etwas schwer zu verstehen...
hier noch ein Artikel zu EXEL (von Prohost):
Following Exelixis (EXEL) press release, which announced that it has regained full rights from Bristol-Myers Squibb (BMS) to its cancer drug XL184, the firm’s stock plummeted. In the press release, Exelixis stated that BMS’ priorities made it difficult for BMS to align on the scope, breadth and pace of the ongoing clinical development of XL184 as agreed upon in the original agreement. Those who instigated the sell-off did not buy Exelixis’ explanation and were more comfortable with the notion that BMS decided to walk away from XL184, probably because of bad news about the drug. Their reasoning was that a giant pharmaceutical company like BMS, which focuses on building an impressive advanced oncology pipeline would not let a promising multi-targeted cancer drug in late phase trials slip from its hands without a significant reason. BMS did not allude to any bad reason or any reason at all for its decision. It did not comment on Exelixis’ explanation and did not deny it.
Although investors’ speculation and their spontaneous response, instigating the selling of the stock, are not irrational, they were not supported by evidence-based knowledge, or facts. Their best speculation was that if BMS doubts the drug’s promises the least of what they can do is the same as BMS did, i.e., abandoning investing in Exelixis. The sell-off was triggered by a pyramid of suppositions that have no concrete proof.
Now that the acute fear has subsided and Exelixis stock price bottomed, other investors and, probably, some of those who sold the stock, might like to take a deep breath and reexamine the whole situation in a more professional and rational way. It is not unreasonable for investors to try to find out good investment opportunities in bottomed stocks.
To find out whether the stock selling based on the sellers’ reading of BMS’ mind is correct, or a mistake, we resorted to history, which tells that many large pharmaceutical firms have abandoned the development of drugs that have become best sellers when other firms continued developing them. Looking at the drug itself and the history of its performance in clinical trials, we see XL184 as not only the most advanced product in Exelixis pipeline, but is also the most advanced MET inhibitor. MET pathway has long been recognized as a master switch and drug target in cancer progression. MET is mutationally activated in hereditary and sporadic papillary renal cell carcinoma and some head and neck cancers. It is either over-expressed or activated in the absence of mutation in glioblastomas, breast carcinomas, gastric cancers and other solid tumors. MET amplification has been demonstrated in some NSCLCs.
XL184 also targets and inhibits VEGFR2, which leads to starving the tumors of oxygen through the antiangiogenic effect. Expression of VEGF occurs in various cancers and has been associated with prognostic significance. Targeting the VEGF receptor has already demonstrated efficacy as anti-cancer strategy in multiple tumors. Dual targeting of MET and VEGFR2 blocks two of the major mechanisms tumors use to overcome hypoxia.
In addition, XL184 inhibits RET, which is identified in multiple endocrine tumors and familial medullary thyroid carcinoma. Activated RET is involved in regulating cell proliferation, migration, differentiation, and survival. It is activated in papillary thyroid cancer (PTC) and in both familial and sporadic forms of medullary thyroid cancer (MTC).
These unique combination of actions makes XL184 capable of causing antiangiogenic, antiproliferative, and antiinvasive effects in tumors – all have been confirmed in preclinical cancer models and in some clinical trial results.
At ASCO 2010 Annual Meeting, the developers reported that in phase 1 trials on patients with medullary thyroid cancer, XL184 demonstrated a 29% response rate, with a median duration of response that had not yet been reached, with a range of 4 to 35+ months. They also reported that in a phase 2 clinical trial on patients with the nasty glioblastoma, XL184 demonstrated a 30% response rate when dosed at 125 mg daily, with a median duration of response of 5.1 months. Data were presented showing that the drug demonstrated objective responses in patients with refractory melanoma, non-small cell lung cancer (NSCLC) (both as a single agent and in combination with erlotinib), hepatocellular carcinoma, prostate and ovarian cancers in an ongoing adaptive randomized discontinuation trial (RDT).
The above results were announced by the developing firms prior to their divorce. They are encouraging. Do we have any concrete reason to doubt them? We personally don’t. All what we see now is a unique drug that targets three very important pathways of a multitude of cancers. About future plans, the drug will be in the hands of the FDA for approval of its first indication, thyroid cancer, in 2011. Exelixis expects to initiate a phase 3 pivotal trial in recurrent glioblastoma in the year-end 2010 time frame and to prioritize tumor types from the RDT in 2011 to add more cancers to the drug indications.
This is how we see the story.
-----------------
Na ja, ich weiß nicht recht...
Hammer hat dazu auch ein kurzes Statement abgegeben:
Alex Says:
June 22nd, 2010 at 1:54 am
HI , why EXEL is so down recently,is it only the XL184 ?
Ohad Hammer Says:
June 22nd, 2010 at 2:25 am
Hi Alex,
I think it’s mostly that. This is not a show stopper for XL184 but it’s certainly bad news for EXEL, as the drug has been returned twice first by GSK and now by BMS).
In general, Exelixis’ compounds did not generate “earth shattering” data at ASCO although there were some nice signals. The randomized discontinuation study looks interesting but we’ll have to wait several months to get a clear picture.
Ohad
Wahrscheinlich hat er da mit 2 Sätzen mehr gesagt, als Prohost mit einem ganzen Artikel...
Alles sehr interessant! Scheint mir aber durchaus so zu sein, dass die letzte Verantwortung für ein vernünftiges Design auch bei einem SPA beim Unternehmen bleibt.
"The bearish counter argument is that while satraplatin demonstrated statistical significance on their designated primary endpoint, the benefit is not clinically significant at less than two weeks median benefit."
Tja, SPA scheint mir sehr wichtig und sehr positiv zu sein - vorausgesetzt, man kann dem Entwickler vertrauen, dass er wirklich die Zulassung im Blick hat und das Design dementsprechend abstimmt. Insofern war der Endpunkt im Fall Satraplatin wahrscheinlich nicht hinreichend sicher ausgewählt.
Ist aber für mich leider trotzdem etwas schwer zu verstehen...
hier noch ein Artikel zu EXEL (von Prohost):
Following Exelixis (EXEL) press release, which announced that it has regained full rights from Bristol-Myers Squibb (BMS) to its cancer drug XL184, the firm’s stock plummeted. In the press release, Exelixis stated that BMS’ priorities made it difficult for BMS to align on the scope, breadth and pace of the ongoing clinical development of XL184 as agreed upon in the original agreement. Those who instigated the sell-off did not buy Exelixis’ explanation and were more comfortable with the notion that BMS decided to walk away from XL184, probably because of bad news about the drug. Their reasoning was that a giant pharmaceutical company like BMS, which focuses on building an impressive advanced oncology pipeline would not let a promising multi-targeted cancer drug in late phase trials slip from its hands without a significant reason. BMS did not allude to any bad reason or any reason at all for its decision. It did not comment on Exelixis’ explanation and did not deny it.
Although investors’ speculation and their spontaneous response, instigating the selling of the stock, are not irrational, they were not supported by evidence-based knowledge, or facts. Their best speculation was that if BMS doubts the drug’s promises the least of what they can do is the same as BMS did, i.e., abandoning investing in Exelixis. The sell-off was triggered by a pyramid of suppositions that have no concrete proof.
Now that the acute fear has subsided and Exelixis stock price bottomed, other investors and, probably, some of those who sold the stock, might like to take a deep breath and reexamine the whole situation in a more professional and rational way. It is not unreasonable for investors to try to find out good investment opportunities in bottomed stocks.
To find out whether the stock selling based on the sellers’ reading of BMS’ mind is correct, or a mistake, we resorted to history, which tells that many large pharmaceutical firms have abandoned the development of drugs that have become best sellers when other firms continued developing them. Looking at the drug itself and the history of its performance in clinical trials, we see XL184 as not only the most advanced product in Exelixis pipeline, but is also the most advanced MET inhibitor. MET pathway has long been recognized as a master switch and drug target in cancer progression. MET is mutationally activated in hereditary and sporadic papillary renal cell carcinoma and some head and neck cancers. It is either over-expressed or activated in the absence of mutation in glioblastomas, breast carcinomas, gastric cancers and other solid tumors. MET amplification has been demonstrated in some NSCLCs.
XL184 also targets and inhibits VEGFR2, which leads to starving the tumors of oxygen through the antiangiogenic effect. Expression of VEGF occurs in various cancers and has been associated with prognostic significance. Targeting the VEGF receptor has already demonstrated efficacy as anti-cancer strategy in multiple tumors. Dual targeting of MET and VEGFR2 blocks two of the major mechanisms tumors use to overcome hypoxia.
In addition, XL184 inhibits RET, which is identified in multiple endocrine tumors and familial medullary thyroid carcinoma. Activated RET is involved in regulating cell proliferation, migration, differentiation, and survival. It is activated in papillary thyroid cancer (PTC) and in both familial and sporadic forms of medullary thyroid cancer (MTC).
These unique combination of actions makes XL184 capable of causing antiangiogenic, antiproliferative, and antiinvasive effects in tumors – all have been confirmed in preclinical cancer models and in some clinical trial results.
At ASCO 2010 Annual Meeting, the developers reported that in phase 1 trials on patients with medullary thyroid cancer, XL184 demonstrated a 29% response rate, with a median duration of response that had not yet been reached, with a range of 4 to 35+ months. They also reported that in a phase 2 clinical trial on patients with the nasty glioblastoma, XL184 demonstrated a 30% response rate when dosed at 125 mg daily, with a median duration of response of 5.1 months. Data were presented showing that the drug demonstrated objective responses in patients with refractory melanoma, non-small cell lung cancer (NSCLC) (both as a single agent and in combination with erlotinib), hepatocellular carcinoma, prostate and ovarian cancers in an ongoing adaptive randomized discontinuation trial (RDT).
The above results were announced by the developing firms prior to their divorce. They are encouraging. Do we have any concrete reason to doubt them? We personally don’t. All what we see now is a unique drug that targets three very important pathways of a multitude of cancers. About future plans, the drug will be in the hands of the FDA for approval of its first indication, thyroid cancer, in 2011. Exelixis expects to initiate a phase 3 pivotal trial in recurrent glioblastoma in the year-end 2010 time frame and to prioritize tumor types from the RDT in 2011 to add more cancers to the drug indications.
This is how we see the story.
-----------------
Na ja, ich weiß nicht recht...
Hammer hat dazu auch ein kurzes Statement abgegeben:
Alex Says:
June 22nd, 2010 at 1:54 am
HI , why EXEL is so down recently,is it only the XL184 ?
Ohad Hammer Says:
June 22nd, 2010 at 2:25 am
Hi Alex,
I think it’s mostly that. This is not a show stopper for XL184 but it’s certainly bad news for EXEL, as the drug has been returned twice first by GSK and now by BMS).
In general, Exelixis’ compounds did not generate “earth shattering” data at ASCO although there were some nice signals. The randomized discontinuation study looks interesting but we’ll have to wait several months to get a clear picture.
Ohad
Wahrscheinlich hat er da mit 2 Sätzen mehr gesagt, als Prohost mit einem ganzen Artikel...
ONXX... auch noch von prohost biotech: ein Artikel über ONXX's Carfilzomib
https://www.prohostbiotech.com/item/432-onyx-onxx-acquiring-…
ONYX (ONXX): ACQUIRING PROTEOLIX COULD BE THE DEAL OF THE YEAR?
Tuesday, 20 October 2009 00:00
Onyx’ (ONXX) stock decline in the past few months puzzled its shareholders. Nothing that has come in the news in the past year has been bad. On the contrary, a string of promising results was incessantly coming from the clinical trials of the firm’s cancer drug Nexavar on a variety of cancers. Now there are sufficient data to get an approval for other malignancies, including breast cancers. The drug was first approved for kidney cancer and then for liver cancer and, for a long while, there was no evidence that it would be approved for other cancers. Shareholders, though, did not lose hope. They were satisfied with the drug sales’ growth with sales that were closing on the $1 billion revenues. How come then that at the moment the drug proved to be useful for breast cancer the stock faltered.
Investors first believed that the ONXX decline was a typical phenomenon that briefly occurs when publicly traded firms raise money. Onyx had indeed raised $300 million, but the stock decline was not brief and has not ceased. Taking a deeper look at what the critics were propagating, informed investors found out that, yes, the financing was, indeed, a trigger for the stock decline but not for the sell-off, which was caused by efforts spent towards not letting the stock recover. Negative analysts kept downgrading the firm for various reasons, mostly irrelevant and included hypothesizing that Onyx might spend the vast amount of money it raised uselessly, acquiring a worthless firm!
It was strange that the critics projected pessimism against Onyx’ possible acquisition of a biotech firm that they, themselves, kept advocating. Stranger is the fact that, at the time, nobody had yet learned about any acquisition, as it was not announced. The analysts’ previous take on Onyx was that, besides the cancer drug Nexavar, the firm’s pipeline lacks mid- and late-phase trial. Their best suggestion was that the firm has to increase its value by adding more drugs to its pipeline through acquisition, which is exactly what the company did.
Onyx decided to acquire the private firm Proteolix, which has a breakthrough cancer drug carfilzomib in advanced-phase trials for multiple myeloma and in various phases of trials for other malignancies, in addition to having other very promising molecules in its pipeline. When the news reached the critics, their opinion changed; they began to see the acquisition only as money to be spent, which deserves a sell-off. The sell-off occurred, bringing wealth to short-sellers and more confusion to long-term positive investors. Following the official announcement of the acquisition, most comments revolved around the $575 million price tag for Proteolix, despite knowing in fact that Onyx will only pay if carfilzomib successfully reaches the stage of guaranteed approval. They cried about the money to spent, but never mentionioned the huge revenues the drug would generate when approved.
The Proteasome
Carfilzomib is a second-generation proteasome inhibitor. The proteasome controls the turnover of proteins in all cells, but cancer cells are more susceptible die when the proteasome is inhibited. The proteasome became one of the most exciting targets for cancer drugs a few years ago when Velcade® (bortezomib), the first proteasome inhibitor, was approved for multiple myeloma, and subsequently, for mantle cell lymphoma. While Velcade® has revolutionized the management of multiple myeloma and mantle cell lymphoma, the two cancers developed resistance to this drug, as they had to conventional drugs, leaving no choice except discovery of other potent therapeutics with improved efficacy, reduced toxicity, and capability to maintain their efficacy in the face of cancer resistance. Moreover, s cientists were confident that blocking proteasome activity would induce cell death in a wide variety of cancer cells. This expectation, though, did not materialize at the hands of Velcade. The drug is still approved for two cancers cited above only.
Proteolix’ drug Carfilzomib seems to meet the criteria required for overcoming cancer resistance and for treating of a variety of cancers. Proteolix has also developed a selective modulator that inhibits the immunoproteasome, which is the predominant form found in immune cells. This drug is expected to be useful in reversing immune disorders, including inflammatory diseases, with fewer side effects than conventional treatments.
Carfilzomib, which will occupy an empty space in Onyx’ pipeline, is a novel highly selective proteasome inhibitor. In scientific terms, the drug targets the unique N-terminal threonine active sites within the proteasome. Like Velcade, carfilzomib is a potent inhibitor of the proteasome chymotrypsin-like activity. Unlike Velcade®, the drug has minimal cross-reactivity with the other catalytic sites within the proteasome, or across other protease classes.
In preclinical, early- and mid-clinical studies, carfilzomib has demonstrated efficacy on myeloma cells resistant to Velcade®. It also demonstrated activity against solid tumors and lymphomas. Phase 1 clinical trial results demonstrate that Carfilzomib is effective in multiple myeloma patients who have relapsed or progressed following treatment, including Velcade®, immunomodulatory agents, and stem cell transplant. Important is that the drug has enabled late-stage cancer patients to continue to achieve durable anti-tumor responses.
Now, carfilzomib is undergoing evaluation as a single agent in two phase 2b clinical trials in patients with relapsed or refractory multiple myeloma and in a phase 2 clinical trial in patients with advanced solid tumors. The drug is also in phase 1 study for lymphoma and a phase 1b trial in combination with Revlimid® and dexamethasone in patients with relapsed multiple myeloma. Following the acquisition, Onyx will conduct a phase 3 study with carfilzomib + dexamethasone + Revlimid in patients with relapsed and refractory multiple myeloma. Onyx might seek regulatory approval by the end of 2010 as a third-line multiple myeloma treatment.
Having said all this, do we believe Onyx has made the right choice?
Our answer is, yes. Data from phase 2 studies presented twice at medical meetings demonstrated many advantages of carfilzomib over Velcade, Revlimid and other drugs used in treating multiple myeloma. The deal, which is expected to close in the fourth quarter of 2009, is now seen to hand Onyx a breakthrough drug in late-phase trials for multiple myeloma on patients who do not respond to other multiple myeloma drugs and patients with recurrences that resist all existing treatment regimens. The drug is also in various phase trials for several other cancers.
In addition, the acquisition will hand Onyx an oral proteasome inhibitor in research, which will be easier for patients to take, especially in long-term maintenance therapy, if requiredand and an immunoproteasome inhibitor for inflammatory and other immune diseases.
The market is large with a very high upside potential space for further growth. Multiple myeloma is the second most common hematologic cancer with 50,000 patients in the U.S. alone, and 20,000 new cases diagnosed annually. Globally 180,000 people suffer from multiple myeloma with over 80,000 new cases being discovered annually. Add these numbers to that of patients suffering from other cancers and inflammatory diseases, the market becomes huge.
mfg ipollit
https://www.prohostbiotech.com/item/432-onyx-onxx-acquiring-…
ONYX (ONXX): ACQUIRING PROTEOLIX COULD BE THE DEAL OF THE YEAR?
Tuesday, 20 October 2009 00:00
Onyx’ (ONXX) stock decline in the past few months puzzled its shareholders. Nothing that has come in the news in the past year has been bad. On the contrary, a string of promising results was incessantly coming from the clinical trials of the firm’s cancer drug Nexavar on a variety of cancers. Now there are sufficient data to get an approval for other malignancies, including breast cancers. The drug was first approved for kidney cancer and then for liver cancer and, for a long while, there was no evidence that it would be approved for other cancers. Shareholders, though, did not lose hope. They were satisfied with the drug sales’ growth with sales that were closing on the $1 billion revenues. How come then that at the moment the drug proved to be useful for breast cancer the stock faltered.
Investors first believed that the ONXX decline was a typical phenomenon that briefly occurs when publicly traded firms raise money. Onyx had indeed raised $300 million, but the stock decline was not brief and has not ceased. Taking a deeper look at what the critics were propagating, informed investors found out that, yes, the financing was, indeed, a trigger for the stock decline but not for the sell-off, which was caused by efforts spent towards not letting the stock recover. Negative analysts kept downgrading the firm for various reasons, mostly irrelevant and included hypothesizing that Onyx might spend the vast amount of money it raised uselessly, acquiring a worthless firm!
It was strange that the critics projected pessimism against Onyx’ possible acquisition of a biotech firm that they, themselves, kept advocating. Stranger is the fact that, at the time, nobody had yet learned about any acquisition, as it was not announced. The analysts’ previous take on Onyx was that, besides the cancer drug Nexavar, the firm’s pipeline lacks mid- and late-phase trial. Their best suggestion was that the firm has to increase its value by adding more drugs to its pipeline through acquisition, which is exactly what the company did.
Onyx decided to acquire the private firm Proteolix, which has a breakthrough cancer drug carfilzomib in advanced-phase trials for multiple myeloma and in various phases of trials for other malignancies, in addition to having other very promising molecules in its pipeline. When the news reached the critics, their opinion changed; they began to see the acquisition only as money to be spent, which deserves a sell-off. The sell-off occurred, bringing wealth to short-sellers and more confusion to long-term positive investors. Following the official announcement of the acquisition, most comments revolved around the $575 million price tag for Proteolix, despite knowing in fact that Onyx will only pay if carfilzomib successfully reaches the stage of guaranteed approval. They cried about the money to spent, but never mentionioned the huge revenues the drug would generate when approved.
The Proteasome
Carfilzomib is a second-generation proteasome inhibitor. The proteasome controls the turnover of proteins in all cells, but cancer cells are more susceptible die when the proteasome is inhibited. The proteasome became one of the most exciting targets for cancer drugs a few years ago when Velcade® (bortezomib), the first proteasome inhibitor, was approved for multiple myeloma, and subsequently, for mantle cell lymphoma. While Velcade® has revolutionized the management of multiple myeloma and mantle cell lymphoma, the two cancers developed resistance to this drug, as they had to conventional drugs, leaving no choice except discovery of other potent therapeutics with improved efficacy, reduced toxicity, and capability to maintain their efficacy in the face of cancer resistance. Moreover, s cientists were confident that blocking proteasome activity would induce cell death in a wide variety of cancer cells. This expectation, though, did not materialize at the hands of Velcade. The drug is still approved for two cancers cited above only.
Proteolix’ drug Carfilzomib seems to meet the criteria required for overcoming cancer resistance and for treating of a variety of cancers. Proteolix has also developed a selective modulator that inhibits the immunoproteasome, which is the predominant form found in immune cells. This drug is expected to be useful in reversing immune disorders, including inflammatory diseases, with fewer side effects than conventional treatments.
Carfilzomib, which will occupy an empty space in Onyx’ pipeline, is a novel highly selective proteasome inhibitor. In scientific terms, the drug targets the unique N-terminal threonine active sites within the proteasome. Like Velcade, carfilzomib is a potent inhibitor of the proteasome chymotrypsin-like activity. Unlike Velcade®, the drug has minimal cross-reactivity with the other catalytic sites within the proteasome, or across other protease classes.
In preclinical, early- and mid-clinical studies, carfilzomib has demonstrated efficacy on myeloma cells resistant to Velcade®. It also demonstrated activity against solid tumors and lymphomas. Phase 1 clinical trial results demonstrate that Carfilzomib is effective in multiple myeloma patients who have relapsed or progressed following treatment, including Velcade®, immunomodulatory agents, and stem cell transplant. Important is that the drug has enabled late-stage cancer patients to continue to achieve durable anti-tumor responses.
Now, carfilzomib is undergoing evaluation as a single agent in two phase 2b clinical trials in patients with relapsed or refractory multiple myeloma and in a phase 2 clinical trial in patients with advanced solid tumors. The drug is also in phase 1 study for lymphoma and a phase 1b trial in combination with Revlimid® and dexamethasone in patients with relapsed multiple myeloma. Following the acquisition, Onyx will conduct a phase 3 study with carfilzomib + dexamethasone + Revlimid in patients with relapsed and refractory multiple myeloma. Onyx might seek regulatory approval by the end of 2010 as a third-line multiple myeloma treatment.
Having said all this, do we believe Onyx has made the right choice?
Our answer is, yes. Data from phase 2 studies presented twice at medical meetings demonstrated many advantages of carfilzomib over Velcade, Revlimid and other drugs used in treating multiple myeloma. The deal, which is expected to close in the fourth quarter of 2009, is now seen to hand Onyx a breakthrough drug in late-phase trials for multiple myeloma on patients who do not respond to other multiple myeloma drugs and patients with recurrences that resist all existing treatment regimens. The drug is also in various phase trials for several other cancers.
In addition, the acquisition will hand Onyx an oral proteasome inhibitor in research, which will be easier for patients to take, especially in long-term maintenance therapy, if requiredand and an immunoproteasome inhibitor for inflammatory and other immune diseases.
The market is large with a very high upside potential space for further growth. Multiple myeloma is the second most common hematologic cancer with 50,000 patients in the U.S. alone, and 20,000 new cases diagnosed annually. Globally 180,000 people suffer from multiple myeloma with over 80,000 new cases being discovered annually. Add these numbers to that of patients suffering from other cancers and inflammatory diseases, the market becomes huge.
mfg ipollit
CBST...
etwas aus einem Biotech-Blog von Jason Chew zum Patentstreit über Cubicin zwischen CBST und Teva
http://biopharmareport.blogspot.com/2010/06/cubist-vs-hatch-…
Tuesday, June 22, 2010
Cubist vs. Hatch-Waxman
In 2003, Cubist was granted approval for its novel antibiotic, Cubicin, for complicated skin and skin structure infections by Gram-positive bacteria, including drug-resistant strains. Over the next couple years, the stock doubled from about $10 to $20. The stock has languished in the $20 trading range for nearly five years now. There have been some pops and drops to be sure, but in general, the stock has barely budged.
At the same time, revenues have grown quite nicely, exceeding even management’s expectations. In 2009, sales reached $562 million; it is expected to reach up to $650 million in 2010 and peak at $1 billion.
Net income for 2009 was about $80 million and in fact, the company has been profitable since the third quarter of 2006. Cash flow is strong; the company currently has nearly $500 million in cash, with $245 million in debt.
Why then is the company valued at a mere $1.2 billion? Cubist has the same problem that faces many small biotechs- almost all its revenue is derived from one source, in this case, Cubicin. Investors initially fretted about the onset of competition from Pfizer’s Dalbavancin, J&J’s Ceftobiprole, and Theravance’s Telavancin. Only Telavancin received approval but is no longer considered competitive.
The larger problem is Cubicin’s IP position. Though Cubicin has three patents listed in the FDA’s Orange Book, (US Patent Nos. 6468967, 6852689) contain only method claims while the remaining patent (US Reissue Pat. No. 39071) contains compound, composition, formulation, and method claims. The first two expire in 2019 while the third expires in 2016. The gold-standard composition of matter patent has already expired. Also, Patent No. 39071 was reissued in 2006 because the original patent contained the incorrect structure. This left Cubist more vulnerable to challenges from generic drug companies. Sure enough, on February 9, 2009, Teva submitted an ANDA with the FDA for marketing approval of generic Cubicin.
This was made possible by the Hatch-Waxman Act, passed in 1984 as a way to promote generics while attempting to retain enough financial incentives for pharmaceutical R&D. Prior to this act, generic drug companies needed to perform clinical trials to prove their drugs’ equivalence to brand name drugs before they could be marketed. The Act allowed for generic companies to file an ANDA whereby the FDA could grant marketing approval based solely on bioequivelence studies.
Because of the long time lag between the issuance of a compound’s patent and its marketing approval, estimated to be 16 years by PhRMA, and a patent life of only 20 years, the Act granted a five extension of exclusivity to the patent holder to compensate for loss of revenue due to generic competition. After this period, the patent holder is open to challenges from generics. If the drug's patent has expired before it comes to market, all that is left is the five-year extension.
To any reasoned mind, this should sound like an incredibly short time to sell a drug that took 16 years and over $1 billion dollars to bring to market. According to a study by the Boston Consulting Group in 1996, “effective intellectual property protection for U.S. pharmaceuticals has deteriorated significantly since passage of the Hatch-Waxman Act” and have “eroded the incentives for pharmaceutical innovation.” The study also noted that prior to the passage of the Act innovator companies held market exclusivity of between 14 to 17 years, decreasing to an average of 11.7 years after its passage. Europe, Canada, and Japan have patent extensions of ten years. Making this change to the Act would significantly enhance the incentives for innovative drug discovery.
It is estimated that the cost of filing an ANDA is $5 million- a far cry from developing a drug. One of four certifications must be made when someone files an ANDA:
1. that the drug has not been patented
2. that the patent has already expired
3. the date on which the patent will expire, and that the generic drug will not go on the market until that date passes
4. that the patent is not infringed or is invalid.
Those certifications are now referred to as the paragraphs I, II, III, and IV certifications.
The ANDA filed against Cubist by Teva is a paragraph IV filing. According to an article by Matthew J. Higgins of the Georgia Institute of Technology and Stuart J.H. Graham of the University of California, Berkeley Law School, between 1992 and 2000, 72% of paragraph IV filings resulted in litigation, with generic companies winning 42% of the cases. Those are pretty good odds for relatively little money spend. After all, the first generic company to receive approval for an ANDA gets 180 days of sales exclusivity on the drug. Not surprisingly, Teva had 92 pending paragraph IV filings in 2007 alone.
Under the Hatch-Waxman Act, Cubist filed suit against Teva for patent infringement, resulting in a 30-month stay, preventing Teva from selling generic Cubicin. It is now awaiting a Markman hearing before a US District Court, a pre-trial hearing where a judge "constructs the claim", examining the evidence from the parties and deciding on the appropriate meaning of the claim's language. This hearing may be more important than the trial itself, as its results may determine the strength of each side’s arguments at trial. These hearings often determine whether the lawsuits get settled or go into litigation. Originally scheduled for June 9, the hearing was canceled and has yet to be rescheduled.
Most analysts believe Cubist has a solid case and will settle with Teva in order to delay entry of generic Cubicin, perhaps until 2018. Teva is expected to attack the reissued Patent No. 39071. Experts are divided on the effectiveness of this stragedy, though it has received considerable attention.
The two remaining patents cover methods of use for Cubicin. These types of patents are often considered relatively weak. In this case, however, I believe they may be the strongest patents and most likely to be upheld.
According to the USPTO’s evidence of nonobviousness is a major factor in determining whether a patent is granted. Several factors that can be used to demonstrate this are:
1. Commercial Success- Success may demonstrate the invention is an important advance in the field that was not obvious to others
2. Long-felt need - If the need had been there for a long time without this invention, it must have been nonobvious
3. The necessity for a Nexus- Demonstrate that commercial success is directly related to the innovative aspect of the patent and not merely marketing
This plays into Cubist’s hand if one follows the history of Cubicin. The drug was originally developed by Eli Lilly. It had good antibacterial activity, but also skeletal muscle toxicity- leading Lilly to give up on the project. Cubist decided to license to compound in 1996 and worked to reduce its toxicity levels. This is where scientists at Cubist succeeded where giant Lilly had failed. They discovered a dosing regimen at which Cubicin was safe and efficacious, getting the drug approved as the first antibiotic in a class called lipopeptides. The drug was added to pharmaceutical formularies in 2004 and studies found that its usage helped limit the occurrence of certain drug resistant bacteria. In 2006, additional indications were approved for Cubicin, further increasing its sales potential. To date 500,000 patients have been treated with Cubicin, with less than 4000 patients reporting side effects. Because of the discoveries by Cubist scientists, half a million patients have benefited, directly resulting in the Cubicin’s commercial success.
With the success of Cubicin, and Cubist’s lack of a pipeline, speculation of acquisitions swirled. It has been suggested Cubist even shopped itself around. Pharma was interested, but there were no takers. They were too risk-adverse to pick up a company with patent issues. They had enough of their own patent problems to deal with.
Cubist did not sit around waiting for an acquirer. Using the strong cash flow from Cubicin, it has slowly built up a pipeline through internal research and strategic deals. From its own labs it now has a Phase I drug for Gram-negative bacteria and a Phase II drug for CDAD infections. Externally, it had licensed a drug from Dyax, but the clinical trial failed and the program was quickly dropped. An RNAi program started with Alnylan that went into the clinic is now back in preclinical studies.
In December 2009, Cubist acquired privately held Calixa for $92.5 million up front and $310 million in milestones. This could be a potential blockbuster. Calixa’s compound, CXA-201, is designed to treat a broad range of Gram-negative infections, including urinary tract infections and nosocomial pneumonia. It is now in Phase II trials; Cubists has very high hopes for it and expects to file an NDA for the drug in 2013.
Cubist’s patent problems have held down its stock for a very long time. Much of the uncertainty should be lifted after the rescheduled Markman hearing. If it goes into litigation, the trial date will be April 25, 2011. Except in the very unlikely event Cubist loses at trial, I expect that within one year’s time, it will be far from a $20 stock, and certainly will not need to shop itself around.
In summary, Cubist’s story makes the perfect case for reform of the Hatch-Waxman Act. Drug development is expensive, risky, and time-consuming. As noted earlier, Cubist was the only company in recent years to develop a financially successful antibiotic. And even in its own studies, it has had multiple failures. The Act hands too much power to generic companies without thoughtfully and adequately compensating the original inventors. It has accomplished half its goal in developing a flourishing generics industry- now it needs to fulfill its other half with the promise to maintain the financial incentives for original research and development. It needs to do this by adding greater protection to pharmaceutical patents, the lifeblood of the industry.
mfg ipollit
etwas aus einem Biotech-Blog von Jason Chew zum Patentstreit über Cubicin zwischen CBST und Teva
http://biopharmareport.blogspot.com/2010/06/cubist-vs-hatch-…
Tuesday, June 22, 2010
Cubist vs. Hatch-Waxman
In 2003, Cubist was granted approval for its novel antibiotic, Cubicin, for complicated skin and skin structure infections by Gram-positive bacteria, including drug-resistant strains. Over the next couple years, the stock doubled from about $10 to $20. The stock has languished in the $20 trading range for nearly five years now. There have been some pops and drops to be sure, but in general, the stock has barely budged.
At the same time, revenues have grown quite nicely, exceeding even management’s expectations. In 2009, sales reached $562 million; it is expected to reach up to $650 million in 2010 and peak at $1 billion.
Net income for 2009 was about $80 million and in fact, the company has been profitable since the third quarter of 2006. Cash flow is strong; the company currently has nearly $500 million in cash, with $245 million in debt.
Why then is the company valued at a mere $1.2 billion? Cubist has the same problem that faces many small biotechs- almost all its revenue is derived from one source, in this case, Cubicin. Investors initially fretted about the onset of competition from Pfizer’s Dalbavancin, J&J’s Ceftobiprole, and Theravance’s Telavancin. Only Telavancin received approval but is no longer considered competitive.
The larger problem is Cubicin’s IP position. Though Cubicin has three patents listed in the FDA’s Orange Book, (US Patent Nos. 6468967, 6852689) contain only method claims while the remaining patent (US Reissue Pat. No. 39071) contains compound, composition, formulation, and method claims. The first two expire in 2019 while the third expires in 2016. The gold-standard composition of matter patent has already expired. Also, Patent No. 39071 was reissued in 2006 because the original patent contained the incorrect structure. This left Cubist more vulnerable to challenges from generic drug companies. Sure enough, on February 9, 2009, Teva submitted an ANDA with the FDA for marketing approval of generic Cubicin.
This was made possible by the Hatch-Waxman Act, passed in 1984 as a way to promote generics while attempting to retain enough financial incentives for pharmaceutical R&D. Prior to this act, generic drug companies needed to perform clinical trials to prove their drugs’ equivalence to brand name drugs before they could be marketed. The Act allowed for generic companies to file an ANDA whereby the FDA could grant marketing approval based solely on bioequivelence studies.
Because of the long time lag between the issuance of a compound’s patent and its marketing approval, estimated to be 16 years by PhRMA, and a patent life of only 20 years, the Act granted a five extension of exclusivity to the patent holder to compensate for loss of revenue due to generic competition. After this period, the patent holder is open to challenges from generics. If the drug's patent has expired before it comes to market, all that is left is the five-year extension.
To any reasoned mind, this should sound like an incredibly short time to sell a drug that took 16 years and over $1 billion dollars to bring to market. According to a study by the Boston Consulting Group in 1996, “effective intellectual property protection for U.S. pharmaceuticals has deteriorated significantly since passage of the Hatch-Waxman Act” and have “eroded the incentives for pharmaceutical innovation.” The study also noted that prior to the passage of the Act innovator companies held market exclusivity of between 14 to 17 years, decreasing to an average of 11.7 years after its passage. Europe, Canada, and Japan have patent extensions of ten years. Making this change to the Act would significantly enhance the incentives for innovative drug discovery.
It is estimated that the cost of filing an ANDA is $5 million- a far cry from developing a drug. One of four certifications must be made when someone files an ANDA:
1. that the drug has not been patented
2. that the patent has already expired
3. the date on which the patent will expire, and that the generic drug will not go on the market until that date passes
4. that the patent is not infringed or is invalid.
Those certifications are now referred to as the paragraphs I, II, III, and IV certifications.
The ANDA filed against Cubist by Teva is a paragraph IV filing. According to an article by Matthew J. Higgins of the Georgia Institute of Technology and Stuart J.H. Graham of the University of California, Berkeley Law School, between 1992 and 2000, 72% of paragraph IV filings resulted in litigation, with generic companies winning 42% of the cases. Those are pretty good odds for relatively little money spend. After all, the first generic company to receive approval for an ANDA gets 180 days of sales exclusivity on the drug. Not surprisingly, Teva had 92 pending paragraph IV filings in 2007 alone.
Under the Hatch-Waxman Act, Cubist filed suit against Teva for patent infringement, resulting in a 30-month stay, preventing Teva from selling generic Cubicin. It is now awaiting a Markman hearing before a US District Court, a pre-trial hearing where a judge "constructs the claim", examining the evidence from the parties and deciding on the appropriate meaning of the claim's language. This hearing may be more important than the trial itself, as its results may determine the strength of each side’s arguments at trial. These hearings often determine whether the lawsuits get settled or go into litigation. Originally scheduled for June 9, the hearing was canceled and has yet to be rescheduled.
Most analysts believe Cubist has a solid case and will settle with Teva in order to delay entry of generic Cubicin, perhaps until 2018. Teva is expected to attack the reissued Patent No. 39071. Experts are divided on the effectiveness of this stragedy, though it has received considerable attention.
The two remaining patents cover methods of use for Cubicin. These types of patents are often considered relatively weak. In this case, however, I believe they may be the strongest patents and most likely to be upheld.
According to the USPTO’s evidence of nonobviousness is a major factor in determining whether a patent is granted. Several factors that can be used to demonstrate this are:
1. Commercial Success- Success may demonstrate the invention is an important advance in the field that was not obvious to others
2. Long-felt need - If the need had been there for a long time without this invention, it must have been nonobvious
3. The necessity for a Nexus- Demonstrate that commercial success is directly related to the innovative aspect of the patent and not merely marketing
This plays into Cubist’s hand if one follows the history of Cubicin. The drug was originally developed by Eli Lilly. It had good antibacterial activity, but also skeletal muscle toxicity- leading Lilly to give up on the project. Cubist decided to license to compound in 1996 and worked to reduce its toxicity levels. This is where scientists at Cubist succeeded where giant Lilly had failed. They discovered a dosing regimen at which Cubicin was safe and efficacious, getting the drug approved as the first antibiotic in a class called lipopeptides. The drug was added to pharmaceutical formularies in 2004 and studies found that its usage helped limit the occurrence of certain drug resistant bacteria. In 2006, additional indications were approved for Cubicin, further increasing its sales potential. To date 500,000 patients have been treated with Cubicin, with less than 4000 patients reporting side effects. Because of the discoveries by Cubist scientists, half a million patients have benefited, directly resulting in the Cubicin’s commercial success.
With the success of Cubicin, and Cubist’s lack of a pipeline, speculation of acquisitions swirled. It has been suggested Cubist even shopped itself around. Pharma was interested, but there were no takers. They were too risk-adverse to pick up a company with patent issues. They had enough of their own patent problems to deal with.
Cubist did not sit around waiting for an acquirer. Using the strong cash flow from Cubicin, it has slowly built up a pipeline through internal research and strategic deals. From its own labs it now has a Phase I drug for Gram-negative bacteria and a Phase II drug for CDAD infections. Externally, it had licensed a drug from Dyax, but the clinical trial failed and the program was quickly dropped. An RNAi program started with Alnylan that went into the clinic is now back in preclinical studies.
In December 2009, Cubist acquired privately held Calixa for $92.5 million up front and $310 million in milestones. This could be a potential blockbuster. Calixa’s compound, CXA-201, is designed to treat a broad range of Gram-negative infections, including urinary tract infections and nosocomial pneumonia. It is now in Phase II trials; Cubists has very high hopes for it and expects to file an NDA for the drug in 2013.
Cubist’s patent problems have held down its stock for a very long time. Much of the uncertainty should be lifted after the rescheduled Markman hearing. If it goes into litigation, the trial date will be April 25, 2011. Except in the very unlikely event Cubist loses at trial, I expect that within one year’s time, it will be far from a $20 stock, and certainly will not need to shop itself around.
In summary, Cubist’s story makes the perfect case for reform of the Hatch-Waxman Act. Drug development is expensive, risky, and time-consuming. As noted earlier, Cubist was the only company in recent years to develop a financially successful antibiotic. And even in its own studies, it has had multiple failures. The Act hands too much power to generic companies without thoughtfully and adequately compensating the original inventors. It has accomplished half its goal in developing a flourishing generics industry- now it needs to fulfill its other half with the promise to maintain the financial incentives for original research and development. It needs to do this by adding greater protection to pharmaceutical patents, the lifeblood of the industry.
mfg ipollit
JAZZ... am 20.8. gibt es eine Empfehlung zu JZP-6 bezügl. einer möglichen FDA-Zulassung
PALO ALTO, Calif., June 24 /PRNewswire-FirstCall/ -- Jazz Pharmaceuticals, Inc. (Nasdaq:JAZZ - News) announced today that the U.S. Food and Drug Administration's (FDA) Arthritis Advisory Committee and Drug Safety and Risk Management Advisory Committee will review JZP-6 (sodium oxybate) for the treatment of fibromyalgia at a joint meeting on August 20, 2010.
In February 2010, the FDA accepted for filing and review the New Drug Application (NDA) of JZP-6 (sodium oxybate) for the treatment of fibromyalgia. The target date for the FDA to complete its review of the NDA under the Prescription Drug User Fee Act (PDUFA) is October 11, 2010.
mfg ipollit
PALO ALTO, Calif., June 24 /PRNewswire-FirstCall/ -- Jazz Pharmaceuticals, Inc. (Nasdaq:JAZZ - News) announced today that the U.S. Food and Drug Administration's (FDA) Arthritis Advisory Committee and Drug Safety and Risk Management Advisory Committee will review JZP-6 (sodium oxybate) for the treatment of fibromyalgia at a joint meeting on August 20, 2010.
In February 2010, the FDA accepted for filing and review the New Drug Application (NDA) of JZP-6 (sodium oxybate) for the treatment of fibromyalgia. The target date for the FDA to complete its review of the NDA under the Prescription Drug User Fee Act (PDUFA) is October 11, 2010.
mfg ipollit
ARNA... hat mit Eisai endlich einen Partner für die Vermarktung von Lorcaserin gefunden. Etwas ungewöhnlich ist, dass keine Royalties gezahlt werden, sondern ARNA Lorcaserin herstellt und Eisai es dann für ca. 32-37% des Umsatzes abkauft. Die Royalties liegen demnach bei ca 32% minus Herstellungskosten. Da die Herstellungskosten relativ gering sind, dürften die Royalties bei über 20% liegen... recht viel, wenn Lorcaserin zu einem Blockbuster wird. Allerdings sieht man an der dazu relativ geringen Vorrauszahlung, dass die Zulassung im Herbst alles andere als sicher ist.
http://finance.yahoo.com/news/Arena-Pharmaceuticals-in-deal-…
Arena Pharmaceuticals in deal for obesity drug
Linda A. Johnson, AP Business Writer, On Thursday July 1, 2010, 3:16 am EDT
Arena Pharmaceuticals Inc. said Thursday it has an agreement with Japan's Eisai Inc. in a deal worth potentially more than $1.3 billion to fund commercialization of an obesity drug that could become Arena's first product.
Arena's lorcaserin is part of a wave of potential new treatments for a tough-to-treat problem that's become an epidemic in developed countries.
San Diego-based Arena said Eisai will pay $50 million upfront for rights to sell lorcaserin in the U.S. In addition, Arena could get as much as $160 million in payments for reaching certain development targets and getting the drug approved by regulators, plus a $1.16 billion one-time payment in the future based on the level of annual sales.
Arena will manufacture the drug and Eisai will distribute it in the U.S.
Arena keeps the rights in other countries, Chief Executive Jack Lief told The Associated Press.
"That allows us to maximize our international opportunities," he said.
Arena, which has no products on sale yet, reported at a medical conference Saturday on a late-stage weight-loss study of lorcaserin that included more than 6,000 patients. The company said that 47 percent of patients in the group taking lorcaserin lost at least 5 percent of their body weight, compared with 23 percent who lost that much in a comparison group taking dummy pills.
Doctors generally say that weight loss of 5 percent or more brings significant health benefits.
"With Esai, we have the right company to market lorcaserin in the United States, the right type of agreement to optimize lorcaserin's medical and commercial potential" at the right time, Lief said.
The drug works by interacting with a brain receptor involved in appetite control and feelings of satiety, according to the company.
Lorcaserin, which has not yet been given a brand name, is part of a new wave of promising experimental obesity drugs. Three drugs are poised to reach what could be a very large and lucrative market around the same time -- late this year or early next year.
The other two medicines are Qnexa, developed by Vivus Inc. of Mountain View, and Contrave, developed by Orexigen Therapeutics, also of San Diego.
All three drug candidates are being developed by small companies and are under review by the U.S. Food and Drug Administration.
Qnexa will be the first to be reviewed by an FDA panel of experts, at a meeting scheduled for July 15.
Lorcaserin is set for review by an FDA panel on Sept. 16, with a tentative deadline of Oct. 22 for the FDA to rule on approval.
Contrave is scheduled for a panel review on Dec. 7, and the agency is set to make a ruling on that drug by Jan. 31, 2011.
Finding a successful obesity treatment has been difficult, with numerous past drugs failing because of limited effectiveness, safety problems or unpleasant side effects such as loose stool.
But with Americans and people in many other countries getting fatter and fatter, any treatment that works well would be a blockbuster.
The market for obesity drugs alone is likely to be worth billions a year, and the new drugs might also turn out to help in treating diseases related to obesity, such as Type 2 diabetes and sleep apnea.
Qnexa, the Vivus drug, has been tested in multiple studies of patients with either diabetes or sleep apnea.
Multiple studies on research by the companies, including the latest on lorcaserin, were presented during the American Diabetes Association medical conference in Orlando, Fla., that wrapped up at the beginning of this week.
*********
http://www.bioworld.com/servlet/com.accumedia.web.Dispatcher…
Arena's U.S. Lorcaserin Deal: Eisai Royalties 'Over the Top'
By Randy Osborne
Staff Writer
Eisai Inc.'s sales force for the acid reflux therapy Aciphex in the U.S. gives Arena Pharmaceuticals Inc. just the oomph needed to push the obesity drug lorcaserin, due for an FDA panel hearing in September. San Diego-based Arena's stock (NASDAQ:ARNA) closed at $3.56, up 49 cents, or 16 percent, on news of the marketing deal disclosed Thursday.
Under the terms of the milestone-loaded arrangement, Eisai, of Woodcliff Lake, N.J., is putting $50 million in Arena's coffers right away. Arena gets $90 million upon approval of lorcaserin and delivery of commercial supply, and will sell lorcaserin to Eisai for a price that starts at 31.5 percent of Eisai's annual net product sales. That price will rise on a tiered basis to as high as 36.5 percent on the portion of annual net sales that go beyond $750 million.
Phil Nadeau, analyst with Cowen & Co., wrote in a research report that "if one assumes a typical small-molecule low single-digit percentage cost of goods, Arena's transfer price equates to an about mid-20 percent to 30 percent royalty" – a rate Nadeau called "attractive."
The deal puts Arena in line for $1.16 billion in one-time purchase price adjustment payments based on annual sales, and as much as $70 million more in regulatory and development milestone payments, noted Jack Lief, Arena's president and CEO.
Some might have viewed the $90 million on approval as less than ideal, but balanced by the royalty structure. "I don't think it's low, first of all," Lief told BioWorld Today. "Secondly, the royalty-like payment is over the top. I'm not aware of another small molecule that has that [size] royalty-like component to it," he added.
Elemer Piros, analyst with Rodman & Renshaw, dubbed the deal "meek" and "fairly modest," noting that the arrangement is "highly back-end loaded, and leaves a lot of lorcaserin commercial risk with Arena shareholders."
Lief said the terms should be viewed holistically. "We look at it as a $140 million deal, with $50 million now and $90 million by the end of the year," he said.
"That's pretty good for us, and it covers our needs, certainly, more than enough. It sets us up really well, not just in the U.S., but it allows us maximum flexibility for the international markets," Lief said.
Lorcaserin, a 5-HT2C serotonin receptor stimulator, is one of three players in the obesity race, and would-be competitors have reported varying trial results. Mountain View, Calif.-based Vivus Inc.'s Qnexa (phentermine/topiramate CR, due for an FDA panel later this month) showed placebo-adjusted weight loss as high as 9.4 percent and excess body weight loss of 42 percent. Orexigen Therapeutics Inc., of San Diego, with Contrave (bupropion SR/naltrexone SR, due for an advisory shakeout in December), hit 5.2 percent and 31 .7 percent on the same outcomes. Lorcaserin tallied 3.6 percent. (See BioWorld Today, Oct. 27, 2009, and June 30, 2010.)
Safety has been the strong suit for lorcaserin, with what Leerink Swann analysts have called "modest, but approvable efficacy." The only ghost of risk is the possibility of valvulopathy – problems with heart valves – but Arena has dispatched most of those worries in trials. In the BLOSSOM pivotal trial, rates of valvulopathy hit only 2 percent in the twice-daily lorcaserin group and the placebo arm. Results from more than 7,000 patients in the BLOOM and BLOSSOM studies, using FDA criteria, effectively ruled out the valvulopathy risk. (See BioWorld Today, Sept. 21 , 2009.)
Lorcaserin is "exactly what physicians are looking for, in terms of this combination of safety, efficacy and tolerability," Lief said. Pooled lorcaserin results detailed at the American Diabetes Association meeting recently showed 47.1 percent lost at least 5 percent of their body weight on the drug vs. 22.6 percent on placebo, with efficacy across demographics including sex, age and ethnicity. Findings in total cholesterol, triglycerides, HDL cholesterol and insulin resistance proved encouraging, too.
Approval seems likely, though time lines are tough to call, with the drug's PDUFA date falling in the last half of October along with three other candidates to be considered by the FDA's endocrinologic and metabolic division. Lorcaserin comes up Oct. 22, with San Diego-based Amylin Pharmaceuticals Inc.'s diabetes therapy Bydureon (exenatide LAR) due for a decision on the same day. Qnexa's date falls one week later, and the PDUFA deadline falls the next day for VIAject, the new formulation of insulin, from Biodel Inc., of Danbury, Conn. The PDUFA date for Contrave is set for the end of January 2011 .
"If the FDA asks us to do any additional studies – and we're not saying they will, but we're going to be ready for it – Eisai will pay 90 percent of the cost," Lief said.
The lorcaserin setup has Vivus investors licking their lips in speculation about partnerships for Vivus around Qnexa, since the candidate garnered better efficacy than Arena's compound.
Jonathan Aschoff, analyst with Brean Murray Carret, estimated that Vivus spent $150 million developing Qnexa and predicted that negotiations on up-front payments will start at that amount, with Vivus securing a 40 percent profit share "at the very least."
Meanwhile, Eisai's sales force, numbering about 500 who could detail lorcaserin, gears up for operations. National Drug Code record keepers say about 34 percent of Eisai's total prescription volume has come through general practitioners and endocrinologists, the sweet spots for lorcaserin.
mfg ipollit
http://finance.yahoo.com/news/Arena-Pharmaceuticals-in-deal-…
Arena Pharmaceuticals in deal for obesity drug
Linda A. Johnson, AP Business Writer, On Thursday July 1, 2010, 3:16 am EDT
Arena Pharmaceuticals Inc. said Thursday it has an agreement with Japan's Eisai Inc. in a deal worth potentially more than $1.3 billion to fund commercialization of an obesity drug that could become Arena's first product.
Arena's lorcaserin is part of a wave of potential new treatments for a tough-to-treat problem that's become an epidemic in developed countries.
San Diego-based Arena said Eisai will pay $50 million upfront for rights to sell lorcaserin in the U.S. In addition, Arena could get as much as $160 million in payments for reaching certain development targets and getting the drug approved by regulators, plus a $1.16 billion one-time payment in the future based on the level of annual sales.
Arena will manufacture the drug and Eisai will distribute it in the U.S.
Arena keeps the rights in other countries, Chief Executive Jack Lief told The Associated Press.
"That allows us to maximize our international opportunities," he said.
Arena, which has no products on sale yet, reported at a medical conference Saturday on a late-stage weight-loss study of lorcaserin that included more than 6,000 patients. The company said that 47 percent of patients in the group taking lorcaserin lost at least 5 percent of their body weight, compared with 23 percent who lost that much in a comparison group taking dummy pills.
Doctors generally say that weight loss of 5 percent or more brings significant health benefits.
"With Esai, we have the right company to market lorcaserin in the United States, the right type of agreement to optimize lorcaserin's medical and commercial potential" at the right time, Lief said.
The drug works by interacting with a brain receptor involved in appetite control and feelings of satiety, according to the company.
Lorcaserin, which has not yet been given a brand name, is part of a new wave of promising experimental obesity drugs. Three drugs are poised to reach what could be a very large and lucrative market around the same time -- late this year or early next year.
The other two medicines are Qnexa, developed by Vivus Inc. of Mountain View, and Contrave, developed by Orexigen Therapeutics, also of San Diego.
All three drug candidates are being developed by small companies and are under review by the U.S. Food and Drug Administration.
Qnexa will be the first to be reviewed by an FDA panel of experts, at a meeting scheduled for July 15.
Lorcaserin is set for review by an FDA panel on Sept. 16, with a tentative deadline of Oct. 22 for the FDA to rule on approval.
Contrave is scheduled for a panel review on Dec. 7, and the agency is set to make a ruling on that drug by Jan. 31, 2011.
Finding a successful obesity treatment has been difficult, with numerous past drugs failing because of limited effectiveness, safety problems or unpleasant side effects such as loose stool.
But with Americans and people in many other countries getting fatter and fatter, any treatment that works well would be a blockbuster.
The market for obesity drugs alone is likely to be worth billions a year, and the new drugs might also turn out to help in treating diseases related to obesity, such as Type 2 diabetes and sleep apnea.
Qnexa, the Vivus drug, has been tested in multiple studies of patients with either diabetes or sleep apnea.
Multiple studies on research by the companies, including the latest on lorcaserin, were presented during the American Diabetes Association medical conference in Orlando, Fla., that wrapped up at the beginning of this week.
*********
http://www.bioworld.com/servlet/com.accumedia.web.Dispatcher…
Arena's U.S. Lorcaserin Deal: Eisai Royalties 'Over the Top'
By Randy Osborne
Staff Writer
Eisai Inc.'s sales force for the acid reflux therapy Aciphex in the U.S. gives Arena Pharmaceuticals Inc. just the oomph needed to push the obesity drug lorcaserin, due for an FDA panel hearing in September. San Diego-based Arena's stock (NASDAQ:ARNA) closed at $3.56, up 49 cents, or 16 percent, on news of the marketing deal disclosed Thursday.
Under the terms of the milestone-loaded arrangement, Eisai, of Woodcliff Lake, N.J., is putting $50 million in Arena's coffers right away. Arena gets $90 million upon approval of lorcaserin and delivery of commercial supply, and will sell lorcaserin to Eisai for a price that starts at 31.5 percent of Eisai's annual net product sales. That price will rise on a tiered basis to as high as 36.5 percent on the portion of annual net sales that go beyond $750 million.
Phil Nadeau, analyst with Cowen & Co., wrote in a research report that "if one assumes a typical small-molecule low single-digit percentage cost of goods, Arena's transfer price equates to an about mid-20 percent to 30 percent royalty" – a rate Nadeau called "attractive."
The deal puts Arena in line for $1.16 billion in one-time purchase price adjustment payments based on annual sales, and as much as $70 million more in regulatory and development milestone payments, noted Jack Lief, Arena's president and CEO.
Some might have viewed the $90 million on approval as less than ideal, but balanced by the royalty structure. "I don't think it's low, first of all," Lief told BioWorld Today. "Secondly, the royalty-like payment is over the top. I'm not aware of another small molecule that has that [size] royalty-like component to it," he added.
Elemer Piros, analyst with Rodman & Renshaw, dubbed the deal "meek" and "fairly modest," noting that the arrangement is "highly back-end loaded, and leaves a lot of lorcaserin commercial risk with Arena shareholders."
Lief said the terms should be viewed holistically. "We look at it as a $140 million deal, with $50 million now and $90 million by the end of the year," he said.
"That's pretty good for us, and it covers our needs, certainly, more than enough. It sets us up really well, not just in the U.S., but it allows us maximum flexibility for the international markets," Lief said.
Lorcaserin, a 5-HT2C serotonin receptor stimulator, is one of three players in the obesity race, and would-be competitors have reported varying trial results. Mountain View, Calif.-based Vivus Inc.'s Qnexa (phentermine/topiramate CR, due for an FDA panel later this month) showed placebo-adjusted weight loss as high as 9.4 percent and excess body weight loss of 42 percent. Orexigen Therapeutics Inc., of San Diego, with Contrave (bupropion SR/naltrexone SR, due for an advisory shakeout in December), hit 5.2 percent and 31 .7 percent on the same outcomes. Lorcaserin tallied 3.6 percent. (See BioWorld Today, Oct. 27, 2009, and June 30, 2010.)
Safety has been the strong suit for lorcaserin, with what Leerink Swann analysts have called "modest, but approvable efficacy." The only ghost of risk is the possibility of valvulopathy – problems with heart valves – but Arena has dispatched most of those worries in trials. In the BLOSSOM pivotal trial, rates of valvulopathy hit only 2 percent in the twice-daily lorcaserin group and the placebo arm. Results from more than 7,000 patients in the BLOOM and BLOSSOM studies, using FDA criteria, effectively ruled out the valvulopathy risk. (See BioWorld Today, Sept. 21 , 2009.)
Lorcaserin is "exactly what physicians are looking for, in terms of this combination of safety, efficacy and tolerability," Lief said. Pooled lorcaserin results detailed at the American Diabetes Association meeting recently showed 47.1 percent lost at least 5 percent of their body weight on the drug vs. 22.6 percent on placebo, with efficacy across demographics including sex, age and ethnicity. Findings in total cholesterol, triglycerides, HDL cholesterol and insulin resistance proved encouraging, too.
Approval seems likely, though time lines are tough to call, with the drug's PDUFA date falling in the last half of October along with three other candidates to be considered by the FDA's endocrinologic and metabolic division. Lorcaserin comes up Oct. 22, with San Diego-based Amylin Pharmaceuticals Inc.'s diabetes therapy Bydureon (exenatide LAR) due for a decision on the same day. Qnexa's date falls one week later, and the PDUFA deadline falls the next day for VIAject, the new formulation of insulin, from Biodel Inc., of Danbury, Conn. The PDUFA date for Contrave is set for the end of January 2011 .
"If the FDA asks us to do any additional studies – and we're not saying they will, but we're going to be ready for it – Eisai will pay 90 percent of the cost," Lief said.
The lorcaserin setup has Vivus investors licking their lips in speculation about partnerships for Vivus around Qnexa, since the candidate garnered better efficacy than Arena's compound.
Jonathan Aschoff, analyst with Brean Murray Carret, estimated that Vivus spent $150 million developing Qnexa and predicted that negotiations on up-front payments will start at that amount, with Vivus securing a 40 percent profit share "at the very least."
Meanwhile, Eisai's sales force, numbering about 500 who could detail lorcaserin, gears up for operations. National Drug Code record keepers say about 34 percent of Eisai's total prescription volume has come through general practitioners and endocrinologists, the sweet spots for lorcaserin.
mfg ipollit
Genmab... der Arzerra-Deal mit GSK wurde geändert: dafür erhält Genmab eine Vorrauszahlung von 134 Mio USD... allerdings werden u.a. die Meilensteinzahlungen um die Hälfte gekürzt. Genmab benötigte dringend Cash.
http://www.fiercebiotech.com/story/genmab-grabs-134m-it-rest…
Genmab grabs $134M as it restructures blockbuster GSK pact
July 1, 2010 — 2:00pm ET | By John Carroll
Just weeks after Lisa Drakeman departed the CEO's post at Genmab, GlaxoSmithKline has engineered a big change to the $2.1 billion deal it struck with the Danish developer more than three years ago on Arzerra (ofatumumab). The troubled Genmab, which has watched its share price slide 90 percent, is getting $134 million upfront on the revised pact, but sees its milestone package cut in half.
In the revised pact GSK takes responsibility for developing the therapy for autoimmune diseases while both companies collaborate on cancer. And GSK's funding commitment for the oncology work is capped at 145 million pounds overall with a 17 million pound annual limit.
"Their (GSK's) flexibility has allowed us to not only ensure that we extract the best possible value from ofatumumab, but also strengthen the financial security of Genmab," said Prof. Jan G.J. van de Winkel, Genmab's new chief. The changes will have a "material impact on Genmab's 2010 financial guidance," the company said in a statement.
Genmab had been one of the bright stars in the European biotech universe when it forged its original deal with GSK back at the end of 2006. That development deal also made Drakeman a star in biotech circles as well. Earlier this year several analysts had attracted attention speculating that GSK might swoop in to buy out Genmab, but those rumors have quieted in the wake of Drakeman's departure.
mfg ipollit
http://www.fiercebiotech.com/story/genmab-grabs-134m-it-rest…
Genmab grabs $134M as it restructures blockbuster GSK pact
July 1, 2010 — 2:00pm ET | By John Carroll
Just weeks after Lisa Drakeman departed the CEO's post at Genmab, GlaxoSmithKline has engineered a big change to the $2.1 billion deal it struck with the Danish developer more than three years ago on Arzerra (ofatumumab). The troubled Genmab, which has watched its share price slide 90 percent, is getting $134 million upfront on the revised pact, but sees its milestone package cut in half.
In the revised pact GSK takes responsibility for developing the therapy for autoimmune diseases while both companies collaborate on cancer. And GSK's funding commitment for the oncology work is capped at 145 million pounds overall with a 17 million pound annual limit.
"Their (GSK's) flexibility has allowed us to not only ensure that we extract the best possible value from ofatumumab, but also strengthen the financial security of Genmab," said Prof. Jan G.J. van de Winkel, Genmab's new chief. The changes will have a "material impact on Genmab's 2010 financial guidance," the company said in a statement.
Genmab had been one of the bright stars in the European biotech universe when it forged its original deal with GSK back at the end of 2006. That development deal also made Drakeman a star in biotech circles as well. Earlier this year several analysts had attracted attention speculating that GSK might swoop in to buy out Genmab, but those rumors have quieted in the wake of Drakeman's departure.
mfg ipollit
Antwort auf Beitrag Nr.: 39.772.978 von ipollit am 03.07.10 15:07:14http://www.businessweek.com/ap/financialnews/D9GMG4R80.htm
The Associated Press July 1, 2010, 5:02PM ET
GlaxoSmithKline amends deal with Genmab
GlaxoSmithKline said Thursday it will take responsibility for the development of ofatumumab as a treatment for autoimmune conditions as part of an amended deal with Genmab.
The companies had been codeveloping the drug candidate and will continue to jointly develop the drug as a potential treatment for cancer.
Genmab will receive an upfront payment of 90 million British pounds ($60 million). Meanwhile, Genmab's future funding commitment for the development of ofatumumab in cancer indications will be capped at 145 million British pounds ($96.7 million), including a yearly spending cap of 17 million British pounds ($11.3 million) for each of the next six years starting with 2010.
Future milestones due to Genmab under the cancer development program will be halved. There will be no change in royalty tiers to the Denmark-based company in the cancer program.
In the autoimmune indication, Genmab will forego development milestones and the first two sales milestones but will keep a double-digit royalty on sales.
GlaxoSmithKline said it is planning a number of midstage studies to take ofatumumab forward in autoimmune indications.
mfg ipollit
The Associated Press July 1, 2010, 5:02PM ET
GlaxoSmithKline amends deal with Genmab
GlaxoSmithKline said Thursday it will take responsibility for the development of ofatumumab as a treatment for autoimmune conditions as part of an amended deal with Genmab.
The companies had been codeveloping the drug candidate and will continue to jointly develop the drug as a potential treatment for cancer.
Genmab will receive an upfront payment of 90 million British pounds ($60 million). Meanwhile, Genmab's future funding commitment for the development of ofatumumab in cancer indications will be capped at 145 million British pounds ($96.7 million), including a yearly spending cap of 17 million British pounds ($11.3 million) for each of the next six years starting with 2010.
Future milestones due to Genmab under the cancer development program will be halved. There will be no change in royalty tiers to the Denmark-based company in the cancer program.
In the autoimmune indication, Genmab will forego development milestones and the first two sales milestones but will keep a double-digit royalty on sales.
GlaxoSmithKline said it is planning a number of midstage studies to take ofatumumab forward in autoimmune indications.
mfg ipollit
sehr erfreuliche "Neuigkeiten" für Immunogen:
Genentech Submits Application to FDA for Trastuzumab-DM1 in Previously Treated Advanced HER2-Positive Breast Cancer
http://www.fiercebiotech.com/press-releases/genentech-submit…
Angekündigt war es ja schon, aber jetzt ist es "amtlich". Natürlich rechnet jeder fest mit einer Zulassung durch die FDA...
Erfreulich auch, dass am Rande das hier nochmals bestätigt wurde:
A planned Phase III study, MARIANNE, will compare both T-DM1 alone and T-DM1 in combination with pertuzumab to Herceptin in combination with a taxane chemotherapy in people with advanced HER2-positive breast cancer who have not been previously treated for advanced disease.
Mit dem MARIANNE-Trial könnte T-DM1 vielleicht eines Tages Herceptin mehr oder weniger komplett ersetzen.
Genentech Submits Application to FDA for Trastuzumab-DM1 in Previously Treated Advanced HER2-Positive Breast Cancer
http://www.fiercebiotech.com/press-releases/genentech-submit…
Angekündigt war es ja schon, aber jetzt ist es "amtlich". Natürlich rechnet jeder fest mit einer Zulassung durch die FDA...
Erfreulich auch, dass am Rande das hier nochmals bestätigt wurde:
A planned Phase III study, MARIANNE, will compare both T-DM1 alone and T-DM1 in combination with pertuzumab to Herceptin in combination with a taxane chemotherapy in people with advanced HER2-positive breast cancer who have not been previously treated for advanced disease.
Mit dem MARIANNE-Trial könnte T-DM1 vielleicht eines Tages Herceptin mehr oder weniger komplett ersetzen.
Genmab... bisher kann ich noch nicht erkennen, wie es wirklich aufwärts gehen kann
http://www.evaluatepharma.com/Universal/View.aspx?type=Story…
EP Vantage Interview – New Genmab CEO aiming to turn tide in its favour
July 13, 2010
Taking a leaf out of any number of management handbooks, Genmab’s new chief executive, Jan van de Winkel, is certainly trying to act fast and decisively.
Appointed last month under somewhat mysterious circumstances following the sudden resignation of Genmab’s long-term chief executive and co-founder, Lisa Drakeman (Mystery and intrigue surround change of helm at Genmab, June 17, 2010), Mr van de Winkel immediately set to work on addressing the Danish biotech’s tricky financial position by restructuring its crucial collaboration with GlaxoSmithKline over Arzerra within just two weeks. The new deal achieves many things, most important being a $135m upfront fee, giving Genmab vital breathing space which Mr van de Winkel sees as drawing “a line in the sand” as the company attempts to leave its recent troubles behind.
Game changer I – Glaxo deal
“I literally started this one day after I took up my new position”, says Mr van de Winkel of the process to renegotiate the Glaxo deal, which was hammered out over two long days followed by four days at the lawyers' offices to amend the terms of the deal.
Aside from the welcome upfront fee, the new terms will see Glaxo take over all responsibility for developing Arzerra in autoimmune indications, such as rheumatoid arthritis and multiple sclerosis, while Genmab’s spending commitments in cancer indications have been capped at cumulative $218m over the next six years, or an annual cap of around $25m.
In return Genmab has naturally had to forego a significant amount of potential milestones under the original $2.1bn deal struck back in 2006. Development milestones in cancer settings have been cut by 50% and removed completely in autoimmune indications. Meanwhile royalty rates for cancer sales are retained at around 20-30% but reduced to low double digits in autoimmune uses.
The mostly give and little take nature of the deal has had a mixed reception amongst analysts, although the overriding theme is the amendment was a necessary evil for the company to address pressing financial concerns.
Investors were clearly encouraged, boosting Genmab’s shares by 46% on the news, although admittedly this was from a seven-year low of DKr40.91 ($6.95) just before the restructured deal was announced.
Nevertheless, it seems it will take a while for investor confidence in Genmab to fully return. The company’s current market capitalisation of $496m is not a lot higher than its cash balance of around $330m after Glaxo’s lump sum payment. Even slashing current royalty estimates in half to broadly reflect the new deal still means revenues of $102m by 2016 which values Arzerra at $695m for Genmab, according to EvaluatePharma’s NPV Analyzer.
Game changer II – sale of manufacturing plant
Another move that will boost investor sentiment is completing the sale of Genmab's Minnesota manufacturing facility, which until the Glaxo deal was restructured was the company’s main cash lifeline, with a disposal expected by the end of the year.
“Two weeks ago I actually needed to sell the facility this year”, says Mr van de Winkel, “we would still like that to happen, but the pressure is now eased to some extent.”
Acquired for $240m from PDL BioPharma in 2008 when Genmab had grand plans to become a fully integrated pharmaceutical company, the plant is now valued at around $150m, a potential loss which has already been written off in its accounts.
As with all sale processes, the stronger the financial position of the seller the better, and following the breathing space afforded by the new Glaxo deal, Mr van de Winkel is hopeful that a good price can be extracted along more creative lines.
Mr van de Winkel claims approaches have been received from big pharma and biotech alike, as well as generic companies considering acquiring the facility for making biosimilar products. Additionally the company is now also considering a sale which may also involve the acquirer gaining access to some of Genmab’s technologies or early stage pipeline assets.
Either way, Genmab plans to stick to its biotech innovation and discovery roots, a change of tack revealed late last year under Ms Drakeman’s leadership (Genmab cuts costs and jobs as it goes back to its roots, November 6, 2009).
Game changer III – pipeline development: partner zalutumumab, develop Arzerra for NHL
Arzerra is currently approved in the US and Europe to treat refractory chronic lymphocytic leukaemia (CLL), sales from which could exceed $200m by 2016. However, many analysts see the real value driver for Arzerra being Non-Hodgkin's lymphoma (NHL), with potential approval in 2013 generating sales of $260m in 2016.
Meanwhile the future of Genmab’s most advanced pipeline candidate, zalutumumab, remains in the balance following largely disappointing phase III results in head and neck cancers earlier this year.
The trial failed to significantly improve overall survival in patients with recurrent or metastatic squamous cell carcinoma of the head and neck, although decent improvement in progression free survival was observed (Genmab's bright spot dims leaving little on the horizon, March 9, 2010).
Nevertheless, Mr van de Winkel remains upbeat about the chances of securing a partner for zalutumumab, although he admits it will depend on the feedback from the FDA and European regulators on a likely path to approval. Full development of zalutumumab is likely to require hundreds of millions of dollars he believes, clearly not within Genmab’s reach.
An update on the partnering scene for zalutumumab will be provided in the autumn, after Mr van de Winkel has completed a 100-day review of the company’s strategic direction. At the same time more detailed guidance on the cash runway could be given. Before the restructured Glaxo deal analysts were forecasting a move into profitability by 2015, it will be interesting if the new deal means this landmark will be brought forward or pushed back.
Although external advisors have been hired to support the strategic review, Mr van de Winkel says the likely outcome “will not be dramatic, more a re-focusing and putting a different emphasis on different programmes.”
This may disappoint some analysts and investors hoping for a more significant change of direction to recapture former glories. For now though Mr van de Winkel seems determined to do it his way, which looks like being a gradual process. Having pulled a rabbit out of the hat with the new Glaxo deal, if he can clinch the deal on the other game changers he might just pull it off.
mfg ipollit
http://www.evaluatepharma.com/Universal/View.aspx?type=Story…
EP Vantage Interview – New Genmab CEO aiming to turn tide in its favour
July 13, 2010
Taking a leaf out of any number of management handbooks, Genmab’s new chief executive, Jan van de Winkel, is certainly trying to act fast and decisively.
Appointed last month under somewhat mysterious circumstances following the sudden resignation of Genmab’s long-term chief executive and co-founder, Lisa Drakeman (Mystery and intrigue surround change of helm at Genmab, June 17, 2010), Mr van de Winkel immediately set to work on addressing the Danish biotech’s tricky financial position by restructuring its crucial collaboration with GlaxoSmithKline over Arzerra within just two weeks. The new deal achieves many things, most important being a $135m upfront fee, giving Genmab vital breathing space which Mr van de Winkel sees as drawing “a line in the sand” as the company attempts to leave its recent troubles behind.
Game changer I – Glaxo deal
“I literally started this one day after I took up my new position”, says Mr van de Winkel of the process to renegotiate the Glaxo deal, which was hammered out over two long days followed by four days at the lawyers' offices to amend the terms of the deal.
Aside from the welcome upfront fee, the new terms will see Glaxo take over all responsibility for developing Arzerra in autoimmune indications, such as rheumatoid arthritis and multiple sclerosis, while Genmab’s spending commitments in cancer indications have been capped at cumulative $218m over the next six years, or an annual cap of around $25m.
In return Genmab has naturally had to forego a significant amount of potential milestones under the original $2.1bn deal struck back in 2006. Development milestones in cancer settings have been cut by 50% and removed completely in autoimmune indications. Meanwhile royalty rates for cancer sales are retained at around 20-30% but reduced to low double digits in autoimmune uses.
The mostly give and little take nature of the deal has had a mixed reception amongst analysts, although the overriding theme is the amendment was a necessary evil for the company to address pressing financial concerns.
Investors were clearly encouraged, boosting Genmab’s shares by 46% on the news, although admittedly this was from a seven-year low of DKr40.91 ($6.95) just before the restructured deal was announced.
Nevertheless, it seems it will take a while for investor confidence in Genmab to fully return. The company’s current market capitalisation of $496m is not a lot higher than its cash balance of around $330m after Glaxo’s lump sum payment. Even slashing current royalty estimates in half to broadly reflect the new deal still means revenues of $102m by 2016 which values Arzerra at $695m for Genmab, according to EvaluatePharma’s NPV Analyzer.
Game changer II – sale of manufacturing plant
Another move that will boost investor sentiment is completing the sale of Genmab's Minnesota manufacturing facility, which until the Glaxo deal was restructured was the company’s main cash lifeline, with a disposal expected by the end of the year.
“Two weeks ago I actually needed to sell the facility this year”, says Mr van de Winkel, “we would still like that to happen, but the pressure is now eased to some extent.”
Acquired for $240m from PDL BioPharma in 2008 when Genmab had grand plans to become a fully integrated pharmaceutical company, the plant is now valued at around $150m, a potential loss which has already been written off in its accounts.
As with all sale processes, the stronger the financial position of the seller the better, and following the breathing space afforded by the new Glaxo deal, Mr van de Winkel is hopeful that a good price can be extracted along more creative lines.
Mr van de Winkel claims approaches have been received from big pharma and biotech alike, as well as generic companies considering acquiring the facility for making biosimilar products. Additionally the company is now also considering a sale which may also involve the acquirer gaining access to some of Genmab’s technologies or early stage pipeline assets.
Either way, Genmab plans to stick to its biotech innovation and discovery roots, a change of tack revealed late last year under Ms Drakeman’s leadership (Genmab cuts costs and jobs as it goes back to its roots, November 6, 2009).
Game changer III – pipeline development: partner zalutumumab, develop Arzerra for NHL
Arzerra is currently approved in the US and Europe to treat refractory chronic lymphocytic leukaemia (CLL), sales from which could exceed $200m by 2016. However, many analysts see the real value driver for Arzerra being Non-Hodgkin's lymphoma (NHL), with potential approval in 2013 generating sales of $260m in 2016.
Meanwhile the future of Genmab’s most advanced pipeline candidate, zalutumumab, remains in the balance following largely disappointing phase III results in head and neck cancers earlier this year.
The trial failed to significantly improve overall survival in patients with recurrent or metastatic squamous cell carcinoma of the head and neck, although decent improvement in progression free survival was observed (Genmab's bright spot dims leaving little on the horizon, March 9, 2010).
Nevertheless, Mr van de Winkel remains upbeat about the chances of securing a partner for zalutumumab, although he admits it will depend on the feedback from the FDA and European regulators on a likely path to approval. Full development of zalutumumab is likely to require hundreds of millions of dollars he believes, clearly not within Genmab’s reach.
An update on the partnering scene for zalutumumab will be provided in the autumn, after Mr van de Winkel has completed a 100-day review of the company’s strategic direction. At the same time more detailed guidance on the cash runway could be given. Before the restructured Glaxo deal analysts were forecasting a move into profitability by 2015, it will be interesting if the new deal means this landmark will be brought forward or pushed back.
Although external advisors have been hired to support the strategic review, Mr van de Winkel says the likely outcome “will not be dramatic, more a re-focusing and putting a different emphasis on different programmes.”
This may disappoint some analysts and investors hoping for a more significant change of direction to recapture former glories. For now though Mr van de Winkel seems determined to do it his way, which looks like being a gradual process. Having pulled a rabbit out of the hat with the new Glaxo deal, if he can clinch the deal on the other game changers he might just pull it off.
mfg ipollit
ARNA... vielleicht leicht positiv für Arena, dass bei der Ablehnung von Vivus Qnexa Sicherheitsbedenken das entscheidende Kriterium waren. Lorcaserin wirkt zwar nicht so gut, scheint aber relativ sicher zu sein, zumal erheblich mehr Studiendaten dafür vorliegen. Positiv vielleicht auch die Einschätzung unten, dass Lorcaserin ähnlich gut wie Xenical oder Meridia wirkt.
http://www.reuters.com/article/idCNN1520175720100715?rpc=44
US advisers reject Vivus' fat pill; shares plunge
Thu Jul 15, 2010 7:12pm EDT
* FDA advisory panel rejects Qnexa drug in 10-6 vote
* Drug works but safety issues concerned experts
* Company says weight loss with drug "unprecedented"
* FDA official surprised by panel vote
* Vivus shares sink 62 percent, stocks of rivals sink
By Susan Heavey and Jon Lentz
GAITHERSBURG, Md., July 15 (Reuters) - The first new prescription weight-loss pill in more than a decade failed to win backing from U.S. health advisers, who said safety concerns about the drug outweighed its ability to help obese patients shed pounds.
Shares of Vivus Inc's (VVUS.O) sank 62 percent on Thursday after U.S. Food and Drug Administration advisers expressed concern the once-a-day pill could cause depression, memory-loss and potential birth defects if used among millions of overweight or obese Americans.
Their decision stunned investors, who had more than doubled the share price of the California biotech in the last year on hopes that safety woes would not keep the drug from market.
An FDA official was also surprised.
"When you listen to even the no votes, you got the sense that a lot of people, they weren't strongly against the drug," said Eric Colman, deputy director of FDA's division that oversees such drugs.
Shares of rival fat-pill hopefuls, Arena Pharmaceuticals Inc (ARNA.O) and Orexigen Therapeutics Inc (OREX.O), also sank after the vote.
Approving the drug would be "a huge public health experiment," said panelist Elaine Morrato of the University of Colorado, one of the 10 panelists who urged against FDA approval. Six others supported the drug, called Qnexa.
The FDA will weigh the vote before making its final decision and usually follows its panelists' advice.
The panel's rejection is a blow to Vivus, which has not had a U.S. product approved since 1996.
Cowen and Co analyst Ian Sanderson said it was possible for Vivus to eventually win U.S. approval, but it would first have to test it in as many as 10,000 patients, at a potential cost of $150 million.
"They'd need a partner," Sanderson said.
The California biotechnology company is seeking the FDA's green light with the hope of beating Arena and Orexigen's rival diet drugs to market.
It told FDA's outside experts that Qnexa helped many patients shed 35 to 40 pounds and that its risks were not major.
There is little doubt the drug works, panelists said, but potential side effects such as depression, memory loss, increased heart rate and birth defects were a top worry.
"This medication, in term of efficacy, is far superior than anything that's on the market. The concerns we have are with safety," said Dr. Abraham Thomas of Henry Ford Hospital in Detroit.
Panelists were also concerned since patients may take Qnexa for years, but Vivus' data only covered about 12 months.
Nearly 70 percent of people in the United States are overweight, and more than a third of them are obese, government statistics show.
"It's like instant willpower," said Erin Aycock, a clinical trial patient who lost 50 pounds on the drug but later regained most of it. "I would do anything to be back on this drug."
In a statement after the meeting, Vivus said the panel's vote was disappointing. The company had been expecting FDA's approval decision by Oct. 28.
Vivus has said it expects data from a longer, 2-year study by the end of this quarter.
Shares of Arena closed down 8.4 percent after the vote, reversing earlier gains. Orexigen closed 10 percent lower. Vivus shares were halted during regular trading on Thursday but fell heavily as they reopened in after-hours trade.
Some analysts saw hope for Arena's candidate, lorcaserin, which tentatively faces FDA's advisers Sept. 16 and has 2-year data. Orexigen's FDA panel is expected Dec. 7.
***************
http://www.thestreet.com/story/10806362/1/wall-st-elite-get-…
Street Elite Get Early Crack at Arena Study
07/14/10 - 10:11 AM EDT
BOSTON (TheStreet) -- On Tuesday afternoon, SummerStreet Research Partners, a healthcare research shop, sent emails informing its institutional investor clients about an important study and related editorial concerning Arena Pharmaceuticals(ARNA) that were published in this week's New England Journal of Medicine.
Here, in full, is the text of the email sent Tuesday to investor clients about Arena and the NEJM by SummerStreet Research:
"ARNA (BUY), The BLOOM trial is published today in the NEJM with an editorial. The editorial points out that lorcaserin has slightly less than or equal efficacy to Orlistat and slightly less efficacy than Meridia (sibutramine). However the justification of approval for lorcaserin is that its safety profile appears to be better than that of Xenical (orlistat) or sibutramine. In addition, lorcaserin appears to result in improvement of all surrogate measures of diabetes and cardiovascular risk: the effect is small however clinically relevant. This is important since sibutramine did not and subsequently went on to demonstrate negative cardiovascular outcome in the SCOUT trial. The SCOUT trial results will be presented tomorrow at the International Obestity conference in Stockholm. The author of the editorial suggests that lorcaserin will probably be used in combination, perhaps with GLP-1s (prior to phentermine). Overall we consider the publication a positive for the shares and it bodes well for the September 16th advisory panel meeting (bolsters the Eisai deal – safety first novel agent)."
mfg ipollit
http://www.reuters.com/article/idCNN1520175720100715?rpc=44
US advisers reject Vivus' fat pill; shares plunge
Thu Jul 15, 2010 7:12pm EDT
* FDA advisory panel rejects Qnexa drug in 10-6 vote
* Drug works but safety issues concerned experts
* Company says weight loss with drug "unprecedented"
* FDA official surprised by panel vote
* Vivus shares sink 62 percent, stocks of rivals sink
By Susan Heavey and Jon Lentz
GAITHERSBURG, Md., July 15 (Reuters) - The first new prescription weight-loss pill in more than a decade failed to win backing from U.S. health advisers, who said safety concerns about the drug outweighed its ability to help obese patients shed pounds.
Shares of Vivus Inc's (VVUS.O) sank 62 percent on Thursday after U.S. Food and Drug Administration advisers expressed concern the once-a-day pill could cause depression, memory-loss and potential birth defects if used among millions of overweight or obese Americans.
Their decision stunned investors, who had more than doubled the share price of the California biotech in the last year on hopes that safety woes would not keep the drug from market.
An FDA official was also surprised.
"When you listen to even the no votes, you got the sense that a lot of people, they weren't strongly against the drug," said Eric Colman, deputy director of FDA's division that oversees such drugs.
Shares of rival fat-pill hopefuls, Arena Pharmaceuticals Inc (ARNA.O) and Orexigen Therapeutics Inc (OREX.O), also sank after the vote.
Approving the drug would be "a huge public health experiment," said panelist Elaine Morrato of the University of Colorado, one of the 10 panelists who urged against FDA approval. Six others supported the drug, called Qnexa.
The FDA will weigh the vote before making its final decision and usually follows its panelists' advice.
The panel's rejection is a blow to Vivus, which has not had a U.S. product approved since 1996.
Cowen and Co analyst Ian Sanderson said it was possible for Vivus to eventually win U.S. approval, but it would first have to test it in as many as 10,000 patients, at a potential cost of $150 million.
"They'd need a partner," Sanderson said.
The California biotechnology company is seeking the FDA's green light with the hope of beating Arena and Orexigen's rival diet drugs to market.
It told FDA's outside experts that Qnexa helped many patients shed 35 to 40 pounds and that its risks were not major.
There is little doubt the drug works, panelists said, but potential side effects such as depression, memory loss, increased heart rate and birth defects were a top worry.
"This medication, in term of efficacy, is far superior than anything that's on the market. The concerns we have are with safety," said Dr. Abraham Thomas of Henry Ford Hospital in Detroit.
Panelists were also concerned since patients may take Qnexa for years, but Vivus' data only covered about 12 months.
Nearly 70 percent of people in the United States are overweight, and more than a third of them are obese, government statistics show.
"It's like instant willpower," said Erin Aycock, a clinical trial patient who lost 50 pounds on the drug but later regained most of it. "I would do anything to be back on this drug."
In a statement after the meeting, Vivus said the panel's vote was disappointing. The company had been expecting FDA's approval decision by Oct. 28.
Vivus has said it expects data from a longer, 2-year study by the end of this quarter.
Shares of Arena closed down 8.4 percent after the vote, reversing earlier gains. Orexigen closed 10 percent lower. Vivus shares were halted during regular trading on Thursday but fell heavily as they reopened in after-hours trade.
Some analysts saw hope for Arena's candidate, lorcaserin, which tentatively faces FDA's advisers Sept. 16 and has 2-year data. Orexigen's FDA panel is expected Dec. 7.
***************
http://www.thestreet.com/story/10806362/1/wall-st-elite-get-…
Street Elite Get Early Crack at Arena Study
07/14/10 - 10:11 AM EDT
BOSTON (TheStreet) -- On Tuesday afternoon, SummerStreet Research Partners, a healthcare research shop, sent emails informing its institutional investor clients about an important study and related editorial concerning Arena Pharmaceuticals(ARNA) that were published in this week's New England Journal of Medicine.
Here, in full, is the text of the email sent Tuesday to investor clients about Arena and the NEJM by SummerStreet Research:
"ARNA (BUY), The BLOOM trial is published today in the NEJM with an editorial. The editorial points out that lorcaserin has slightly less than or equal efficacy to Orlistat and slightly less efficacy than Meridia (sibutramine). However the justification of approval for lorcaserin is that its safety profile appears to be better than that of Xenical (orlistat) or sibutramine. In addition, lorcaserin appears to result in improvement of all surrogate measures of diabetes and cardiovascular risk: the effect is small however clinically relevant. This is important since sibutramine did not and subsequently went on to demonstrate negative cardiovascular outcome in the SCOUT trial. The SCOUT trial results will be presented tomorrow at the International Obestity conference in Stockholm. The author of the editorial suggests that lorcaserin will probably be used in combination, perhaps with GLP-1s (prior to phentermine). Overall we consider the publication a positive for the shares and it bodes well for the September 16th advisory panel meeting (bolsters the Eisai deal – safety first novel agent)."
mfg ipollit
Antwort auf Beitrag Nr.: 39.831.237 von ipollit am 16.07.10 11:28:56ARNA - hier noch eine Meinung gegen eine Lorcaserin-Phentermine Kombination (das ja dem wohlbekannten Fen-Phen entsprechen würde)
http://seekingalpha.com/instablog/646558-jason-chew/80595-wh…
Why Arena’s Lorcaserin Will Not Be Used In Combination With Phentermine
Jul 7, 2010 7:55 PM
It is tempting to believe that once Lorcaserin has been approved, it will be used off-label in combination with phentermine to create a safe and efficacious version of Fen-Phen, the once popular diet drug. This is not to be the case.
Some history:
Fenfluramine, the “Fen” in Fen-Phen, a potent agonist of 5-HT2B receptors, a type of serotonin receptor found in the brain as well as in high quantities on heart valves. It was approved in 1973, its enantiomer, dexfenfluramine was approved by the FDA in 1996 to Wyeth. Neither fenfluramine nor dexfenfluramine were popular diet drugs due to their mediocre efficacy. It wasn’t until 1992 when an article was published showing fenfluramine combined with Phentermine led to 15% weight loss when the Fen-Phen diet craze began. While Fenfluramine and Phentermine each on their own led to similar weight losses of around 8%, the synergistic effect of the combination allowed for use of lower doses of both drugs, but resulted in a significantly better effect.
In 1997, the Mayo Clinic documented 24 cases of women with valvular heart defects who were taking Fen-Phen, playing large role in the recall of fenfluramine and dexfenfluramine from the market. Further testing by the Mayo Clinic found about 30% of patients on Fen-Phen who were asymtomatic for heart disease were found to had abnormal echocardiographic results.
To this day, scientists are still debating how exactly fenfluramine (fenfluramine will refer to both it and dexfenfluramine) causes the heart defects, but revolves around stimulation of the 5-HT2B receptors, leading to excess cell proliferation and vascular remodeling.
Lorcaserin:
Lorcaserin is designed as a potent agonist of 5-HT2C, with greater than 100 fold selectivity over 5-HT2B, the apparent culprit in Fen-Phen. It has been tested in thousands of patients and shows no signs of causing heart defects. Lorcaserin has been shown to have mild side-effects, however, its efficacy is similar to that of fenfluramine- modest. Would it not make perfect sense to boost its effects with a dose of phentermine? After all, phentermine was not taken off the market, in fact, it is currently the number one prescribed stimulant obesity drug in the US.
Not so simple. As previously mentioned, the exact mechanism of Fen-Phen induced heart valve defects has not been deduced. It is known that Fenfluramine by itself is a culprit; it is also known that Phentermine on its own is not. But the combination of the two may increase the risk of heart problems through multiple mechanisms. Though this is far from proven, Phentermine faces guilt by association- cases of heart defects were not noticed until the two drugs were used together. A large part of this is because relatively few people used fenfluramine before the study documenting the superior effects of its combined use with phentermine. Also, in a retrospective JAMA, 2000 study, researchers showed 4.1% of untreated patients had Aortic Regurgitation, compared to 8.9% of patients who took fenfluramine only, and 13.7% of patients on the Fen-Phen combo. (Note: this was a Wyeth sponsored paper.)
Arena, being smart, has not tested Lorcaserin in combination with phentermine. If it did, even preclinical, it would have to report the data to the FDA. There is just no way to predict exactly what will happen when you put two compounds together. Arena will not be able to promote Lorcaserin as a combination pill without the studies, it will depend entirely on doctors prescribing the combo off-label. Aggressive doctors may be tempted to do just that, but I believe the FDA, with its emphasis on safety, already has that in mind. Until there is evidence to the contrary, there is likely to be a warning against the use of Lorcaserin in combination with phentermine. This warning would deter most doctors from prescribing a Lorcaserin-Phentermine combo.
mfg ipollit
http://seekingalpha.com/instablog/646558-jason-chew/80595-wh…
Why Arena’s Lorcaserin Will Not Be Used In Combination With Phentermine
Jul 7, 2010 7:55 PM
It is tempting to believe that once Lorcaserin has been approved, it will be used off-label in combination with phentermine to create a safe and efficacious version of Fen-Phen, the once popular diet drug. This is not to be the case.
Some history:
Fenfluramine, the “Fen” in Fen-Phen, a potent agonist of 5-HT2B receptors, a type of serotonin receptor found in the brain as well as in high quantities on heart valves. It was approved in 1973, its enantiomer, dexfenfluramine was approved by the FDA in 1996 to Wyeth. Neither fenfluramine nor dexfenfluramine were popular diet drugs due to their mediocre efficacy. It wasn’t until 1992 when an article was published showing fenfluramine combined with Phentermine led to 15% weight loss when the Fen-Phen diet craze began. While Fenfluramine and Phentermine each on their own led to similar weight losses of around 8%, the synergistic effect of the combination allowed for use of lower doses of both drugs, but resulted in a significantly better effect.
In 1997, the Mayo Clinic documented 24 cases of women with valvular heart defects who were taking Fen-Phen, playing large role in the recall of fenfluramine and dexfenfluramine from the market. Further testing by the Mayo Clinic found about 30% of patients on Fen-Phen who were asymtomatic for heart disease were found to had abnormal echocardiographic results.
To this day, scientists are still debating how exactly fenfluramine (fenfluramine will refer to both it and dexfenfluramine) causes the heart defects, but revolves around stimulation of the 5-HT2B receptors, leading to excess cell proliferation and vascular remodeling.
Lorcaserin:
Lorcaserin is designed as a potent agonist of 5-HT2C, with greater than 100 fold selectivity over 5-HT2B, the apparent culprit in Fen-Phen. It has been tested in thousands of patients and shows no signs of causing heart defects. Lorcaserin has been shown to have mild side-effects, however, its efficacy is similar to that of fenfluramine- modest. Would it not make perfect sense to boost its effects with a dose of phentermine? After all, phentermine was not taken off the market, in fact, it is currently the number one prescribed stimulant obesity drug in the US.
Not so simple. As previously mentioned, the exact mechanism of Fen-Phen induced heart valve defects has not been deduced. It is known that Fenfluramine by itself is a culprit; it is also known that Phentermine on its own is not. But the combination of the two may increase the risk of heart problems through multiple mechanisms. Though this is far from proven, Phentermine faces guilt by association- cases of heart defects were not noticed until the two drugs were used together. A large part of this is because relatively few people used fenfluramine before the study documenting the superior effects of its combined use with phentermine. Also, in a retrospective JAMA, 2000 study, researchers showed 4.1% of untreated patients had Aortic Regurgitation, compared to 8.9% of patients who took fenfluramine only, and 13.7% of patients on the Fen-Phen combo. (Note: this was a Wyeth sponsored paper.)
Arena, being smart, has not tested Lorcaserin in combination with phentermine. If it did, even preclinical, it would have to report the data to the FDA. There is just no way to predict exactly what will happen when you put two compounds together. Arena will not be able to promote Lorcaserin as a combination pill without the studies, it will depend entirely on doctors prescribing the combo off-label. Aggressive doctors may be tempted to do just that, but I believe the FDA, with its emphasis on safety, already has that in mind. Until there is evidence to the contrary, there is likely to be a warning against the use of Lorcaserin in combination with phentermine. This warning would deter most doctors from prescribing a Lorcaserin-Phentermine combo.
mfg ipollit
Ohad Hammer zur Situation bei Exelixis:
Exelixis – What’s Next?
Exelixis (EXEL) came under a lot of pressure recently and the stock is now trading near its 52 week low following two unrelated events that took place last month. In late June, the company announced the resignation of its CEO, George Scangos, who was appointed as Biogen Idec’s (BIIB) CEO. Earlier that month, the company announced BMS (BMY) decided to give back rights for Exelixis’ flagship product, XL184. Adding to the pressure are interesting but not stellar clinical results for some of the company’s compounds at ASCO and potentially an additional partnership termination.
...
hier gehts weiter:
http://www.hammerstockblog.com/exelixis-%e2%80%93-what%e2%80…
Exelixis – What’s Next?
Exelixis (EXEL) came under a lot of pressure recently and the stock is now trading near its 52 week low following two unrelated events that took place last month. In late June, the company announced the resignation of its CEO, George Scangos, who was appointed as Biogen Idec’s (BIIB) CEO. Earlier that month, the company announced BMS (BMY) decided to give back rights for Exelixis’ flagship product, XL184. Adding to the pressure are interesting but not stellar clinical results for some of the company’s compounds at ASCO and potentially an additional partnership termination.
...
hier gehts weiter:
http://www.hammerstockblog.com/exelixis-%e2%80%93-what%e2%80…
zu Onyx:
Onyx Pharmaceuticals Announces Positive Top-Line Carfilzomib Data From Phase 2b Study
24 Percent Overall Response Rate and Duration of Response Exceeding Seven Months in Heavily Pretreated Advanced Multiple Myeloma Patients
Onyx to Host Investor Teleconference and Webcast Today at 8:00 a.m. Eastern Time to Review Top-Line Data
EMERYVILLE, Calif., July 26 /PRNewswire-FirstCall/ -- Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX) today announced positive top-line results from the Phase 2b 003-A1 study of single-agent carfilzomib, a selective, next generation proteasome inhibitor, in patients with relapsed and refractory multiple myeloma. In an independent review of the data, carfilzomib achieved an overall response rate (partial response or greater) of 24 percent and a median duration of response of 7.4 months in patients who entered the study after receiving a median of five prior lines of therapy (corresponding to a median of 13 anti-myeloma agents) and whose disease was refractory to their last therapeutic regimen. The clinical benefit rate (minimal response or greater) in the study population was 36 percent. Carfilzomib was well-tolerated and there were no new or unexpected toxicities observed. Full results of the trial will be presented at an upcoming scientific meeting. Based on these results, Onyx is continuing discussions with the U.S. Food and Drug Administration (FDA) regarding next steps in filing a new drug application (NDA) for carfilzomib, which the company expects to submit by year-end 2010 for potential accelerated approval in the U.S.
"Despite recent advances in treating multiple myeloma, all patients eventually relapse. The unmet medical need remains great, as the outlook for patients with relapsed and refractory disease is grim," said Michael G. Kauffman, M.D., Ph.D., Chief Medical Officer of Onyx Pharmaceuticals. "According to a study from the International Myeloma Working Group, patients, such as those enrolled in the 003-A1 study, can expect to respond to therapy only 11 percent of the time and survive for only six to 10 months.(i) The single-agent activity with durable disease control and favorable tolerability observed in this study indicate that carfilzomib has the potential to alter the natural course of this deadly disease."
"Carfilzomib has the potential to be an important therapy in multiple myeloma and exemplifies the Onyx vision to build a leading oncology company by developing innovative targeted therapies," said N. Anthony Coles, M.D., President and Chief Executive Officer of Onyx Pharmaceuticals. "We are committed to bringing this promising treatment to patients as quickly as possible by pursuing an accelerated approval pathway in the U.S., while simultaneously moving forward with two Phase 3 studies. The first study, ASPIRE, is designed to support full carfilzomib registration in the U.S. in earlier-stage patients who have relapsed following initial lines of therapy, and the second study is designed to support approval in relapsed and refractory patients in Europe."
Trial Design
The 003-A1 study was an open-label, single-arm Phase 2b trial. The trial evaluated 266 heavily-pretreated patients with relapsed and refractory multiple myeloma whose disease was refractory to their last treatment regimen and who had received at least two prior therapies, including bortezomib, either thalidomide or lenalidomide, an alkylating agent, glucocorticoids and an anthracycline. Refractory disease was defined as < 25% response or progression during therapy or within 60 days after completion of therapy.(ii) Patients enrolled in the 003-A1 trial had received a median of five prior therapeutic regimens, corresponding to a median of 13 anti-myeloma agents. Patients received carfilzomib at 20mg/m2 for the first cycle followed by 27mg/m2 thereafter for up to 12 cycles. Patients who completed the 12 cycles were eligible to enter an extension study. Responses and progression were determined according to the International Myeloma Working Group (IMWG) criteria. The trial was conducted in collaboration with the Multiple Myeloma Research Consortium (MMRC) and at additional sites in the U.S. and Canada.
"The patients in the 003-A1 trial represent an advanced population with significant unmet medical need who had received many lines of therapy and had limited options available to them outside of a clinical trial, strongly underscoring the need for new treatments. We are proud to have worked with the Onyx team that is developing carfilzomib and are encouraged by these results," said Kathy Giusti, Founder and CEO of the MMRC and a multiple myeloma patient. The MMRC initiated a relationship with the company (then Proteolix) in 2006, which included the participation of 11 MMRC Member Institutions in the 003-A1 trial, representing 36 percent of the total trial centers and 60 percent of enrolled patients.
"There is a high unmet need for treatment options for patients with relapsed and refractory multiple myeloma who are no longer responding to available therapies," said Brian G.M. Durie, M.D., Cofounder and Chair of the Board of Directors of the International Myeloma Foundation (IMF).
Onyx Pharmaceuticals Announces Positive Top-Line Carfilzomib Data From Phase 2b Study
24 Percent Overall Response Rate and Duration of Response Exceeding Seven Months in Heavily Pretreated Advanced Multiple Myeloma Patients
Onyx to Host Investor Teleconference and Webcast Today at 8:00 a.m. Eastern Time to Review Top-Line Data
EMERYVILLE, Calif., July 26 /PRNewswire-FirstCall/ -- Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX) today announced positive top-line results from the Phase 2b 003-A1 study of single-agent carfilzomib, a selective, next generation proteasome inhibitor, in patients with relapsed and refractory multiple myeloma. In an independent review of the data, carfilzomib achieved an overall response rate (partial response or greater) of 24 percent and a median duration of response of 7.4 months in patients who entered the study after receiving a median of five prior lines of therapy (corresponding to a median of 13 anti-myeloma agents) and whose disease was refractory to their last therapeutic regimen. The clinical benefit rate (minimal response or greater) in the study population was 36 percent. Carfilzomib was well-tolerated and there were no new or unexpected toxicities observed. Full results of the trial will be presented at an upcoming scientific meeting. Based on these results, Onyx is continuing discussions with the U.S. Food and Drug Administration (FDA) regarding next steps in filing a new drug application (NDA) for carfilzomib, which the company expects to submit by year-end 2010 for potential accelerated approval in the U.S.
"Despite recent advances in treating multiple myeloma, all patients eventually relapse. The unmet medical need remains great, as the outlook for patients with relapsed and refractory disease is grim," said Michael G. Kauffman, M.D., Ph.D., Chief Medical Officer of Onyx Pharmaceuticals. "According to a study from the International Myeloma Working Group, patients, such as those enrolled in the 003-A1 study, can expect to respond to therapy only 11 percent of the time and survive for only six to 10 months.(i) The single-agent activity with durable disease control and favorable tolerability observed in this study indicate that carfilzomib has the potential to alter the natural course of this deadly disease."
"Carfilzomib has the potential to be an important therapy in multiple myeloma and exemplifies the Onyx vision to build a leading oncology company by developing innovative targeted therapies," said N. Anthony Coles, M.D., President and Chief Executive Officer of Onyx Pharmaceuticals. "We are committed to bringing this promising treatment to patients as quickly as possible by pursuing an accelerated approval pathway in the U.S., while simultaneously moving forward with two Phase 3 studies. The first study, ASPIRE, is designed to support full carfilzomib registration in the U.S. in earlier-stage patients who have relapsed following initial lines of therapy, and the second study is designed to support approval in relapsed and refractory patients in Europe."
Trial Design
The 003-A1 study was an open-label, single-arm Phase 2b trial. The trial evaluated 266 heavily-pretreated patients with relapsed and refractory multiple myeloma whose disease was refractory to their last treatment regimen and who had received at least two prior therapies, including bortezomib, either thalidomide or lenalidomide, an alkylating agent, glucocorticoids and an anthracycline. Refractory disease was defined as < 25% response or progression during therapy or within 60 days after completion of therapy.(ii) Patients enrolled in the 003-A1 trial had received a median of five prior therapeutic regimens, corresponding to a median of 13 anti-myeloma agents. Patients received carfilzomib at 20mg/m2 for the first cycle followed by 27mg/m2 thereafter for up to 12 cycles. Patients who completed the 12 cycles were eligible to enter an extension study. Responses and progression were determined according to the International Myeloma Working Group (IMWG) criteria. The trial was conducted in collaboration with the Multiple Myeloma Research Consortium (MMRC) and at additional sites in the U.S. and Canada.
"The patients in the 003-A1 trial represent an advanced population with significant unmet medical need who had received many lines of therapy and had limited options available to them outside of a clinical trial, strongly underscoring the need for new treatments. We are proud to have worked with the Onyx team that is developing carfilzomib and are encouraged by these results," said Kathy Giusti, Founder and CEO of the MMRC and a multiple myeloma patient. The MMRC initiated a relationship with the company (then Proteolix) in 2006, which included the participation of 11 MMRC Member Institutions in the 003-A1 trial, representing 36 percent of the total trial centers and 60 percent of enrolled patients.
"There is a high unmet need for treatment options for patients with relapsed and refractory multiple myeloma who are no longer responding to available therapies," said Brian G.M. Durie, M.D., Cofounder and Chair of the Board of Directors of the International Myeloma Foundation (IMF).
Antwort auf Beitrag Nr.: 39.876.399 von SLGramann am 26.07.10 14:42:10
Onyx will seek early approval on blockbuster cancer candidate
July 26, 2010 — 8:34am ET | By John Carroll
The multiple myeloma therapy carfilzomib registered a response in about one of four drug-resistant patients in a mid-stage trial, reports Onyx Pharmaceuticals, which the biotech believes is solid enough data to win an accelerated approval for a therapy with clear blockbuster potential. Onyx CEO Anthony Coles told reporters that a regulatory filing will be made before the end of this year.
The Phase IIb data on the proteasome inhibitor is a big win for Onyx, which scooped up the therapy when it acquired Proteolix in an $816 million buyout deal--with $276 million up front--just a year ago. And Coles says the drug could be on the market a year from now for the lethal cancer. "The unmet need in the population is so huge and the disease is almost uniformly fatal," Coles told the San Francisco Business Times.
Researchers recruited 266 patients for the trial and found that 24 percent registered at least a partial response to the therapy. The median duration of response in patients was 7.4 months. But the company declined to say for now just how long patients taking the drug survived, notes the New York Times, waiting for the right scientific conference to detail that data. For now, the company will only say that it believes carfilzomib will prolong survival. Now Onyx's blockbuster hopes will shift to the Phase III stage. Two late-stage trials are planned for the drug, including one head-to-head study that will examine the drug in combination with a low dose of the steroid dexamethasone against a Revlimid/dexamethasone combo. A European Phase III study is also planned.
As Forbes' Robert Langreth notes today, Onyx is anxious to score another blockbuster cancer drug approval to complement its success with Nexavar. Onyx has a unique status as the only sizeable standalone cancer drug developer, he adds, making the company a likely candidate for a rich buyout offer if it can come up with a new blockbuster therapy.
Onyx will seek early approval on blockbuster cancer candidate
July 26, 2010 — 8:34am ET | By John Carroll
The multiple myeloma therapy carfilzomib registered a response in about one of four drug-resistant patients in a mid-stage trial, reports Onyx Pharmaceuticals, which the biotech believes is solid enough data to win an accelerated approval for a therapy with clear blockbuster potential. Onyx CEO Anthony Coles told reporters that a regulatory filing will be made before the end of this year.
The Phase IIb data on the proteasome inhibitor is a big win for Onyx, which scooped up the therapy when it acquired Proteolix in an $816 million buyout deal--with $276 million up front--just a year ago. And Coles says the drug could be on the market a year from now for the lethal cancer. "The unmet need in the population is so huge and the disease is almost uniformly fatal," Coles told the San Francisco Business Times.
Researchers recruited 266 patients for the trial and found that 24 percent registered at least a partial response to the therapy. The median duration of response in patients was 7.4 months. But the company declined to say for now just how long patients taking the drug survived, notes the New York Times, waiting for the right scientific conference to detail that data. For now, the company will only say that it believes carfilzomib will prolong survival. Now Onyx's blockbuster hopes will shift to the Phase III stage. Two late-stage trials are planned for the drug, including one head-to-head study that will examine the drug in combination with a low dose of the steroid dexamethasone against a Revlimid/dexamethasone combo. A European Phase III study is also planned.
As Forbes' Robert Langreth notes today, Onyx is anxious to score another blockbuster cancer drug approval to complement its success with Nexavar. Onyx has a unique status as the only sizeable standalone cancer drug developer, he adds, making the company a likely candidate for a rich buyout offer if it can come up with a new blockbuster therapy.
Antwort auf Beitrag Nr.: 39.876.608 von SLGramann am 26.07.10 15:16:43
Onyx gekauft. Obwohl mir der Aufschlag heute weh tut. Hammer hats rechtzeitig gesehen und hatte Onyx gerade eben ins Depot genommen.
Onyx gekauft. Obwohl mir der Aufschlag heute weh tut. Hammer hats rechtzeitig gesehen und hatte Onyx gerade eben ins Depot genommen.
Antwort auf Beitrag Nr.: 39.876.399 von SLGramann am 26.07.10 14:42:10
hier noch ein Artikel, um die Daten besser einordnen zu können (und dann reichts damit von meiner Seite auch ):
Monday, July 26, 2010, 4:00am PDT
Onyx seeks quick OK of blood cancer drug
San Francisco Business Times - by Ron Leuty
Nearly one in four multiple myeloma patients responded to an experimental therapy from Onyx Pharmaceuticals Inc., according to initial data from a mid-stage trial, the company said.
Onyx (NASDAQ: ONXX) said results from the 266-patient trial are encouraging enough that the Emeryville-based company will seek accelerated approval from the Food and Drug Administration for the potential blockbuster drug, carfilzomib. That means the drug, which Onyx CEO Anthony Coles said could hit $1 billion in annual sales, could reach the market next year.
Although Onyx’s trial focused on the blood cancer’s sickest of the sick — those who had failed a median of five previous drug regimens that included a median of 13 anti-myeloma agents — 24 percent had a “partial response” or better. The median duration of the response was 7.4 months.
Although that means only one in four trial participants responded to carfilzomib, in the oncology world that is akin to a baseball hitter batting .400. Multiple myeloma patients typically respond to therapy only 11 percent of the time and survive for only six to 10 months.
“The unmet need in the population is so huge and the disease is almost uniformly fatal,” Coles told the San Francisco Business Times.
Patients received carfilzomib for up to 13 cycles of roughly 28 days each.
“There just aren’t enough approved therapies for this disease,” Coles said.
Millennium Pharmaceuticals’ Velcade, approved in 2003 as the first proteasome inhibitor, failed in 99 percent of those patients in Onyx’s trial, Coles said.
Carfilzomib also is a proteasome inhibitor, meaning that it blocks a large protein complex that controls the turnover of cellular proteins. Researchers found that cancer cells are especially sensitive to proteasomes being blocked.
Other approved multiple myeloma treatments include Revlimid and Thalomid, both from Celgene Corp., as well as steroids.
“Many of these patients have failed all therapies,” said Dr. Ken Anderson, chief of the division of hematologic neoplasia at Dana-Farber Cancer Center in Boston.
Coles, Anderson and officials with the Multiple Myeloma Research Foundation and Multiple Myeloma Research Consortium did not say how already-marketed drugs like Velcade performed in similar trials.
Coles said the patients in Onyx’s trial were “much sicker” than those in Velcade’s pivotal trial.
Carfilzomib’s results are significant, Anderson said in an e-mail to the San Francisco Business Times, because of the “rate and durability” of patients’ responses. The drug also appears to be safe, Anderson said.
Anderson has not been involved with carfilzomib trials, he said.
Onyx last week started a Phase III trial that will compare patients taking carfilzomib in combination with Revlimid and a low dose of the steroid dexamethasone to those taking only Revlimid and dexamethasone. That 700-patient trial is focusing on patients who had failed one to three previous drug regimens.
Onyx also intends to launch an 84-patient Phase III trial in Europe, Coles said.
If all of those trials are successful and carfilzomib wins approval in the United States and Europe for very sick multiple myeloma patients and those with an earlier stage of the disease, the drug could capture annual sales of more than $1 billion, Coles said.
Carfilzomib would be Onyx’s second drug. Nexavar, which it developed with Bayer HealthCare Pharmaceuticals, is sold in 90 countries to treat liver cancer and advanced kidney cancer and has sales of nearly $1 billion.
Carfilzomib’s success also is noteworthy, Coles said, because Onyx acquired the drug in the company’s $276 million deal in November for Proteolix Inc. of South San Francisco. That was part of Onyx’s attempt to build a richer portfolio of potential drugs.
"It's a great local story," Coles said.
hier noch ein Artikel, um die Daten besser einordnen zu können (und dann reichts damit von meiner Seite auch
):Monday, July 26, 2010, 4:00am PDT
Onyx seeks quick OK of blood cancer drug
San Francisco Business Times - by Ron Leuty
Nearly one in four multiple myeloma patients responded to an experimental therapy from Onyx Pharmaceuticals Inc., according to initial data from a mid-stage trial, the company said.
Onyx (NASDAQ: ONXX) said results from the 266-patient trial are encouraging enough that the Emeryville-based company will seek accelerated approval from the Food and Drug Administration for the potential blockbuster drug, carfilzomib. That means the drug, which Onyx CEO Anthony Coles said could hit $1 billion in annual sales, could reach the market next year.
Although Onyx’s trial focused on the blood cancer’s sickest of the sick — those who had failed a median of five previous drug regimens that included a median of 13 anti-myeloma agents — 24 percent had a “partial response” or better. The median duration of the response was 7.4 months.
Although that means only one in four trial participants responded to carfilzomib, in the oncology world that is akin to a baseball hitter batting .400. Multiple myeloma patients typically respond to therapy only 11 percent of the time and survive for only six to 10 months.
“The unmet need in the population is so huge and the disease is almost uniformly fatal,” Coles told the San Francisco Business Times.
Patients received carfilzomib for up to 13 cycles of roughly 28 days each.
“There just aren’t enough approved therapies for this disease,” Coles said.
Millennium Pharmaceuticals’ Velcade, approved in 2003 as the first proteasome inhibitor, failed in 99 percent of those patients in Onyx’s trial, Coles said.
Carfilzomib also is a proteasome inhibitor, meaning that it blocks a large protein complex that controls the turnover of cellular proteins. Researchers found that cancer cells are especially sensitive to proteasomes being blocked.
Other approved multiple myeloma treatments include Revlimid and Thalomid, both from Celgene Corp., as well as steroids.
“Many of these patients have failed all therapies,” said Dr. Ken Anderson, chief of the division of hematologic neoplasia at Dana-Farber Cancer Center in Boston.
Coles, Anderson and officials with the Multiple Myeloma Research Foundation and Multiple Myeloma Research Consortium did not say how already-marketed drugs like Velcade performed in similar trials.
Coles said the patients in Onyx’s trial were “much sicker” than those in Velcade’s pivotal trial.
Carfilzomib’s results are significant, Anderson said in an e-mail to the San Francisco Business Times, because of the “rate and durability” of patients’ responses. The drug also appears to be safe, Anderson said.
Anderson has not been involved with carfilzomib trials, he said.
Onyx last week started a Phase III trial that will compare patients taking carfilzomib in combination with Revlimid and a low dose of the steroid dexamethasone to those taking only Revlimid and dexamethasone. That 700-patient trial is focusing on patients who had failed one to three previous drug regimens.
Onyx also intends to launch an 84-patient Phase III trial in Europe, Coles said.
If all of those trials are successful and carfilzomib wins approval in the United States and Europe for very sick multiple myeloma patients and those with an earlier stage of the disease, the drug could capture annual sales of more than $1 billion, Coles said.
Carfilzomib would be Onyx’s second drug. Nexavar, which it developed with Bayer HealthCare Pharmaceuticals, is sold in 90 countries to treat liver cancer and advanced kidney cancer and has sales of nearly $1 billion.
Carfilzomib’s success also is noteworthy, Coles said, because Onyx acquired the drug in the company’s $276 million deal in November for Proteolix Inc. of South San Francisco. That was part of Onyx’s attempt to build a richer portfolio of potential drugs.
"It's a great local story," Coles said.
REGN heute mit Q-Zahlen, aber vor allem mit einer Kooperationsverlängerung:
TARRYTOWN, N.Y. and TOKYO, July 28, 2010 /PRNewswire via COMTEX News Network/ -- Regeneron Pharmaceuticals, Inc. ("Regeneron"; Nasdaq: REGN) and Astellas Pharma Inc. ("Astellas"; Headquarters: Tokyo, Japan; President & CEO: Masafumi Nogimori) announced today that Astellas has extended through 2023 the non-exclusive license agreement that allows Astellas to utilize Regeneron's VelocImmune(R) technology in its internal research programs to discover fully human monoclonal antibody product candidates.
Astellas will pay $165 million upfront and another $130 million in June 2018 unless it terminates the agreement prior to that date. Upon commercialization of any antibody products discovered utilizing VelocImmune, Astellas will pay a mid-single-digit royalty on product sales.
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=49…
TARRYTOWN, N.Y. and TOKYO, July 28, 2010 /PRNewswire via COMTEX News Network/ -- Regeneron Pharmaceuticals, Inc. ("Regeneron"; Nasdaq: REGN) and Astellas Pharma Inc. ("Astellas"; Headquarters: Tokyo, Japan; President & CEO: Masafumi Nogimori) announced today that Astellas has extended through 2023 the non-exclusive license agreement that allows Astellas to utilize Regeneron's VelocImmune(R) technology in its internal research programs to discover fully human monoclonal antibody product candidates.
Astellas will pay $165 million upfront and another $130 million in June 2018 unless it terminates the agreement prior to that date. Upon commercialization of any antibody products discovered utilizing VelocImmune, Astellas will pay a mid-single-digit royalty on product sales.
http://newsroom.regeneron.com/releasedetail.cfm?ReleaseID=49…
ArQule geht mit ARQ197 nun in die P III. Das war natürlich erwartet, aber dennoch eine schöne Nachricht. Außerdem konnte ein SPA vereinbart werden. Ich finde, dass das alles sehr zügig ging und glaube, dass bei der FDA gute Stimmung in Bezug auf ARQ197 herrscht:
(und weil ich gerade so viel glaube, glaube ich gleich noch dazu, dass im Erfolgsfall ArQule bei der Marktkapitalisierung die Milliarde knacken würde)
Press Release Source: ArQule, Inc. On Tuesday August 3, 2010, 7:00 am EDT
WOBURN, Mass. and TOKYO--(BUSINESS WIRE)--ArQule, Inc. (Nasdaq: ARQL - News) and Daiichi Sankyo Co., Ltd. (TSE 4568) today announced that they will move forward with a Phase 3 clinical trial of ARQ 197, a small molecule inhibitor of the c-Met receptor tyrosine kinase, in patients with non-small cell lung cancer (NSCLC). In connection with this decision, the sponsor company, Daiichi Sankyo, will file a Special Protocol Assessment (SPA) with the U.S. Food and Drug Administration (FDA) for a trial comparing ARQ 197 plus erlotinib against erlotinib plus placebo.
“The decision to advance ARQ 197 into Phase 3 clinical testing underscores the success of our partnership with Daiichi Sankyo,” said Paolo Pucci, chief executive officer of ArQule. “ArQule and Daiichi Sankyo signed the ARQ 197 partnership in December 2008, and in less than two years, we are requesting an SPA for a Phase 3 trial with this potential first-in-class molecule.”
“The efficacy observed among patients with NSCLC who received ARQ 197 provides us with encouraging evidence that ARQ 197 may be beneficial to this patient population. Based on these results, we are developing plans to support a Phase 3 clinical program and bring new hope to patients with this disease,” said Dr. Kazunori Hirokawa, global head of R&D Unit, Daiichi Sankyo.
An SPA is an agreement with the FDA establishing the design, endpoints and statistical analysis of a clinical trial intended to provide the necessary data to support a New Drug Application (NDA). Following FDA review of the SPA, the two companies will implement the protocol for the Phase 3 trial and commence patient enrollment. Details regarding the trial design will be communicated at that time.
This decision follows the completion of a comprehensive review of clinical and pre-clinical data, including discussions with key opinion leaders and a meeting with the FDA following the recently completed Phase 2 trial. In this trial, treatment with ARQ 197 in combination with erlotinib showed promising overall survival and progression-free survival among patients with advanced, refractory NSCLC. Data from this trial related to overall survival and progression-free survival were statistically significant in patients with non-squamous cell histology when adjusted for imbalances in key prognostic factors.
(und weil ich gerade so viel glaube, glaube ich gleich noch dazu, dass im Erfolgsfall ArQule bei der Marktkapitalisierung die Milliarde knacken würde)
Press Release Source: ArQule, Inc. On Tuesday August 3, 2010, 7:00 am EDT
WOBURN, Mass. and TOKYO--(BUSINESS WIRE)--ArQule, Inc. (Nasdaq: ARQL - News) and Daiichi Sankyo Co., Ltd. (TSE 4568) today announced that they will move forward with a Phase 3 clinical trial of ARQ 197, a small molecule inhibitor of the c-Met receptor tyrosine kinase, in patients with non-small cell lung cancer (NSCLC). In connection with this decision, the sponsor company, Daiichi Sankyo, will file a Special Protocol Assessment (SPA) with the U.S. Food and Drug Administration (FDA) for a trial comparing ARQ 197 plus erlotinib against erlotinib plus placebo.
“The decision to advance ARQ 197 into Phase 3 clinical testing underscores the success of our partnership with Daiichi Sankyo,” said Paolo Pucci, chief executive officer of ArQule. “ArQule and Daiichi Sankyo signed the ARQ 197 partnership in December 2008, and in less than two years, we are requesting an SPA for a Phase 3 trial with this potential first-in-class molecule.”
“The efficacy observed among patients with NSCLC who received ARQ 197 provides us with encouraging evidence that ARQ 197 may be beneficial to this patient population. Based on these results, we are developing plans to support a Phase 3 clinical program and bring new hope to patients with this disease,” said Dr. Kazunori Hirokawa, global head of R&D Unit, Daiichi Sankyo.
An SPA is an agreement with the FDA establishing the design, endpoints and statistical analysis of a clinical trial intended to provide the necessary data to support a New Drug Application (NDA). Following FDA review of the SPA, the two companies will implement the protocol for the Phase 3 trial and commence patient enrollment. Details regarding the trial design will be communicated at that time.
This decision follows the completion of a comprehensive review of clinical and pre-clinical data, including discussions with key opinion leaders and a meeting with the FDA following the recently completed Phase 2 trial. In this trial, treatment with ARQ 197 in combination with erlotinib showed promising overall survival and progression-free survival among patients with advanced, refractory NSCLC. Data from this trial related to overall survival and progression-free survival were statistically significant in patients with non-squamous cell histology when adjusted for imbalances in key prognostic factors.
Incyte hat bei INCB28050 die co-development-Option ausgeübt. Damit ist dieses Projekt für die künftige Entwicklung von INCY mindestens so wichtig geworden, wie INCB18424:
Paul A. Friedman, M.D., Incyte's President and Chief Executive Officer, stated, "Given the positive Phase IIa results in rheumatoid arthritis patients that we have seen for our oral JAK1/JAK2 inhibitor, INCB28050, we have elected to co-develop the compound with Lilly for this indication. As a result of this decision, we will now fund 30 percent of future development costs through regulatory approval and look forward to initiating the Phase IIb trial with Lilly later this year. This will substantially increase our royalty rate across all tiers, resulting in effective royalty rates ranging up to the high twenties on potential future global sales of INCB28050 in this indication. The commercial opportunity for a new oral therapy for rheumatoid arthritis is significant and our decision to exercise the co-development option is an important mechanism for building future shareholder value.
Paul A. Friedman, M.D., Incyte's President and Chief Executive Officer, stated, "Given the positive Phase IIa results in rheumatoid arthritis patients that we have seen for our oral JAK1/JAK2 inhibitor, INCB28050, we have elected to co-develop the compound with Lilly for this indication. As a result of this decision, we will now fund 30 percent of future development costs through regulatory approval and look forward to initiating the Phase IIb trial with Lilly later this year. This will substantially increase our royalty rate across all tiers, resulting in effective royalty rates ranging up to the high twenties on potential future global sales of INCB28050 in this indication. The commercial opportunity for a new oral therapy for rheumatoid arthritis is significant and our decision to exercise the co-development option is an important mechanism for building future shareholder value.
Ohad Hammer hatte zuletzt neben Onyx auch Synta[/b] in sein Depot aufgenommen und hat heute dazu die Begründung nachgeliefert:
http://www.hammerstockblog.com/synta-is-heating-up/
Für mich ist Synta der letzte Biotechwert, der in meinem Depot auftaucht, denn in den kommenden Jahren will ich meine inzwischen sehr umfangreiche Biotechsammlung wieder rückbauen und mich eines Tages konservativen Investments widmen.
http://www.hammerstockblog.com/synta-is-heating-up/
Für mich ist Synta der letzte Biotechwert, der in meinem Depot auftaucht, denn in den kommenden Jahren will ich meine inzwischen sehr umfangreiche Biotechsammlung wieder rückbauen und mich eines Tages konservativen Investments widmen.
zu AVEO:
Press Release Source: AVEO Pharmaceuticals, Inc. On Thursday August 12, 2010, 6:30 am EDT
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO - News), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced that it has achieved its enrollment target for TIVO-1, its global Phase 3 clinical trial for its oral, triple VEGF receptor inhibitor, tivozanib, in patients with advanced renal cell carcinoma (RCC). In February of this year, AVEO initiated enrollment in TIVO-1, and has successfully reached the target enrollment of 500 patients.
“The enrollment in this trial underscores the need for improved treatment options for this difficult-to-treat cancer,” stated Robert Motzer, M.D., attending physician at Memorial Sloan-Kettering Cancer Center and global lead TIVO-1 study investigator. “The results from this trial have the potential to provide the medical community with an understanding of tivozanib’s efficacy and tolerability in patients with advanced renal cell carcinoma.”
TIVO-1 is a global, randomized (1:1), controlled trial evaluating tivozanib compared to sorafenib. The primary endpoint of the trial is to compare the progression-free survival (PFS) of tivozanib vs. sorafenib. Secondary endpoints include overall survival, objective response rate, duration of response and quality of life. Patients with RCC of clear cell histology that have had a prior nephrectomy and that have not received prior VEGF-targeted therapy are eligible for this trial. Prior to entering the trial, independent, central review of the CT scans was required for all patients to ensure patients met the eligibility criteria regarding measureable disease. During treatment, scans are being obtained every eight weeks, and, again, are centrally reviewed by blinded independent reviewers. Patients who demonstrate disease progression during treatment with sorafenib will have the opportunity to be treated with tivozanib by participating in a separate long-term treatment protocol.
“We are very pleased to see TIVO-1 reach our enrollment target six months ahead of schedule, and I want to thank our investigators for their enthusiastic support for and tremendous commitment to the development of tivozanib,” said Tuan Ha-Ngoc, president and chief executive officer of AVEO. “We believe this successful enrollment is a tribute to the differentiated safety and efficacy data of tivozanib observed in the Phase 2 trial. Based on achieving this milestone this early, we expect data from TIVO-1 to be available in mid-2011.”
Prior to launching TIVO-1, AVEO successfully completed a 272-patient Phase 2 clinical trial of tivozanib in patients with advanced RCC. Data from the Phase 2 trial showed that the median PFS achieved by patients with advanced clear cell RCC who had undergone a prior nephrectomy, the patient population also being studied in TIVO-1, was 14.8 months – comparing favorably to historical data from trials testing other currently approved multikinase inhibitors in RCC. Hypertension, which has been proposed as a biomarker of clinical effect among agents targeting the VEGF receptor tyrosine kinases in RCC, was the most commonly reported treatment-related adverse effect, and was observed in 50% of treated patients. In the Phase 2 study, development of hypertension was directly associated with improved clinical outcomes among patients overall and in the subset of patients with clear cell RCC who had undergone a prior nephrectomy. Off-target toxicities commonly associated with other targeted therapies, such as mucositis, fatigue and hand-foot syndrome, were notably low in the tivozanib group, which AVEO believes underscores a favorable tolerability profile and potential for combinability with other therapeutic agents.
---------------------
Unglaublich, wie schnell das ging! Ein schlechtes Zeichen ist das erst mal nicht...
Press Release Source: AVEO Pharmaceuticals, Inc. On Thursday August 12, 2010, 6:30 am EDT
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO - News), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced that it has achieved its enrollment target for TIVO-1, its global Phase 3 clinical trial for its oral, triple VEGF receptor inhibitor, tivozanib, in patients with advanced renal cell carcinoma (RCC). In February of this year, AVEO initiated enrollment in TIVO-1, and has successfully reached the target enrollment of 500 patients.
“The enrollment in this trial underscores the need for improved treatment options for this difficult-to-treat cancer,” stated Robert Motzer, M.D., attending physician at Memorial Sloan-Kettering Cancer Center and global lead TIVO-1 study investigator. “The results from this trial have the potential to provide the medical community with an understanding of tivozanib’s efficacy and tolerability in patients with advanced renal cell carcinoma.”
TIVO-1 is a global, randomized (1:1), controlled trial evaluating tivozanib compared to sorafenib. The primary endpoint of the trial is to compare the progression-free survival (PFS) of tivozanib vs. sorafenib. Secondary endpoints include overall survival, objective response rate, duration of response and quality of life. Patients with RCC of clear cell histology that have had a prior nephrectomy and that have not received prior VEGF-targeted therapy are eligible for this trial. Prior to entering the trial, independent, central review of the CT scans was required for all patients to ensure patients met the eligibility criteria regarding measureable disease. During treatment, scans are being obtained every eight weeks, and, again, are centrally reviewed by blinded independent reviewers. Patients who demonstrate disease progression during treatment with sorafenib will have the opportunity to be treated with tivozanib by participating in a separate long-term treatment protocol.
“We are very pleased to see TIVO-1 reach our enrollment target six months ahead of schedule, and I want to thank our investigators for their enthusiastic support for and tremendous commitment to the development of tivozanib,” said Tuan Ha-Ngoc, president and chief executive officer of AVEO. “We believe this successful enrollment is a tribute to the differentiated safety and efficacy data of tivozanib observed in the Phase 2 trial. Based on achieving this milestone this early, we expect data from TIVO-1 to be available in mid-2011.”
Prior to launching TIVO-1, AVEO successfully completed a 272-patient Phase 2 clinical trial of tivozanib in patients with advanced RCC. Data from the Phase 2 trial showed that the median PFS achieved by patients with advanced clear cell RCC who had undergone a prior nephrectomy, the patient population also being studied in TIVO-1, was 14.8 months – comparing favorably to historical data from trials testing other currently approved multikinase inhibitors in RCC. Hypertension, which has been proposed as a biomarker of clinical effect among agents targeting the VEGF receptor tyrosine kinases in RCC, was the most commonly reported treatment-related adverse effect, and was observed in 50% of treated patients. In the Phase 2 study, development of hypertension was directly associated with improved clinical outcomes among patients overall and in the subset of patients with clear cell RCC who had undergone a prior nephrectomy. Off-target toxicities commonly associated with other targeted therapies, such as mucositis, fatigue and hand-foot syndrome, were notably low in the tivozanib group, which AVEO believes underscores a favorable tolerability profile and potential for combinability with other therapeutic agents.
---------------------
Unglaublich, wie schnell das ging! Ein schlechtes Zeichen ist das erst mal nicht...
zu ARQL:
Ohad Hammer hat sich zu dem Komplex ARQ 197 vs. Stutent + Tarceva geäußert:
Yesterday Pfizer (PFE) announced that Sutent failed to prolong survival of lung cancer patients when given in combination with Tarceva. The drug led to an increase in progression free survival (PFS), which was the secondary endpoint of the study. The results are not published and there are several open questions such as the extent of PFS improvement, benefit across different subtypes and use of Sutent in patients from the placebo cohort after progressing on Tarceva. Nevertheless, chances to see Sutent+Tarceva as a standard of care for 2nd/3rd line NSCLC are slim.
Pfizer’s failure provided some clarity for Arqule (ARQL) and its partner Daiichi Sankyo, who plan to initiate a phase III study for ARQ-197 in combination with Tarceva. The indication Arqule is pursuing is very similar to that pursued by Pfizer, so had the Sutent trial been successful, it would have adversely affect ARQ-197’s prospects in general and potentially the required clinical route.
...
hier gehts weiter:
http://www.hammerstockblog.com/pfizer%E2%80%99s-failure-prov…
Ohad Hammer hat sich zu dem Komplex ARQ 197 vs. Stutent + Tarceva geäußert:
Yesterday Pfizer (PFE) announced that Sutent failed to prolong survival of lung cancer patients when given in combination with Tarceva. The drug led to an increase in progression free survival (PFS), which was the secondary endpoint of the study. The results are not published and there are several open questions such as the extent of PFS improvement, benefit across different subtypes and use of Sutent in patients from the placebo cohort after progressing on Tarceva. Nevertheless, chances to see Sutent+Tarceva as a standard of care for 2nd/3rd line NSCLC are slim.
Pfizer’s failure provided some clarity for Arqule (ARQL) and its partner Daiichi Sankyo, who plan to initiate a phase III study for ARQ-197 in combination with Tarceva. The indication Arqule is pursuing is very similar to that pursued by Pfizer, so had the Sutent trial been successful, it would have adversely affect ARQ-197’s prospects in general and potentially the required clinical route.
...
hier gehts weiter:
http://www.hammerstockblog.com/pfizer%E2%80%99s-failure-prov…
zu IMGN:
Ablehnung der beschleunigten Zulassung von T-DM1 in der 3rd-line führte zu einem Kurseinbruch von fast 40%.
Aus meiner Sicht eher eine gute Gelegenheit, dort eine Position auf- oder auszubauen.
Hier ein Artikel dazu, den der User wachholder ausgegraben hat und der das Thema "beschleunigte Zulassung" generell behandelt:
ImmunoGen Cancer Drug Setback May Signal Delays for Onyx, Seattle Genetics
By Ellen Gibson and Catherine Larkin - Aug 27, 2010
ImmunoGen Inc.’s failure to win an accelerated U.S. review for its cancer drug sent shares down the most in eight years, and may signal delays for biotechnology companies led by Onyx Pharmaceuticals Inc., analysts say.
ImmunoGen shares plunged $3.23, or 39 percent, to $5.16 at 4 p.m. New York time in Nasdaq Stock Market composite trading after the Food and Drug Administration rejected a request from the company’s partner, Swiss drugmaker Roche Holding AG, to consider clearing the breast-cancer drug trastuzumab-DM1 based on results from mid-stage human trials.
The FDA has reconsidered fast-track approval from three marketed medicines in as many months, suggesting drugmakers may have a harder time getting new treatments cleared until longer, larger human tests are completed, said Jason Kantor, an analyst at RBC Capital Markets. Onyx and Seattle Genetics Inc. have said they will seek drug approvals from early results and may be the next companies delayed by regulators.
Today’s announcement “signals a harder line stance from the FDA against accelerated approval and approvals based on small patient numbers,” Kantor said in a telephone interview. “It’s a symptom of something that’s been going on for a while at the FDA, and it highlights the importance of defining the patient population ahead of time.”
Accelerated approval was authorized in 1992 to help spur development of medicines for HIV. The program allows companies to win conditional marketing clearance of drugs for serious illnesses based on preliminary evidence showing they work. It has been popular for makers of cancer drugs because tumor shrinkage or other physical signs that cancer is being defeated can be measured more quickly than prolonged survival.
Phase Two Results
Onyx plans to file an application for accelerated approval of its experimental tumor treatment carfilzomib this year, based on data from the second of three rounds of testing usually required by regulators, Chief Operating Officer Laure Brege said in a conference with investors on Aug. 10. In the study of 266 people with the blood cancer multiple myeloma, the drug lowered the blood concentration of a protein produced by cancer cells in 36 percent of patients.
“Today’s news about trastuzumab-DM1 does not change our plans,” Onyx spokeswoman Lori Murray said today in an e-mail. “We continue to believe there is a clear unmet medical need for patients with relapsed and refractory multiple myeloma.”
Seattle Genetics
Seattle Genetics Inc. said in a July 27 conference call with investors that it intends to apply for accelerated approval in the first half of next year for its experimental treatment for Hodgkin lymphoma. The Bothell, Washington-based company is testing its product, brentuximab vedotin, in 100 patients in collaboration with Takeda Pharmaceutical Co., of Osaka, Japan.
The shares of Onyx rose 33 cents, or 1.3 percent, to $25.18 in Nasdaq trading. Seattle Genetics increased 35 cents, or 3 percent, to $12.
The targeted antibody brentuximab, also known as SGN-35, homes in on the CD30 protein that is the marker of a Hodgkin lymphoma cell, where it unloads a cell-killing toxin. The drug uses a method similar to that employed by ImmunoGen’s treatment, known as T-DM1.
Seattle Genetics faces less approval risk than its competitors because the FDA preapproved its phase-two study design in what’s known as a special protocol assessment, Kantor said.
‘Treatment Landscape’
“We do not believe the FDA’s decision on T-DM1 has implications for Seattle Genetics,” Eric Dobmeier, a company spokesman, said in an e-mail. “In our view, the decision appears to relate to the treatment landscape for metastatic breast cancer, rather than more generally applicable principles.”
T-DM1 is an “armed antibody” that fuses Roche’s Herceptin breast-tumor medicine with a potent cancer-killing chemotherapy developed by Waltham, Massachusetts-based ImmunoGen. Herceptin acts as the guidance system, delivering the drug directly to its target.
A study last year found that T-DM1 reduced tumors in one- third of critically ill patients. Roche filed for approval of its breast-cancer drug in July using results from this trial, the second of three stages of testing usually required. The FDA denied the request, saying patients in the study had not exhausted all treatment options, Roche said in its statement.
‘By the Book’
“It seems to indicate that the FDA is playing by the book, that it’s really enforcing the regulatory standard by trying to hold accelerated approval up to the standard,” Howard Liang, an analyst with Leerink Swann & Co. in New York, said today in a telephone interview. “It’s not going to cut slack for a drug just because it’s active.”
Roche, based in Basel, Switzerland, said today that the companies expect to submit an updated application for the breast-cancer drug in mid-2012.
Companies that receive accelerated approval are required to conduct additional tests after their products are on the market to prove that they work and are safe.
Pfizer Inc. said June 21 that it would withdraw its blood cancer drug Mylotarg after 10 years on the market because studies found it didn’t work and was tied to deaths from liver and lung complications. Mylotarg was the first drug with accelerated approval to be pulled from the market, the FDA said.
Roche’s Avastin
An advisory panel recommended July 20 that the FDA rescind Roche’s accelerated approval for Avastin in breast cancer after follow-up tests failed to prove the drug slowed progression of the disease. The agency’s decision, scheduled to come by Sept. 17, may cost Roche $1 billion in annual sales, according to Sanford C. Bernstein & Co. analyst Tim Anderson.
Shire Plc said Aug. 17 that it will stop selling the blood- pressure drug ProAmatine at the end of September because studies haven’t been completed to show the treatment works. The medicine was cleared in 1996 through the accelerated approval program.
“It’s going to take a while to know if there’s some real climate trend that should cause us to be more concerned,” Ira Loss, an analyst at Washington Analysis who has followed the FDA for more than three decades, said today in a telephone interview.
The agency is “absolutely not” backtracking on the concept of accelerated approval, Loss said. “Each of these circumstances has its own characteristics and it’s not a sign of abandonment of this approach,” he said.
To contact the reporters on this story: Ellen Gibson in New York at egibson9@bloomberg.net; Catherine Larkin in Washington at clarkin4@bloomberg.net.
Ablehnung der beschleunigten Zulassung von T-DM1 in der 3rd-line führte zu einem Kurseinbruch von fast 40%.
Aus meiner Sicht eher eine gute Gelegenheit, dort eine Position auf- oder auszubauen.
Hier ein Artikel dazu, den der User wachholder ausgegraben hat und der das Thema "beschleunigte Zulassung" generell behandelt:
ImmunoGen Cancer Drug Setback May Signal Delays for Onyx, Seattle Genetics
By Ellen Gibson and Catherine Larkin - Aug 27, 2010
ImmunoGen Inc.’s failure to win an accelerated U.S. review for its cancer drug sent shares down the most in eight years, and may signal delays for biotechnology companies led by Onyx Pharmaceuticals Inc., analysts say.
ImmunoGen shares plunged $3.23, or 39 percent, to $5.16 at 4 p.m. New York time in Nasdaq Stock Market composite trading after the Food and Drug Administration rejected a request from the company’s partner, Swiss drugmaker Roche Holding AG, to consider clearing the breast-cancer drug trastuzumab-DM1 based on results from mid-stage human trials.
The FDA has reconsidered fast-track approval from three marketed medicines in as many months, suggesting drugmakers may have a harder time getting new treatments cleared until longer, larger human tests are completed, said Jason Kantor, an analyst at RBC Capital Markets. Onyx and Seattle Genetics Inc. have said they will seek drug approvals from early results and may be the next companies delayed by regulators.
Today’s announcement “signals a harder line stance from the FDA against accelerated approval and approvals based on small patient numbers,” Kantor said in a telephone interview. “It’s a symptom of something that’s been going on for a while at the FDA, and it highlights the importance of defining the patient population ahead of time.”
Accelerated approval was authorized in 1992 to help spur development of medicines for HIV. The program allows companies to win conditional marketing clearance of drugs for serious illnesses based on preliminary evidence showing they work. It has been popular for makers of cancer drugs because tumor shrinkage or other physical signs that cancer is being defeated can be measured more quickly than prolonged survival.
Phase Two Results
Onyx plans to file an application for accelerated approval of its experimental tumor treatment carfilzomib this year, based on data from the second of three rounds of testing usually required by regulators, Chief Operating Officer Laure Brege said in a conference with investors on Aug. 10. In the study of 266 people with the blood cancer multiple myeloma, the drug lowered the blood concentration of a protein produced by cancer cells in 36 percent of patients.
“Today’s news about trastuzumab-DM1 does not change our plans,” Onyx spokeswoman Lori Murray said today in an e-mail. “We continue to believe there is a clear unmet medical need for patients with relapsed and refractory multiple myeloma.”
Seattle Genetics
Seattle Genetics Inc. said in a July 27 conference call with investors that it intends to apply for accelerated approval in the first half of next year for its experimental treatment for Hodgkin lymphoma. The Bothell, Washington-based company is testing its product, brentuximab vedotin, in 100 patients in collaboration with Takeda Pharmaceutical Co., of Osaka, Japan.
The shares of Onyx rose 33 cents, or 1.3 percent, to $25.18 in Nasdaq trading. Seattle Genetics increased 35 cents, or 3 percent, to $12.
The targeted antibody brentuximab, also known as SGN-35, homes in on the CD30 protein that is the marker of a Hodgkin lymphoma cell, where it unloads a cell-killing toxin. The drug uses a method similar to that employed by ImmunoGen’s treatment, known as T-DM1.
Seattle Genetics faces less approval risk than its competitors because the FDA preapproved its phase-two study design in what’s known as a special protocol assessment, Kantor said.
‘Treatment Landscape’
“We do not believe the FDA’s decision on T-DM1 has implications for Seattle Genetics,” Eric Dobmeier, a company spokesman, said in an e-mail. “In our view, the decision appears to relate to the treatment landscape for metastatic breast cancer, rather than more generally applicable principles.”
T-DM1 is an “armed antibody” that fuses Roche’s Herceptin breast-tumor medicine with a potent cancer-killing chemotherapy developed by Waltham, Massachusetts-based ImmunoGen. Herceptin acts as the guidance system, delivering the drug directly to its target.
A study last year found that T-DM1 reduced tumors in one- third of critically ill patients. Roche filed for approval of its breast-cancer drug in July using results from this trial, the second of three stages of testing usually required. The FDA denied the request, saying patients in the study had not exhausted all treatment options, Roche said in its statement.
‘By the Book’
“It seems to indicate that the FDA is playing by the book, that it’s really enforcing the regulatory standard by trying to hold accelerated approval up to the standard,” Howard Liang, an analyst with Leerink Swann & Co. in New York, said today in a telephone interview. “It’s not going to cut slack for a drug just because it’s active.”
Roche, based in Basel, Switzerland, said today that the companies expect to submit an updated application for the breast-cancer drug in mid-2012.
Companies that receive accelerated approval are required to conduct additional tests after their products are on the market to prove that they work and are safe.
Pfizer Inc. said June 21 that it would withdraw its blood cancer drug Mylotarg after 10 years on the market because studies found it didn’t work and was tied to deaths from liver and lung complications. Mylotarg was the first drug with accelerated approval to be pulled from the market, the FDA said.
Roche’s Avastin
An advisory panel recommended July 20 that the FDA rescind Roche’s accelerated approval for Avastin in breast cancer after follow-up tests failed to prove the drug slowed progression of the disease. The agency’s decision, scheduled to come by Sept. 17, may cost Roche $1 billion in annual sales, according to Sanford C. Bernstein & Co. analyst Tim Anderson.
Shire Plc said Aug. 17 that it will stop selling the blood- pressure drug ProAmatine at the end of September because studies haven’t been completed to show the treatment works. The medicine was cleared in 1996 through the accelerated approval program.
“It’s going to take a while to know if there’s some real climate trend that should cause us to be more concerned,” Ira Loss, an analyst at Washington Analysis who has followed the FDA for more than three decades, said today in a telephone interview.
The agency is “absolutely not” backtracking on the concept of accelerated approval, Loss said. “Each of these circumstances has its own characteristics and it’s not a sign of abandonment of this approach,” he said.
To contact the reporters on this story: Ellen Gibson in New York at egibson9@bloomberg.net; Catherine Larkin in Washington at clarkin4@bloomberg.net.
wie hat sich das depot bisher in 2010 entwickelt?
schade dass du die geneart (akt. Börsenweret ca. 100 mio usd ) nicht aufgenommen hast! das war wohl der renner in 2010 bisher. auch mologen ( Börsenwert ca 125 mio) hat sich 2010 nicht schlecht entwickelt.
schade dass du die geneart (akt. Börsenweret ca. 100 mio usd ) nicht aufgenommen hast! das war wohl der renner in 2010 bisher. auch mologen ( Börsenwert ca 125 mio) hat sich 2010 nicht schlecht entwickelt.
finde deine postings sehr gut. ein dank von einem aufmerkasamen auge 

Antwort auf Beitrag Nr.: 40.063.921 von pokemon am 29.08.10 19:51:50naja, dank der Mühe von SLGramann steht in diesem Thread hin und wieder wenigstens noch etwas Neues.
die Werte, die ich hier im Depot habe, sind von der Entwicklung recht gemischt. Besonders negativ sind EXEL und ALTH gelaufen. Der Biotech-Index ist auf Jahressicht eigentlich leicht im Minus... dank der Euroschwäche reicht es aber auf Euro-Basis zu einem Plus. Unterm Strich ist die Performance des Depots nicht sonderlich erwähnenswert. Ich sehe in den einzelnen Werten noch Potential und hoffe, dass der Gesamtmarkt einmal deutlicher nach oben geht.
wie gesagt alles auf Euro-Basis (das macht bei den US-Werten jeweils ca. 13% zusätzlichen Gewinn aus) und die Angaben beziehen sich jeweils auf den Stand zu Jahresanfang...
+5,6% Depot
+10,5% NBI
-0,1% Dax
+12,8% USD
Entwicklung seit Jahresanfang (Depotanteil)
+114,5% Arena ( 4,3% )
+68,4% ViroPharma ( 3,2% )
+61,1% ArQule ( 5,3% )
+56,3% Incyte ( 10,1% )
+33,9% Cubist ( 8,5% )
+33,3% Seattle Genetics ( 4,5% )
+26,0% Ziopharm ( 3,0% )
+23,1% JAZZ ( 2,9% )
+15,5% NeuroSearch ( 3,2% )
+12,8% Array ( 3,3% )
+6,7% Micromet ( 4,8% )
+6,3% Regeneron ( 9,6% )
-3,2% Onyx ( 5,1% )
-4,3% Rigel ( 5,1% )
-8,8% AVEO ( 3,8% )
-11,3% NeurogesX ( 3,1% )
-19,2% Isis ( 3,8% )
-23,7% Medigene ( 2,4% )
-30,5% Genmab ( 4,9% )
-31,3% AMAG ( 2,9% )
-31,4% Allos ( 3,5% )
-50,1% Exelixis ( 2,7% )
werde die Tage wohl auch ein paar IMGN kaufen.
mfg ipollit
die Werte, die ich hier im Depot habe, sind von der Entwicklung recht gemischt. Besonders negativ sind EXEL und ALTH gelaufen. Der Biotech-Index ist auf Jahressicht eigentlich leicht im Minus... dank der Euroschwäche reicht es aber auf Euro-Basis zu einem Plus. Unterm Strich ist die Performance des Depots nicht sonderlich erwähnenswert. Ich sehe in den einzelnen Werten noch Potential und hoffe, dass der Gesamtmarkt einmal deutlicher nach oben geht.
wie gesagt alles auf Euro-Basis (das macht bei den US-Werten jeweils ca. 13% zusätzlichen Gewinn aus) und die Angaben beziehen sich jeweils auf den Stand zu Jahresanfang...
+5,6% Depot
+10,5% NBI
-0,1% Dax
+12,8% USD
Entwicklung seit Jahresanfang (Depotanteil)
+114,5% Arena ( 4,3% )
+68,4% ViroPharma ( 3,2% )
+61,1% ArQule ( 5,3% )
+56,3% Incyte ( 10,1% )
+33,9% Cubist ( 8,5% )
+33,3% Seattle Genetics ( 4,5% )
+26,0% Ziopharm ( 3,0% )
+23,1% JAZZ ( 2,9% )
+15,5% NeuroSearch ( 3,2% )
+12,8% Array ( 3,3% )
+6,7% Micromet ( 4,8% )
+6,3% Regeneron ( 9,6% )
-3,2% Onyx ( 5,1% )
-4,3% Rigel ( 5,1% )
-8,8% AVEO ( 3,8% )
-11,3% NeurogesX ( 3,1% )
-19,2% Isis ( 3,8% )
-23,7% Medigene ( 2,4% )
-30,5% Genmab ( 4,9% )
-31,3% AMAG ( 2,9% )
-31,4% Allos ( 3,5% )
-50,1% Exelixis ( 2,7% )
werde die Tage wohl auch ein paar IMGN kaufen.
mfg ipollit
Hier aus unserem Depotspiel die Vergleichswerte aller NBI Werte. Ebenfalls mit € Umrechnung. Der DBI bildet die wichtigsten deutschen Biotechs ab. Leider haben wir in Deutschland nur sehr wenige Werte. Der gute Schnitt vom DBI in Deutschland wird getragen durch den außerordentlichen Gewinn und Übernahme von Geneart. Ohne Geneart hätte der DBI einen Wert von ca. -11%
Biotechspiel Statistik KW 34
DBI Deutscher Biotechindex
NBI Nasdaq Biotechindex
NBI in € Nasdaq Biotechindex mit Euro-Dollar Währungsumrechnung
DAX Deutscher Aktienindex

Biotechspiel Statistik KW 34
DBI Deutscher Biotechindex
NBI Nasdaq Biotechindex
NBI in € Nasdaq Biotechindex mit Euro-Dollar Währungsumrechnung
DAX Deutscher Aktienindex
Antwort auf Beitrag Nr.: 40.064.553 von BICYPAPA am 30.08.10 06:43:31mein depot hat keine geneart, leider! 
60% MOLOGEN
20% WILEX
20% 4 SC
im spiel wäre depot 2010 auf platz 4! wobei es bei den starten vom jan. keine umschichtungen gibt.

60% MOLOGEN
20% WILEX
20% 4 SC
im spiel wäre depot 2010 auf platz 4! wobei es bei den starten vom jan. keine umschichtungen gibt.
Einige Gedanken zu AVEO, die kürzlich aus einem Tal der Depression wieder etwas aufgetaucht sind:
Aveo Pharma Stock Jumps 40 Percent in A Week, CEO Notes Progress in Pivotal Kidney Cancer Trial
Ryan McBride 8/31/10
Aveo Pharmaceuticals has grabbed the attention of investors, who have driven its stock price up by 40 percent over the past week to $8.67 per share at yesterday’s close. The Cambridge, MA-based biotech firm’s (NASDAQ:AVEO) stock is heating up as much of the rest of the market remains cool, and Aveo CEO Tuan Ha-Ngoc provided some insights about his firm that help explain why its thermostat seems to be set on high.
In recent weeks, Ha-Ngoc has been out talking to investors about his firm’s rapid progress in its Phase III clinical trial of the drug tivozanib in patients with advanced kidney cancer. Aveo issued a statement on August 12 to say that enrollment of patients into the trial was wrapped up six months ahead of the original one-year plan. This is no small deal for Aveo and its investors; the company, which has no products on the market, is now expecting to provide initial data from the pivotal study of its lead cancer drug in mid-2011 rather than late next year.
The price jump is a nice lift for Aveo, which went public in a March initial public offering priced at $9 per share rather than the earlier estimated $13-15 per share range. Aveo brought in $72.2 million in the IPO, selling investors on the promise of tivozanib and its technology that uses genetically engineered mice to help drug developers discern whether an experimental cancer drug might work in humans. It’s also the first time since late April that the small biotech firm’s stock has approached its IPO selling price. Ha-Ngoc, who is proud that his is one of the few biotech companies to break into the public markets in recent years, gave some color on the nod his firm has garnered from Wall Street in recent trading days.
“Lately, you see our stock finally get some recognition as the rest of the market goes sideways or downwards, as people start to see that maybe the company isn’t as risky as some had feared,” Ha-Ngoc said. He added that investors would rather buy into his firm’s stock now rather than wait until after the company reveals initial results of its lead kidney cancer trial, which, if positive, could drive up the price of its shares.
To talk to Ha-Ngoc is to hear insights from a biotech executive who understands how past and future events are tied together. He explained how his company stock’s upward price swing might be linked to his initial talks with public buyers—some of which occurred years before he and his colleagues officially took to the IPO trail late last year.
For example, public stock buyers always asked the CEO why they should buy Aveo’s stock in the IPO rather than waiting until the firm needs cash in a follow-on financing, when its shares could potentially be scooped up at a lower cost. Ha-Ngoc’s answer was that in the IPO the firm planned to raise (and now in fact has) enough money to operate through mid-2012, and its next stock sale wouldn’t be until after it reports top-line data from the late-stage study of tivozanib. In some cases, companies spend up to 18 months recruiting patients into large cancer drug trials, which can force some firms to go back for follow-on financing before they reach a binary event like reporting results from the trial, Ha-Ngoc says.
That won’t be the case for Aveo, though. The firm has finished recruitment for its 500-patient study (which might actually end up a 515- or 520-person trial), and its first results are expected to be available about a year before the company’s estimated current war chest of roughly $90 million is depleted. In the study, the firm is testing its drug head-to-head with Bayer’s and Onyx Pharmaceuticals’s sorafenib (Nexavar). Aveo says that patients who took its drug in its mid-stage study, which included 272 participants, had median progression-free survival of 11.8 months. Compare that with 5.5 months of medial progression-free survival for patients who took Nexavar, according to data from Onyx. Still, Aveo faces the challenge of providing similar benefits to patients in its larger Phase III trial.
The promise of tivozanib—which Aveo licensed from Japan’s Kyowa Hakko Kirin in December 2006—is to thwart a tumor’s ability to develop the blood vessels it needs to grow without causing lots of side effects. In the Phase II trial, which was completed last year, Aveo found that there were relatively low numbers of patients who experienced side effects usually associated with targeted cancer drugs, such as fatigue and digestive tract inflammation.
Presumably, investors are banking on positive results from the firm’s lead kidney cancer drug trial to drive up the value of their Aveo shares further still. Until the data is reported, we won’t know whether the stock price is destined make an even bigger jump or fall hard.
Ryan McBride is Xconomy's correspondent. You can reach him at rmcbride@xconomy.com, or follow him on Twitter at http://twitter.com/Ryan_McBride.
http://www.xconomy.com/boston/2010/08/31/aveo-pharma-stock-j…
Aveo Pharma Stock Jumps 40 Percent in A Week, CEO Notes Progress in Pivotal Kidney Cancer Trial
Ryan McBride 8/31/10
Aveo Pharmaceuticals has grabbed the attention of investors, who have driven its stock price up by 40 percent over the past week to $8.67 per share at yesterday’s close. The Cambridge, MA-based biotech firm’s (NASDAQ:AVEO) stock is heating up as much of the rest of the market remains cool, and Aveo CEO Tuan Ha-Ngoc provided some insights about his firm that help explain why its thermostat seems to be set on high.
In recent weeks, Ha-Ngoc has been out talking to investors about his firm’s rapid progress in its Phase III clinical trial of the drug tivozanib in patients with advanced kidney cancer. Aveo issued a statement on August 12 to say that enrollment of patients into the trial was wrapped up six months ahead of the original one-year plan. This is no small deal for Aveo and its investors; the company, which has no products on the market, is now expecting to provide initial data from the pivotal study of its lead cancer drug in mid-2011 rather than late next year.
The price jump is a nice lift for Aveo, which went public in a March initial public offering priced at $9 per share rather than the earlier estimated $13-15 per share range. Aveo brought in $72.2 million in the IPO, selling investors on the promise of tivozanib and its technology that uses genetically engineered mice to help drug developers discern whether an experimental cancer drug might work in humans. It’s also the first time since late April that the small biotech firm’s stock has approached its IPO selling price. Ha-Ngoc, who is proud that his is one of the few biotech companies to break into the public markets in recent years, gave some color on the nod his firm has garnered from Wall Street in recent trading days.
“Lately, you see our stock finally get some recognition as the rest of the market goes sideways or downwards, as people start to see that maybe the company isn’t as risky as some had feared,” Ha-Ngoc said. He added that investors would rather buy into his firm’s stock now rather than wait until after the company reveals initial results of its lead kidney cancer trial, which, if positive, could drive up the price of its shares.
To talk to Ha-Ngoc is to hear insights from a biotech executive who understands how past and future events are tied together. He explained how his company stock’s upward price swing might be linked to his initial talks with public buyers—some of which occurred years before he and his colleagues officially took to the IPO trail late last year.
For example, public stock buyers always asked the CEO why they should buy Aveo’s stock in the IPO rather than waiting until the firm needs cash in a follow-on financing, when its shares could potentially be scooped up at a lower cost. Ha-Ngoc’s answer was that in the IPO the firm planned to raise (and now in fact has) enough money to operate through mid-2012, and its next stock sale wouldn’t be until after it reports top-line data from the late-stage study of tivozanib. In some cases, companies spend up to 18 months recruiting patients into large cancer drug trials, which can force some firms to go back for follow-on financing before they reach a binary event like reporting results from the trial, Ha-Ngoc says.
That won’t be the case for Aveo, though. The firm has finished recruitment for its 500-patient study (which might actually end up a 515- or 520-person trial), and its first results are expected to be available about a year before the company’s estimated current war chest of roughly $90 million is depleted. In the study, the firm is testing its drug head-to-head with Bayer’s and Onyx Pharmaceuticals’s sorafenib (Nexavar). Aveo says that patients who took its drug in its mid-stage study, which included 272 participants, had median progression-free survival of 11.8 months. Compare that with 5.5 months of medial progression-free survival for patients who took Nexavar, according to data from Onyx. Still, Aveo faces the challenge of providing similar benefits to patients in its larger Phase III trial.
The promise of tivozanib—which Aveo licensed from Japan’s Kyowa Hakko Kirin in December 2006—is to thwart a tumor’s ability to develop the blood vessels it needs to grow without causing lots of side effects. In the Phase II trial, which was completed last year, Aveo found that there were relatively low numbers of patients who experienced side effects usually associated with targeted cancer drugs, such as fatigue and digestive tract inflammation.
Presumably, investors are banking on positive results from the firm’s lead kidney cancer drug trial to drive up the value of their Aveo shares further still. Until the data is reported, we won’t know whether the stock price is destined make an even bigger jump or fall hard.
Ryan McBride is Xconomy's correspondent. You can reach him at rmcbride@xconomy.com, or follow him on Twitter at http://twitter.com/Ryan_McBride.
http://www.xconomy.com/boston/2010/08/31/aveo-pharma-stock-j…
Ohad Hammer hat so ziemlich alles, was man derzeit über IMGN wissen muss zu zwei Artikeln verarbeitet:
Teil 1:
http://www.hammerstockblog.com/immunogen-what-really-happene…
Teil 2:
http://www.hammerstockblog.com/immunogen-beyond-the-t-dm1-de…
Teil 1:
http://www.hammerstockblog.com/immunogen-what-really-happene…
Teil 2:
http://www.hammerstockblog.com/immunogen-beyond-the-t-dm1-de…
ONXX... erhält ca. 60 Mio USD für die Lizenz zur Entwicklung und Vermarktung von Carfilzomib im japanischen Markt
Onyx licenses to Ono Japanese rights to cancer drug
Wed Sep 8, 2010 3:00am EDT
* Japan rights go for $59 mln upfront, total exceeds $300 mln
* Onyx to retain rights outside of Japan
By Deena Beasley
LOS ANGELES, Sept 8 (Reuters) - Onyx Pharmaceuticals Inc (ONXX.O) has sold the Japanese marketing rights for its experimental multiple myeloma drug and a related compound to Ono Pharmaceutical Inc (4528.OS) in a deal valued at more than $300 million, plus potential royalties.
The licensing agreement, which has an upfront payment of about $59 million, covers carfilzomib, which Onyx expects will soon be reviewed by U.S. regulators as a treatment for multiple myeloma, and a related drug called ONX0912.
Ono has exclusive rights to develop and commercialize both compounds for all oncology indications in Japan, while Onyx retains commercialization rights in all other regions of the world, the U.S. company said on Wednesday.
Onyx Chief Executive Officer Tony Coles said the company expects to keep rights to the drugs in the United States and Europe, but retains the flexibility to carry out licensing deals in other parts of Asia and the world.
"We like the access this provides for us in Japan," he told Reuters in a telephone interview.
In July, Onyx unveiled data in which nearly a quarter of extremely ill multiple myeloma patients who were no longer being helped by current medicines responded to carfilzomib -- a far higher rate than would have been expected.
ONX0912, an oral drug with the same mechanism of action as carfilzomib, which is given intravenously, is currently in Phase 1 testing.
Onyx said it expects to submit a new drug application for carfilzomib to the U.S. Food and Drug Administration by year-end, with a request for six-month accelerated approval.
mfg ipollit
Onyx licenses to Ono Japanese rights to cancer drug
Wed Sep 8, 2010 3:00am EDT
* Japan rights go for $59 mln upfront, total exceeds $300 mln
* Onyx to retain rights outside of Japan
By Deena Beasley
LOS ANGELES, Sept 8 (Reuters) - Onyx Pharmaceuticals Inc (ONXX.O) has sold the Japanese marketing rights for its experimental multiple myeloma drug and a related compound to Ono Pharmaceutical Inc (4528.OS) in a deal valued at more than $300 million, plus potential royalties.
The licensing agreement, which has an upfront payment of about $59 million, covers carfilzomib, which Onyx expects will soon be reviewed by U.S. regulators as a treatment for multiple myeloma, and a related drug called ONX0912.
Ono has exclusive rights to develop and commercialize both compounds for all oncology indications in Japan, while Onyx retains commercialization rights in all other regions of the world, the U.S. company said on Wednesday.
Onyx Chief Executive Officer Tony Coles said the company expects to keep rights to the drugs in the United States and Europe, but retains the flexibility to carry out licensing deals in other parts of Asia and the world.
"We like the access this provides for us in Japan," he told Reuters in a telephone interview.
In July, Onyx unveiled data in which nearly a quarter of extremely ill multiple myeloma patients who were no longer being helped by current medicines responded to carfilzomib -- a far higher rate than would have been expected.
ONX0912, an oral drug with the same mechanism of action as carfilzomib, which is given intravenously, is currently in Phase 1 testing.
Onyx said it expects to submit a new drug application for carfilzomib to the U.S. Food and Drug Administration by year-end, with a request for six-month accelerated approval.
mfg ipollit
zu SGEN:
Lintuzumab ist gescheitert. Da mit dem Programm eh kaum Hoffnungen verbunden waren, hat der Kurs nur wenig gelitten.
Bis Ende Oktober werden wir die entscheidenden Daten zu Brentuximab haben. Das darf natürlich wirklich nicht schief gehen, sonst wirds finster.
(Damit sind übrigens beide "naked" mabs bei SGEN gescheitert, wenn man Dacetuzumab als tot ansehen will, was wohl vernünftig ist. SGEN ist jetzt ein reiner ADC-Play. Ohne die Misserfolge SGENs jetzt zu sehr zu verallgemeinern zu wollen, habe ich schon den Eindruck, dass die ganz großen Erfolge der herkömmlichen mab-Technologie schon Geschichte sind... oder glaubt jemand an ein zweites Avastin oder Herceptin auf dieser Basis?)
UPDATE 1-Seattle Genetics says leukemia drug fails trial
Mon Sep 13, 2010 11:18am EDT
* Lintuzumab development program dropped
* Results of lymphoma trials expected within next 6 weeks
* Shares fall 4 percent (Adds analyst comment, share price, byline)
By Deena Beasley
LOS ANGELES, Sept 13 (Reuters) - Seattle Genetics (SGEN.O) said a key trial of its experimental drug lintuzumab showed that the biotechnology treatment did not extend the lives of patients with acute myeloid leukemia, sending its shares down 4 percent.
The company, based in Bothell, Washington, said on Monday it would discontinue development of the drug.
Wall Street analysts had modest expectations for lintuzumab, which was licensed by Seattle Genetics in 2005 after PDL BioPharma Inc (PDLI.O) decided not to pursue the antibody following disappointing results from leukemia trials using a lower dose of the drug.
"The bottom line is that this is a minor disappointment that removes a potentially high-reward call option from the story," JP Morgan analyst Cory Kasimov said in a research note.
The Phase IIb study was designed to show whether lintuzumab, or SGN33, in combination with low-dose chemotherapy, improved survival for people over the age of 60 who have acute myeloid leukemia, the most common type of the blood cancer in adults, compared to chemotherapy alone.
Seattle Genetics said the difference in survival between the treatment arms was not statistically significant, and patients in both groups lived longer than had been seen in previous studies of the chemotherapy drug used in the trial.
"We continue to focus on advancing our lead program, brentuximab vedotin, or SGN35," Clay Siegall, the company's chief executive officer, told Reuters in a telephone interview.
He expects to have data from a pivotal trial of brentuximab vedotin in patients with Hodgkin lymphoma within the next few weeks, followed a few weeks later by results from a different Phase 3 trial in patients with anaplastic large cell lymphoma (ALCL).
Kasimov at JP Morgan said any near-term selling pressure on Seattle Genetics would be mitigated by greater optimism for pivotal SGN35 data due by the end of October.
Unlike lintuzumab, which is an older "naked" antibody, SGN35 is a conjugate made up of a tumor-targeting antibody linked to a chemotherapy drug called monomethyl auristatin E.
It targets an antigen expressed on Hodgkin lymphoma (HL), several types of T-cell lymphoma and other hematologic malignancies.
The company is developing the drug in partnership with Takeda Pharmaceutical Co Ltd (4502.T).
Seattle Genetics plans to file for U.S. regulatory approval of brentuximab vedotin as a treatment for HL in the first half of 2011 and may also file for ALCL at the same time, depending on results from the upcoming trial.
Shares of Seattle Genetics were down 51 cents at $12.21 in morning trading on Nasdaq. (Reporting by Deena Beasley, editing by Derek Caney, Dave Zimmerman)
Lintuzumab ist gescheitert. Da mit dem Programm eh kaum Hoffnungen verbunden waren, hat der Kurs nur wenig gelitten.
Bis Ende Oktober werden wir die entscheidenden Daten zu Brentuximab haben. Das darf natürlich wirklich nicht schief gehen, sonst wirds finster.
(Damit sind übrigens beide "naked" mabs bei SGEN gescheitert, wenn man Dacetuzumab als tot ansehen will, was wohl vernünftig ist. SGEN ist jetzt ein reiner ADC-Play. Ohne die Misserfolge SGENs jetzt zu sehr zu verallgemeinern zu wollen, habe ich schon den Eindruck, dass die ganz großen Erfolge der herkömmlichen mab-Technologie schon Geschichte sind... oder glaubt jemand an ein zweites Avastin oder Herceptin auf dieser Basis?)
UPDATE 1-Seattle Genetics says leukemia drug fails trial
Mon Sep 13, 2010 11:18am EDT
* Lintuzumab development program dropped
* Results of lymphoma trials expected within next 6 weeks
* Shares fall 4 percent (Adds analyst comment, share price, byline)
By Deena Beasley
LOS ANGELES, Sept 13 (Reuters) - Seattle Genetics (SGEN.O) said a key trial of its experimental drug lintuzumab showed that the biotechnology treatment did not extend the lives of patients with acute myeloid leukemia, sending its shares down 4 percent.
The company, based in Bothell, Washington, said on Monday it would discontinue development of the drug.
Wall Street analysts had modest expectations for lintuzumab, which was licensed by Seattle Genetics in 2005 after PDL BioPharma Inc (PDLI.O) decided not to pursue the antibody following disappointing results from leukemia trials using a lower dose of the drug.
"The bottom line is that this is a minor disappointment that removes a potentially high-reward call option from the story," JP Morgan analyst Cory Kasimov said in a research note.
The Phase IIb study was designed to show whether lintuzumab, or SGN33, in combination with low-dose chemotherapy, improved survival for people over the age of 60 who have acute myeloid leukemia, the most common type of the blood cancer in adults, compared to chemotherapy alone.
Seattle Genetics said the difference in survival between the treatment arms was not statistically significant, and patients in both groups lived longer than had been seen in previous studies of the chemotherapy drug used in the trial.
"We continue to focus on advancing our lead program, brentuximab vedotin, or SGN35," Clay Siegall, the company's chief executive officer, told Reuters in a telephone interview.
He expects to have data from a pivotal trial of brentuximab vedotin in patients with Hodgkin lymphoma within the next few weeks, followed a few weeks later by results from a different Phase 3 trial in patients with anaplastic large cell lymphoma (ALCL).
Kasimov at JP Morgan said any near-term selling pressure on Seattle Genetics would be mitigated by greater optimism for pivotal SGN35 data due by the end of October.
Unlike lintuzumab, which is an older "naked" antibody, SGN35 is a conjugate made up of a tumor-targeting antibody linked to a chemotherapy drug called monomethyl auristatin E.
It targets an antigen expressed on Hodgkin lymphoma (HL), several types of T-cell lymphoma and other hematologic malignancies.
The company is developing the drug in partnership with Takeda Pharmaceutical Co Ltd (4502.T).
Seattle Genetics plans to file for U.S. regulatory approval of brentuximab vedotin as a treatment for HL in the first half of 2011 and may also file for ALCL at the same time, depending on results from the upcoming trial.
Shares of Seattle Genetics were down 51 cents at $12.21 in morning trading on Nasdaq. (Reporting by Deena Beasley, editing by Derek Caney, Dave Zimmerman)
zu INCY:
Die bauen 18424 systematisch aus und haben jetzt ein SPA für die P III in Polycythemia Vera bekommen.
Hoffentlich werden sie nächstes Jahr auch eine P III bei Essential thrombocythemia (ET) auflegen. Bis Ende des Jahres könnten zunächst aber mal die Daten für Myelofibrose vorliegen.
Press Release Source: Incyte Corporation On Monday September 13, 2010, 7:00 am EDT
WILMINGTON, Del.--(BUSINESS WIRE)--Incyte Corporation (Nasdaq:INCY - News) announced today that it has reached agreement with the U.S. Food and Drug Administration (FDA) regarding a Special Protocol Assessment (SPA) for the design of a pivotal Phase III trial for its JAK1 and JAK2 inhibitor, INCB18424 in patients with polycythemia vera (PV), a blood cancer that belongs to a group of diseases known as myeloproliferative neoplasms (MPNs). Two Phase III trials of INCB18424 in myelofibrosis, also an MPN, COMFORT-I and COMFORT-II, are already fully enrolled and are expected to be completed later this year.
RESPONSE (Randomized, open label, multicenter phase III study of Efficacy and Safety in POlycythemia vera subjects who are resistant to or intolerant of hydroxyurea: JAK iNhibitor INC424 tablets verSus bEst available care) is a global study conducted by Incyte in the US and Novartis in rest of world and is expected to enroll approximately 300 patients with PV who are resistant to or intolerant of hydroxyurea (HU). RESPONSE will compare the efficacy and safety of INCB18424 to the physician’s choice of best available therapy. Patient enrollment in the US is expected to begin in October (www.responsetrial.com).
“Securing the SPA for INCB18424 in PV establishes the requirements we must meet to obtain approval in this second indication and supports our objective to expand beyond myelofibrosis,” stated Paul A. Friedman, M.D., Incyte’s President and CEO. “The encouraging results from the ongoing Phase II PV trial, combined with the data we anticipate achieving from RESPONSE, have the potential to clearly establish the long-term safety and efficacy of INCB18424 and optimally position the compound for use in these underserved patients.”
Srdan Verstovsek, M.D., Ph.D., Associate Professor, Leukemia Department, Myeloproliferative Disorders Program Leader, University of Texas M.D. Anderson Cancer Center, and the US principal investigator for RESPONSE, stated, “Patients with advanced PV represent a particularly high-risk group with few therapeutic options for long-term care. These patients would benefit from new chronic therapies that safely and effectively address the aberrant hematological measures, enlarged spleens and significant disease-related symptoms that so negatively impact their lives. In previous trials, INCB18424 has been well tolerated in patients with advanced PV, and much sicker patients with myelofibrosis for as long as 30 months. In the Phase II trial involving 34 PV patients who were HU resistant or HU intolerant, INCB18424 provided rapid and durable activity in these patients. We look forward to evaluating INCB18424 in this Phase III trial designed in agreement with FDA.”
RESPONSE Trial
Design
RESPONSE is a global, randomized, open-label, multi-center Phase III trial that will compare the efficacy and safety of the oral JAK1 and JAK2 inhibitor INCB18424 to best available therapy in approximately 300 patients with PV. To be eligible for the study, patients must be resistant to or intolerant of HU, require phlebotomy based on inadequate hematocrit control at least once every three months, possess an elevated white blood cell count and/or platelet count and exhibit a palpated spleen length of 5 cm or greater.
Primary efficacy endpoint
The proportion of patients treated with INCB18424 versus best available therapy achieving a response at week 32, with response defined as having achieved both of the following:
* the absence of phlebotomy and
* a ≥35% reduction in spleen volume as assessed by imaging
Key secondary efficacy endpoints
* Proportion of patients treated with INCB18424 versus best available therapy who both achieve the week 32 primary response endpoint and maintain response for 48 weeks from the time that response was initially documented
* Proportion of patients treated with INCB18424 versus best available therapy achieving complete hematologic remission at week 32
Duration of trial
The randomized, controlled period of the trial is 32 weeks. Data are scheduled to be analyzed when the last patient remaining in the study has completed week 80 (i.e. 48 weeks following the primary endpoint at week 32) to enable the assessment of response durability. Patients receiving benefit from INCB18424 at week 80 will be eligible to continue to receive INCB18424 in a separate open-label extension study.
Best available therapy
Therapy will be selected by the investigator on a patient-by-patient basis among the following six options: interferon/pegylated interferon, pipobroman, anagrelide, immunomodulators (imids such as lenalidomide and thalidomide), HU (at a tolerated dose if, in the opinion of the investigator, the patient is likely to receive benefit), and observation only (i.e. absence of treatment apart from aspirin and phlebotomy when the subject is eligible). The dose and administration schedule of initial best available therapy may be changed at any time, however the initial therapy may only be changed if specific treatment discontinuation or disease progression criteria are met.
Cross-over from best available therapy to INCB18424
Patients randomized to best available therapy, who fail to meet the primary endpoint at week 32 or whose response ends subsequent to week 32, may be eligible to cross over to receive INCB18424.
Current Approach to Treating PV
The current therapeutic approach in PV focuses on lowering the risk for thrombotic events without exposing patients to increased risk of leukemic transformation1. While phlebotomy and low dose aspirin are accepted as the standard of care for initial therapy, cytoreductive therapy is recommended to aid in the control of erythrocytosis in the presence of poor tolerance to phlebotomy, symptomatic or progressive splenomegaly, or evidence of high thrombotic risk2. Though not FDA approved for use in PV, HU is the most frequently used myelosuppressive agent for initial cytoreductive therapy2. HU treatment is associated with cytopenias and often unsatisfactory hematological control over time, aphthous and leg ulcers, multiple other toxicities, and risk of leukemia estimated at up to 10% at the 13th year3,4. Therapeutic options beyond HU are limited, and as a consequence, it is estimated that up to 20% of patients may continue on HU even though response is less than satisfactory4.
About Special Protocol Assessment
The SPA is a process that allows for official FDA evaluation of the clinical protocols of a Phase III clinical trial intended to form the primary basis for an efficacy claim and provides trial sponsors with a written agreement that the design and analysis of the trial are adequate to support a New Drug Application (NDA) if the trial is performed according to the SPA. Final marketing approval depends on the results of efficacy, the adverse event profile and on an evaluation of the benefit/risk of treatment demonstrated in the Phase III trial. The SPA agreement may only be changed through a written agreement between the sponsor and the FDA, or if the FDA becomes aware of a substantial scientific issue essential to product efficacy or safety. For more information on Special Protocol Assessment, please visit the FDA web site at www.FDA.gov.
About PV and INCB18424
PV is a characterized by the overproduction of red blood cells which increases blood viscosity and leads to elevated thromboembolic risk. Increased levels of white blood cells and platelets are also common. In advanced disease, patients frequently exhibit spleen enlargement and debilitating symptoms including pruritus, night sweats, fatigue, and muscle and bone pain. In the Phase II trial, as presented at the American Society of Hematology Annual Meeting, December 2009, clinical benefits associated with INCB18424 treatment in the 34 PV patients enrolled in the study included hematocrit control without the need for phlebotomy, normalization of white blood cell and platelet counts, and rapid and durable reductions in enlarged spleens and symptoms, particularly pruritus. INCB18424 was generally well tolerated in these patients, and the most common adverse events observed involved low blood cell counts which, in the majority of patients, were reversed by dose reduction.
About the Incyte Novartis Alliance
In November 2009, Incyte and Novartis announced a major collaboration and license agreement for two hematology-oncology programs in which Incyte retained exclusive rights to develop and commercialize INCB18424 in the US and Novartis received exclusive rights to develop and commercialize INCB18424 for territories outside the US. Novartis also received worldwide rights for Incyte’s cMET Inhibitor, INCB28060.
Die bauen 18424 systematisch aus und haben jetzt ein SPA für die P III in Polycythemia Vera bekommen.
Hoffentlich werden sie nächstes Jahr auch eine P III bei Essential thrombocythemia (ET) auflegen. Bis Ende des Jahres könnten zunächst aber mal die Daten für Myelofibrose vorliegen.
Press Release Source: Incyte Corporation On Monday September 13, 2010, 7:00 am EDT
WILMINGTON, Del.--(BUSINESS WIRE)--Incyte Corporation (Nasdaq:INCY - News) announced today that it has reached agreement with the U.S. Food and Drug Administration (FDA) regarding a Special Protocol Assessment (SPA) for the design of a pivotal Phase III trial for its JAK1 and JAK2 inhibitor, INCB18424 in patients with polycythemia vera (PV), a blood cancer that belongs to a group of diseases known as myeloproliferative neoplasms (MPNs). Two Phase III trials of INCB18424 in myelofibrosis, also an MPN, COMFORT-I and COMFORT-II, are already fully enrolled and are expected to be completed later this year.
RESPONSE (Randomized, open label, multicenter phase III study of Efficacy and Safety in POlycythemia vera subjects who are resistant to or intolerant of hydroxyurea: JAK iNhibitor INC424 tablets verSus bEst available care) is a global study conducted by Incyte in the US and Novartis in rest of world and is expected to enroll approximately 300 patients with PV who are resistant to or intolerant of hydroxyurea (HU). RESPONSE will compare the efficacy and safety of INCB18424 to the physician’s choice of best available therapy. Patient enrollment in the US is expected to begin in October (www.responsetrial.com).
“Securing the SPA for INCB18424 in PV establishes the requirements we must meet to obtain approval in this second indication and supports our objective to expand beyond myelofibrosis,” stated Paul A. Friedman, M.D., Incyte’s President and CEO. “The encouraging results from the ongoing Phase II PV trial, combined with the data we anticipate achieving from RESPONSE, have the potential to clearly establish the long-term safety and efficacy of INCB18424 and optimally position the compound for use in these underserved patients.”
Srdan Verstovsek, M.D., Ph.D., Associate Professor, Leukemia Department, Myeloproliferative Disorders Program Leader, University of Texas M.D. Anderson Cancer Center, and the US principal investigator for RESPONSE, stated, “Patients with advanced PV represent a particularly high-risk group with few therapeutic options for long-term care. These patients would benefit from new chronic therapies that safely and effectively address the aberrant hematological measures, enlarged spleens and significant disease-related symptoms that so negatively impact their lives. In previous trials, INCB18424 has been well tolerated in patients with advanced PV, and much sicker patients with myelofibrosis for as long as 30 months. In the Phase II trial involving 34 PV patients who were HU resistant or HU intolerant, INCB18424 provided rapid and durable activity in these patients. We look forward to evaluating INCB18424 in this Phase III trial designed in agreement with FDA.”
RESPONSE Trial
Design
RESPONSE is a global, randomized, open-label, multi-center Phase III trial that will compare the efficacy and safety of the oral JAK1 and JAK2 inhibitor INCB18424 to best available therapy in approximately 300 patients with PV. To be eligible for the study, patients must be resistant to or intolerant of HU, require phlebotomy based on inadequate hematocrit control at least once every three months, possess an elevated white blood cell count and/or platelet count and exhibit a palpated spleen length of 5 cm or greater.
Primary efficacy endpoint
The proportion of patients treated with INCB18424 versus best available therapy achieving a response at week 32, with response defined as having achieved both of the following:
* the absence of phlebotomy and
* a ≥35% reduction in spleen volume as assessed by imaging
Key secondary efficacy endpoints
* Proportion of patients treated with INCB18424 versus best available therapy who both achieve the week 32 primary response endpoint and maintain response for 48 weeks from the time that response was initially documented
* Proportion of patients treated with INCB18424 versus best available therapy achieving complete hematologic remission at week 32
Duration of trial
The randomized, controlled period of the trial is 32 weeks. Data are scheduled to be analyzed when the last patient remaining in the study has completed week 80 (i.e. 48 weeks following the primary endpoint at week 32) to enable the assessment of response durability. Patients receiving benefit from INCB18424 at week 80 will be eligible to continue to receive INCB18424 in a separate open-label extension study.
Best available therapy
Therapy will be selected by the investigator on a patient-by-patient basis among the following six options: interferon/pegylated interferon, pipobroman, anagrelide, immunomodulators (imids such as lenalidomide and thalidomide), HU (at a tolerated dose if, in the opinion of the investigator, the patient is likely to receive benefit), and observation only (i.e. absence of treatment apart from aspirin and phlebotomy when the subject is eligible). The dose and administration schedule of initial best available therapy may be changed at any time, however the initial therapy may only be changed if specific treatment discontinuation or disease progression criteria are met.
Cross-over from best available therapy to INCB18424
Patients randomized to best available therapy, who fail to meet the primary endpoint at week 32 or whose response ends subsequent to week 32, may be eligible to cross over to receive INCB18424.
Current Approach to Treating PV
The current therapeutic approach in PV focuses on lowering the risk for thrombotic events without exposing patients to increased risk of leukemic transformation1. While phlebotomy and low dose aspirin are accepted as the standard of care for initial therapy, cytoreductive therapy is recommended to aid in the control of erythrocytosis in the presence of poor tolerance to phlebotomy, symptomatic or progressive splenomegaly, or evidence of high thrombotic risk2. Though not FDA approved for use in PV, HU is the most frequently used myelosuppressive agent for initial cytoreductive therapy2. HU treatment is associated with cytopenias and often unsatisfactory hematological control over time, aphthous and leg ulcers, multiple other toxicities, and risk of leukemia estimated at up to 10% at the 13th year3,4. Therapeutic options beyond HU are limited, and as a consequence, it is estimated that up to 20% of patients may continue on HU even though response is less than satisfactory4.
About Special Protocol Assessment
The SPA is a process that allows for official FDA evaluation of the clinical protocols of a Phase III clinical trial intended to form the primary basis for an efficacy claim and provides trial sponsors with a written agreement that the design and analysis of the trial are adequate to support a New Drug Application (NDA) if the trial is performed according to the SPA. Final marketing approval depends on the results of efficacy, the adverse event profile and on an evaluation of the benefit/risk of treatment demonstrated in the Phase III trial. The SPA agreement may only be changed through a written agreement between the sponsor and the FDA, or if the FDA becomes aware of a substantial scientific issue essential to product efficacy or safety. For more information on Special Protocol Assessment, please visit the FDA web site at www.FDA.gov.
About PV and INCB18424
PV is a characterized by the overproduction of red blood cells which increases blood viscosity and leads to elevated thromboembolic risk. Increased levels of white blood cells and platelets are also common. In advanced disease, patients frequently exhibit spleen enlargement and debilitating symptoms including pruritus, night sweats, fatigue, and muscle and bone pain. In the Phase II trial, as presented at the American Society of Hematology Annual Meeting, December 2009, clinical benefits associated with INCB18424 treatment in the 34 PV patients enrolled in the study included hematocrit control without the need for phlebotomy, normalization of white blood cell and platelet counts, and rapid and durable reductions in enlarged spleens and symptoms, particularly pruritus. INCB18424 was generally well tolerated in these patients, and the most common adverse events observed involved low blood cell counts which, in the majority of patients, were reversed by dose reduction.
About the Incyte Novartis Alliance
In November 2009, Incyte and Novartis announced a major collaboration and license agreement for two hematology-oncology programs in which Incyte retained exclusive rights to develop and commercialize INCB18424 in the US and Novartis received exclusive rights to develop and commercialize INCB18424 for territories outside the US. Novartis also received worldwide rights for Incyte’s cMET Inhibitor, INCB28060.
obschon Synta nicht in ipollits Depot ist, eine kleine Anmerkung. Die haben gestern einen netten Sprung getan aufgrund dieser Meldung:
LEXINGTON, Mass. (AP) -- Shares of Synta Pharmaceuticals Corp. surged 15 percent on Monday after the company said it is expanding a clinical trial of a lung cancer drug after an initial stage of testing produced encouraging results.
Shares added 48 cents to $3.65 in afternoon trading. The stock has ranged between $2.55 and $6.95 over the past 52 weeks.
Synta said it will broaden its phase-two clinical trial of STA-9090 to 146 patients from 69. The participants in the trial are in certain stages of non-small-cell lung cancer.
STA-9090 is a potent, second-generation, small-molecule Hsp90 inhibitor, the company said.
The company aims to identify cancer types that will be especially responsive to treatment with STA-9090.
In the first phase of clinical trials, more than 70 percent of patients showed "a high disease-control rate," said Vojo Vukovic, Synta Pharmaceutical's chief medical officer.
"This early signal, combined with the objective responses seen following treatment with STA-9090, is very encouraging, particularly as the patients have been heavily pretreated and are refractory to many standard-of-care drugs," he added.
Die Hintergründe zum Unternehmen hatte Hammer hier dargestellt:
http://www.hammerstockblog.com/synta-is-heating-up/
Irgendwie sieht das schon "lustig" aus, mit welcher Gewalt Synta das HSP90-Projekt vorantreibt. Da laufen jetzt 12 Indikationen parallel und bis zum Jahresende sollen es bis zu 15 Trials sein!?

STA-9090 is currently being evaluated in eleven clinical trials: seven Phase 2 trials in solid tumor cancers - non-small cell lung cancer, gastrointestinal stromal tumors, colon cancer, gastric cancer, and small cell lung cancer; and five Phase 1 and Phase 1/2 trials in hematologic and solid tumor cancers.
LEXINGTON, Mass. (AP) -- Shares of Synta Pharmaceuticals Corp. surged 15 percent on Monday after the company said it is expanding a clinical trial of a lung cancer drug after an initial stage of testing produced encouraging results.
Shares added 48 cents to $3.65 in afternoon trading. The stock has ranged between $2.55 and $6.95 over the past 52 weeks.
Synta said it will broaden its phase-two clinical trial of STA-9090 to 146 patients from 69. The participants in the trial are in certain stages of non-small-cell lung cancer.
STA-9090 is a potent, second-generation, small-molecule Hsp90 inhibitor, the company said.
The company aims to identify cancer types that will be especially responsive to treatment with STA-9090.
In the first phase of clinical trials, more than 70 percent of patients showed "a high disease-control rate," said Vojo Vukovic, Synta Pharmaceutical's chief medical officer.
"This early signal, combined with the objective responses seen following treatment with STA-9090, is very encouraging, particularly as the patients have been heavily pretreated and are refractory to many standard-of-care drugs," he added.
Die Hintergründe zum Unternehmen hatte Hammer hier dargestellt:
http://www.hammerstockblog.com/synta-is-heating-up/
Irgendwie sieht das schon "lustig" aus, mit welcher Gewalt Synta das HSP90-Projekt vorantreibt. Da laufen jetzt 12 Indikationen parallel und bis zum Jahresende sollen es bis zu 15 Trials sein!?
STA-9090 is currently being evaluated in eleven clinical trials: seven Phase 2 trials in solid tumor cancers - non-small cell lung cancer, gastrointestinal stromal tumors, colon cancer, gastric cancer, and small cell lung cancer; and five Phase 1 and Phase 1/2 trials in hematologic and solid tumor cancers.
Antwort auf Beitrag Nr.: 40.144.981 von SLGramann am 14.09.10 07:52:22
zu INCYs 18424 ist ein Artikel bei Pharma Strategy Blog erschienen:
http://www.pharmastrategyblog.com/2010/09/incytes-incb018424…
Die allgemein sehr positive Einstellung zu dem Projekt wird hier geteilt. Im Kurs von Incyte ist ein Erfolg stark eingepreist. Das "darf" echt nicht schiefgehen...
zu INCYs 18424 ist ein Artikel bei Pharma Strategy Blog erschienen:
http://www.pharmastrategyblog.com/2010/09/incytes-incb018424…
Die allgemein sehr positive Einstellung zu dem Projekt wird hier geteilt. Im Kurs von Incyte ist ein Erfolg stark eingepreist. Das "darf" echt nicht schiefgehen...
Erfolg für SGEN mit Brentuximab!
Seattle Genetics and Millennium Announce Positive Top-Line Brentuximab Vedotin (SGN-35) Data from Pivotal Trial in Relapsed and Refractory Hodgkin Lymphoma
-Objective Responses Achieved in 75 percent of Patients; Median Duration of Response Greater than Six Months-
-BLA Submission Planned in First Half of 2011--Seattle Genetics to Host Conference Call and Webcast Today at 8:30 a.m. ET-
BOTHELL, Wash. & CAMBRIDGE, Mass., Sep 27, 2010 (BUSINESS WIRE) --
Seattle Genetics, Inc. (Nasdaq: SGEN) and Millennium: The Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited (TSE:4502), today announced positive top-line results from the pivotal trial of single-agent brentuximab vedotin (SGN-35), an antibody-drug conjugate (ADC) targeted to CD30. The trial was conducted in 102 relapsed or refractory Hodgkin lymphoma (HL) patients.
Seventy-five percent of patients in the pivotal trial achieved an objective response as assessed by an independent central review, the primary endpoint of the trial. The median duration of response was greater than six months. The safety profile of brentuximab vedotin in this trial was generally consistent with prior clinical trial experience. A more complete data set will be presented at an upcoming scientific meeting.
"We are extremely excited with the top-line results, as they move us one step closer to our goal of bringing brentuximab vedotin to patients with relapsed or refractory Hodgkin lymphoma," said Clay B. Siegall, Ph.D., President and Chief Executive Officer of Seattle Genetics. "We are positioned for a Biologics License Application (BLA) submission to the U.S. Food and Drug Administration (FDA) in the first half of 2011. In addition, we plan to report top-line data from our phase II trial of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma (ALCL) within the next few weeks."
"The lack of adequate therapies for the treatment of relapsed and refractory Hodgkin lymphoma represents a substantial unmet medical need worldwide, with almost a third of the 30,000 newly diagnosed patients relapsing or becoming refractory to front-line therapy annually," said Nancy Simonian, M.D., Chief Medical Officer of Millennium. "These data have the potential to provide an important advance in therapy for Hodgkin lymphoma. We intend to discuss these results with European regulators to support our goal of submitting a Marketing Authorization Application to the European Medicines Agency (EMA) in 2011."
Pivotal Trial Design
The single-arm pivotal trial assessed efficacy and safety of single-agent brentuximab vedotin in relapsed or refractory, post-autologous stem cell transplant (ASCT) HL patients. Patients received 1.8 milligrams per kilogram of brentuximab vedotin every three weeks for up to 16 total doses. The primary endpoint of the trial was objective response rate as assessed by an independent review facility. Response assessments were based on the rigorous and internationally established Revised Response Criteria for Malignant Lymphoma (Cheson, 2007). Secondary endpoints included complete response rate, duration of response, progression-free survival, overall survival and tolerability. The trial was conducted under a Special Protocol Assessment (SPA) with the FDA and was discussed with the EMA during the process of obtaining EU Centralized Scientific Advice on the brentuximab vedotin development program. Brentuximab vedotin has been granted orphan drug designation by the FDA and EMA for the treatment of HL and ALCL and has been granted fast track designation by the FDA for HL.
About Brentuximab Vedotin
Brentuximab vedotin is an ADC comprising an anti-CD30 monoclonal antibody attached by an enzyme cleavable linker to a potent, synthetic drug payload, monomethyl auristatin E (MMAE) utilizing Seattle Genetics' proprietary technology. The ADC employs a novel linker system that is designed to be stable in the bloodstream but to release MMAE upon internalization into CD30-expressing tumor cells. This approach is intended to spare non-targeted cells and thus may help minimize the potential toxic effects of traditional chemotherapy while allowing for the selective targeting of CD30-expressing cancer cells, thus potentially enhancing the antitumor activity.
In addition to the pivotal HL trial, Seattle Genetics and Millennium are conducting a phase II trial for relapsed and refractory systemic ALCL, a phase III clinical trial (the AETHERA trial) for patients at high risk of residual HL following autologous stem cell transplant, a phase II retreatment trial for relapsed patients who previously responded to brentuximab vedotin, and a phase I combination trial for front-line treatment of HL.
About Hodgkin Lymphoma
Lymphoma is a general term for a group of cancers that originate in the lymphatic system. There are two major categories of lymphoma: Hodgkin lymphoma and non-Hodgkin lymphoma. Hodgkin lymphoma is distinguished from other types of lymphoma by the presence of one characteristic type of cell, known as the Reed-Sternberg cell. A defining attribute of the Reed-Sternberg cell is its expression of the CD30 antigen.
According to the American Cancer Society, approximately 8,500 cases of Hodgkin lymphoma will be diagnosed in the United States during 2010 and more than 1,300 will die from the disease. Globally, there are more than 30,000 cases of Hodgkin lymphoma diagnosed each year. Although front-line combination chemotherapy can result in durable response rates, up to 30 percent of these patients relapse or are refractory to front-line treatment and have few therapeutic options beyond ASCT.
About the Seattle Genetics/Millennium Collaboration
Seattle Genetics and Millennium are jointly developing brentuximab vedotin. Under the terms of the collaboration agreement, Seattle Genetics has U.S. and Canadian commercialization rights and the Takeda Group has rights to commercialize brentuximab vedotin in the rest of the world. Seattle Genetics and the Takeda Group are funding joint development costs for brentuximab vedotin on a 50:50 basis, except in Japan where the Takeda Group will be solely responsible for development costs.
Seattle Genetics and Millennium Announce Positive Top-Line Brentuximab Vedotin (SGN-35) Data from Pivotal Trial in Relapsed and Refractory Hodgkin Lymphoma
-Objective Responses Achieved in 75 percent of Patients; Median Duration of Response Greater than Six Months-
-BLA Submission Planned in First Half of 2011--Seattle Genetics to Host Conference Call and Webcast Today at 8:30 a.m. ET-
BOTHELL, Wash. & CAMBRIDGE, Mass., Sep 27, 2010 (BUSINESS WIRE) --
Seattle Genetics, Inc. (Nasdaq: SGEN) and Millennium: The Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited (TSE:4502), today announced positive top-line results from the pivotal trial of single-agent brentuximab vedotin (SGN-35), an antibody-drug conjugate (ADC) targeted to CD30. The trial was conducted in 102 relapsed or refractory Hodgkin lymphoma (HL) patients.
Seventy-five percent of patients in the pivotal trial achieved an objective response as assessed by an independent central review, the primary endpoint of the trial. The median duration of response was greater than six months. The safety profile of brentuximab vedotin in this trial was generally consistent with prior clinical trial experience. A more complete data set will be presented at an upcoming scientific meeting.
"We are extremely excited with the top-line results, as they move us one step closer to our goal of bringing brentuximab vedotin to patients with relapsed or refractory Hodgkin lymphoma," said Clay B. Siegall, Ph.D., President and Chief Executive Officer of Seattle Genetics. "We are positioned for a Biologics License Application (BLA) submission to the U.S. Food and Drug Administration (FDA) in the first half of 2011. In addition, we plan to report top-line data from our phase II trial of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma (ALCL) within the next few weeks."
"The lack of adequate therapies for the treatment of relapsed and refractory Hodgkin lymphoma represents a substantial unmet medical need worldwide, with almost a third of the 30,000 newly diagnosed patients relapsing or becoming refractory to front-line therapy annually," said Nancy Simonian, M.D., Chief Medical Officer of Millennium. "These data have the potential to provide an important advance in therapy for Hodgkin lymphoma. We intend to discuss these results with European regulators to support our goal of submitting a Marketing Authorization Application to the European Medicines Agency (EMA) in 2011."
Pivotal Trial Design
The single-arm pivotal trial assessed efficacy and safety of single-agent brentuximab vedotin in relapsed or refractory, post-autologous stem cell transplant (ASCT) HL patients. Patients received 1.8 milligrams per kilogram of brentuximab vedotin every three weeks for up to 16 total doses. The primary endpoint of the trial was objective response rate as assessed by an independent review facility. Response assessments were based on the rigorous and internationally established Revised Response Criteria for Malignant Lymphoma (Cheson, 2007). Secondary endpoints included complete response rate, duration of response, progression-free survival, overall survival and tolerability. The trial was conducted under a Special Protocol Assessment (SPA) with the FDA and was discussed with the EMA during the process of obtaining EU Centralized Scientific Advice on the brentuximab vedotin development program. Brentuximab vedotin has been granted orphan drug designation by the FDA and EMA for the treatment of HL and ALCL and has been granted fast track designation by the FDA for HL.
About Brentuximab Vedotin
Brentuximab vedotin is an ADC comprising an anti-CD30 monoclonal antibody attached by an enzyme cleavable linker to a potent, synthetic drug payload, monomethyl auristatin E (MMAE) utilizing Seattle Genetics' proprietary technology. The ADC employs a novel linker system that is designed to be stable in the bloodstream but to release MMAE upon internalization into CD30-expressing tumor cells. This approach is intended to spare non-targeted cells and thus may help minimize the potential toxic effects of traditional chemotherapy while allowing for the selective targeting of CD30-expressing cancer cells, thus potentially enhancing the antitumor activity.
In addition to the pivotal HL trial, Seattle Genetics and Millennium are conducting a phase II trial for relapsed and refractory systemic ALCL, a phase III clinical trial (the AETHERA trial) for patients at high risk of residual HL following autologous stem cell transplant, a phase II retreatment trial for relapsed patients who previously responded to brentuximab vedotin, and a phase I combination trial for front-line treatment of HL.
About Hodgkin Lymphoma
Lymphoma is a general term for a group of cancers that originate in the lymphatic system. There are two major categories of lymphoma: Hodgkin lymphoma and non-Hodgkin lymphoma. Hodgkin lymphoma is distinguished from other types of lymphoma by the presence of one characteristic type of cell, known as the Reed-Sternberg cell. A defining attribute of the Reed-Sternberg cell is its expression of the CD30 antigen.
According to the American Cancer Society, approximately 8,500 cases of Hodgkin lymphoma will be diagnosed in the United States during 2010 and more than 1,300 will die from the disease. Globally, there are more than 30,000 cases of Hodgkin lymphoma diagnosed each year. Although front-line combination chemotherapy can result in durable response rates, up to 30 percent of these patients relapse or are refractory to front-line treatment and have few therapeutic options beyond ASCT.
About the Seattle Genetics/Millennium Collaboration
Seattle Genetics and Millennium are jointly developing brentuximab vedotin. Under the terms of the collaboration agreement, Seattle Genetics has U.S. and Canadian commercialization rights and the Takeda Group has rights to commercialize brentuximab vedotin in the rest of the world. Seattle Genetics and the Takeda Group are funding joint development costs for brentuximab vedotin on a 50:50 basis, except in Japan where the Takeda Group will be solely responsible for development costs.
Antwort auf Beitrag Nr.: 40.215.696 von SLGramann am 27.09.10 13:15:17sieht gut aus... interessant besonders zur Validierung der ADC-Technologie. Da der reine AK keine Wirkung gezeigt hat und nun mit der chemischen Beladung so wirksam ist, spricht das dafür, dass ADCs generell die bisherigen AKs verbessern können.
http://blogs.forbes.com/matthewherper/2010/09/27/seattle-gen…
Seattle Genetics Scores Win For Smart-Bomb Cancer Drugs
By MATTHEW HERPER
A seductive but elusive cancer-fighting idea may have hit pay dirt.
In a result eagerly awaited by Wall Street, drug developer Seattle Genetics announced that its experimental cancer medicine shrank tumors in 75% of patients with Hodkin lymphoma that had relapsed despite rounds of chemotherapy and bone marrow transplants. Historically, most drugs in this patient population only shrink tumors in only 30% of patients.
The exciting thing is that Seattle’s drug, brentuximab vedotin, combines an antibody with a chemotherapy drug, meaning it can seek out tumor cells and then poison them.This has been a tough thing to pull off but Seattle Genetics says it has been told by the Food and Drug Administration that its study could be sufficient for an approval, despite its small size. The company expects to submit a marketing application to the FDA in the early part of next year. A regulatory decision could come six months after that.
The idea of ’smart bomb’ tumor drugs has long been a mainstay of biotech, and antibodies, lab-made versions of the immune particles the body uses as one way to fight germs, are one of the most-promising approaches. These latch on to a protein that is expressed often in cancer cell but not in normal ones. Big selling cancer drugs like Rituxan, Avastin and Erbitux are antibodies against signaling molecules that spur tumor growth.
But in many cases, an antibody that can find tumor cells doesn’t do enough to kill them. “Most naked antibodies find tumors very well but they just don’t kill them,” says Clay B. Siegall, chief executive of Seattle Genetics. “You’re taking a tumor and you’re decorating it with antibodies.” Brentuximab, the antibody component of Seattle Genetics’ drug, was tried by itself and did little.
So why not attach a payload – a killer chemotherapy drug – to these homing antibodies so that they can actually do some damage? It’s an obvious idea, but one that has proved very hard to execute. The first such drug, Mylotarg, was recently withdrawn from the market after it failed to show a clinical benefit in studies. Medicines that pair antibodies with radioactive molecules have proven to be a tough sell for logistical reasons, even though they work. Last month, the FDA refused to even consider a breast cancer drug that combined Roche’s Herceptin with a chemotherapy using a linking technology developed by Immunogen, causing shares of Immunogen to plummet.
Seattle Genetics says it expects to avoid Immunogen’s fate, because it has an agreement called a Special Protocol Assessment with the FDA. “The FDA has agreed that our pivotal trial design meets requirements for accelerated approval, including agreement that our pivotal trial population represents an unmet medical need,” a spokeswoman writes. She adds that the company believes the FDA’s decision on the Immunogen drug “relates to the evolving landscape and many therapies approved for the treatment of metastatic breast cancer, rather than any more broadly applicable principles.”
If the study results in approval for brentuximab vedotin, it will also be proof that the FDA does sometimes approve drug based on small studies, or without placebo groups. There is no placebo group on the trial, and it involved only 102 patients. A second study of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma (ALCL) will be released in the next few weeks, and might also serve as the basis for and FDA filing. It is also a small, early stage trial.
Siegall says he is “extremely excited” by the data, which represent strong proof of efficacy of the drug. In the patients who responded to the treatment, the tumor shrinkage persisted for at least six months. He hopes to test the medicine in less sick populations who might be impacted even more. If it is approved, Seattle Genetics will market brentuximab vedotin usin a sales force of only sixty or so sales representatives in the U.S. and Canada. Overseas, marketing will be handled by its partner, Takeda. The company will host a conference call for investors this morning.
mfg ipollit
http://blogs.forbes.com/matthewherper/2010/09/27/seattle-gen…
Seattle Genetics Scores Win For Smart-Bomb Cancer Drugs
By MATTHEW HERPER
A seductive but elusive cancer-fighting idea may have hit pay dirt.
In a result eagerly awaited by Wall Street, drug developer Seattle Genetics announced that its experimental cancer medicine shrank tumors in 75% of patients with Hodkin lymphoma that had relapsed despite rounds of chemotherapy and bone marrow transplants. Historically, most drugs in this patient population only shrink tumors in only 30% of patients.
The exciting thing is that Seattle’s drug, brentuximab vedotin, combines an antibody with a chemotherapy drug, meaning it can seek out tumor cells and then poison them.This has been a tough thing to pull off but Seattle Genetics says it has been told by the Food and Drug Administration that its study could be sufficient for an approval, despite its small size. The company expects to submit a marketing application to the FDA in the early part of next year. A regulatory decision could come six months after that.
The idea of ’smart bomb’ tumor drugs has long been a mainstay of biotech, and antibodies, lab-made versions of the immune particles the body uses as one way to fight germs, are one of the most-promising approaches. These latch on to a protein that is expressed often in cancer cell but not in normal ones. Big selling cancer drugs like Rituxan, Avastin and Erbitux are antibodies against signaling molecules that spur tumor growth.
But in many cases, an antibody that can find tumor cells doesn’t do enough to kill them. “Most naked antibodies find tumors very well but they just don’t kill them,” says Clay B. Siegall, chief executive of Seattle Genetics. “You’re taking a tumor and you’re decorating it with antibodies.” Brentuximab, the antibody component of Seattle Genetics’ drug, was tried by itself and did little.
So why not attach a payload – a killer chemotherapy drug – to these homing antibodies so that they can actually do some damage? It’s an obvious idea, but one that has proved very hard to execute. The first such drug, Mylotarg, was recently withdrawn from the market after it failed to show a clinical benefit in studies. Medicines that pair antibodies with radioactive molecules have proven to be a tough sell for logistical reasons, even though they work. Last month, the FDA refused to even consider a breast cancer drug that combined Roche’s Herceptin with a chemotherapy using a linking technology developed by Immunogen, causing shares of Immunogen to plummet.
Seattle Genetics says it expects to avoid Immunogen’s fate, because it has an agreement called a Special Protocol Assessment with the FDA. “The FDA has agreed that our pivotal trial design meets requirements for accelerated approval, including agreement that our pivotal trial population represents an unmet medical need,” a spokeswoman writes. She adds that the company believes the FDA’s decision on the Immunogen drug “relates to the evolving landscape and many therapies approved for the treatment of metastatic breast cancer, rather than any more broadly applicable principles.”
If the study results in approval for brentuximab vedotin, it will also be proof that the FDA does sometimes approve drug based on small studies, or without placebo groups. There is no placebo group on the trial, and it involved only 102 patients. A second study of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma (ALCL) will be released in the next few weeks, and might also serve as the basis for and FDA filing. It is also a small, early stage trial.
Siegall says he is “extremely excited” by the data, which represent strong proof of efficacy of the drug. In the patients who responded to the treatment, the tumor shrinkage persisted for at least six months. He hopes to test the medicine in less sick populations who might be impacted even more. If it is approved, Seattle Genetics will market brentuximab vedotin usin a sales force of only sixty or so sales representatives in the U.S. and Canada. Overseas, marketing will be handled by its partner, Takeda. The company will host a conference call for investors this morning.
mfg ipollit
Antwort auf Beitrag Nr.: 40.215.816 von ipollit am 27.09.10 13:39:28
Hi ipollit, das ist genau das, was ich hoffe: Dass wir mit der ADC-Technologie die große Zeit der mabs erst wirklich einläuten. Es gibt viele mabs, die an ihr Target binden, aber wirkungslos sind. Da können ADCs plötzlich für Wirksamkeit sorgen und das schöne ist, dass die mabs schon da sind. Man muss sie "nur" bewaffnen. Und selbst naked mabs, die wirksam sind, können als ADC ihre Wirkung steigern und/oder die begleitende Chemotherapie ersetzen (siehe dazu T-DM1, der eines Tages genau das leisten soll).
Ich finde, dass es sehr gute Gründe gibt, bei SGEN und IMGN dabei zu sein und ich glaube, dass da in der vorklinischen Entwicklung der Zug voll unter Dampf ist (man siehts ja an den ganzen SGEN-Partnerschaften)!
Hier noch ein schöner Artikel von Xconomy:
Seattle Genetics, Millennium Generate “Dream” Data With Empowered Antibody Drug for Cancer
Luke Timmerman 9/27/10
Seattle Genetics is making history today, after a dozen years of effort, with groundbreaking clinical trial results in the field of cancer drugs. The results pave the way for the first successful “empowered antibody” drug for cancer, and a new therapy for people who have run out of options for fighting Hodgkin’s disease.
http://www.xconomy.com/seattle/2010/09/27/seattle-genetics-m…
Hi ipollit, das ist genau das, was ich hoffe: Dass wir mit der ADC-Technologie die große Zeit der mabs erst wirklich einläuten. Es gibt viele mabs, die an ihr Target binden, aber wirkungslos sind. Da können ADCs plötzlich für Wirksamkeit sorgen und das schöne ist, dass die mabs schon da sind. Man muss sie "nur" bewaffnen. Und selbst naked mabs, die wirksam sind, können als ADC ihre Wirkung steigern und/oder die begleitende Chemotherapie ersetzen (siehe dazu T-DM1, der eines Tages genau das leisten soll).
Ich finde, dass es sehr gute Gründe gibt, bei SGEN und IMGN dabei zu sein und ich glaube, dass da in der vorklinischen Entwicklung der Zug voll unter Dampf ist (man siehts ja an den ganzen SGEN-Partnerschaften)!
Hier noch ein schöner Artikel von Xconomy:
Seattle Genetics, Millennium Generate “Dream” Data With Empowered Antibody Drug for Cancer
Luke Timmerman 9/27/10
Seattle Genetics is making history today, after a dozen years of effort, with groundbreaking clinical trial results in the field of cancer drugs. The results pave the way for the first successful “empowered antibody” drug for cancer, and a new therapy for people who have run out of options for fighting Hodgkin’s disease.
http://www.xconomy.com/seattle/2010/09/27/seattle-genetics-m…
zu REGN:
Die Trap-Eye Daten kommen offenbar im Dezember.
PiperJaffray says it is a buyer of Regeneron Pharmaceuticals (NASDAQ: REGN) ahead of pivotal VEGF Trap-Eye data in Age-related Macular Degeneration (AMD) in December.
If approved, we believe VEGF Trap-Eye could be a game changer for Regeneron pushing the company to profitability in 2012. Regeneron retains U.S. rights where there are >200,000 new cases of AMD each year. PiperJaffray conservatively forecasts sales of $710 million by 2015 with only 25% penetration. If VEGF Trap-Eye proves superior to Lucentis on either metric, we could see more rapid uptake and increased peak penetration. Regeneron is partnered with Bayer overseas and receives 50% of the profits providing further upside to its revenue forecast and price target.
Including $165 million from Astellas, Regeneron holds proforma cash of $545 million and no debt. Trading at a market cap of $2.1 billion and an enterprise value of $1.6 billion, Piper sees shares of REGN as attractive.
PiperJaffray reiterates its Overweight rating on REGN and a $37 price target.
--------------
Tja, und was ist, wenn kein Vorteil gegenüber Lucentis besteht? Wie viel der Marktcap von 2,2 Mrd. Dollar hängt an Trap-Eye? Bestimmt nicht wenig...
ipollit, was glaubst Du, wie das ausgeht und wie die Konsequenzen in der einen und in der anderen Richtung wären?
Die Trap-Eye Daten kommen offenbar im Dezember.
PiperJaffray says it is a buyer of Regeneron Pharmaceuticals (NASDAQ: REGN) ahead of pivotal VEGF Trap-Eye data in Age-related Macular Degeneration (AMD) in December.
If approved, we believe VEGF Trap-Eye could be a game changer for Regeneron pushing the company to profitability in 2012. Regeneron retains U.S. rights where there are >200,000 new cases of AMD each year. PiperJaffray conservatively forecasts sales of $710 million by 2015 with only 25% penetration. If VEGF Trap-Eye proves superior to Lucentis on either metric, we could see more rapid uptake and increased peak penetration. Regeneron is partnered with Bayer overseas and receives 50% of the profits providing further upside to its revenue forecast and price target.
Including $165 million from Astellas, Regeneron holds proforma cash of $545 million and no debt. Trading at a market cap of $2.1 billion and an enterprise value of $1.6 billion, Piper sees shares of REGN as attractive.
PiperJaffray reiterates its Overweight rating on REGN and a $37 price target.
--------------
Tja, und was ist, wenn kein Vorteil gegenüber Lucentis besteht? Wie viel der Marktcap von 2,2 Mrd. Dollar hängt an Trap-Eye? Bestimmt nicht wenig...
ipollit, was glaubst Du, wie das ausgeht und wie die Konsequenzen in der einen und in der anderen Richtung wären?
Prohost hat sich zu INCYs 18424 eingelassen:
INCYTE’S (INCY) DRUG NBC 18424
NCB18424 is topping the news in the category of drugs that have chance of approval. The drug, which is partnered with Novartis is for the treatment of myeloproliferative diseases, which include myelofibrosis, polycythemia vera, and essential thrombocythemia. Recent promising results of phase 1/2 studies of NCB18424 on myelofibrosis were published in The New England Journal of Medicine. This rare, life-threatening hematologic disease is typified by malfunctioning fibrosed bone marrow that causes the formation of abnormally shaped red blood cells. The disease is characterized by bone marrow failure, anemia, enlarged spleen (splenomegaly), and debilitating symptoms, including extreme fatigue, poor quality of life, pancytopenia, weight loss and shortened survival, among other symptoms.
INCB18424 is a janus kinase (JAK) inhibitor. There are four Janus kinase families. JAK1 and JAK2 are involved in interferon-gamma signalling, whereas JAK1 and tyrosine kinase 2 (TYK2) are involved in type I interferon signaling. Mice that do not express TYK2 have defective natural cell killer function. Transgenic mice that do not express JAK1 have defective responses to some cytokines such as interferon gamma, which is critical for innate and adoptive immunity against viral and intracellular bacterial infections and for tumor control. Aberrant interferon gamma expression is associated with a number of autoinflammatory and autoimmune diseases. The cytokine inhibits viral replication directly, and has immunostimulatory and immunomodulatory effects. There is a strong association between abnormal JAK signaling and the development of myeloproliferative diseases.
Trial results of INCB18424 on myelofibrosis demonstrate that 70% to 82% of patients receiving oral INCB18424 twice daily had marked reduction i(25% or more) in palpable spleen size that was durable for more than a year of follow-up. More than half the subjects treated with an optimized dose regimen, which began with 15 mg twice daily, achieved at least a 50% reduction in palpable spleen size. This marked reduction was confirmed using objective measurements by MRI, where 48% of patients on optimized regimens had at least a 35% reduction in spleen volume. After one month of therapy, patients with symptoms including fatigue, night sweats and pruritus achieved more than 50% improvement in symptom scores.
Patients with prior weight loss, experienced a clinically meaningful gain in total body weight following treatment. The drug caused improvement in patients’ performance status, increased exercise capacity, reductions of circulating cytokines (inflammation-causing proteins in the blood, which are markedly elevated in patients with myelofibrosis.
Two Phase 3 clinical trials, COMFORT-I in the US, Canada and Australia, and COMFORT-II in Europe, have completed enrollment and are evaluating the benefits of treatment with INCB18424 compared to either placebo or best available treatment.
Conclusion: The discovery of JAK mutations common to myelofibrosis, polycythemia vera and essential thrombocythemia, has linked them on a molecular level. Together with the fact that the patients tend to have elevated inflammatory cytokines that signal through JAK1 and JAK2, led Incyte to the discovery and development of INCB18424, a potent, selective inhibitor of the JAK1 and JAK2 tyrosine kinases. So, there exist a solid rationale behind the development of the drug for the selected diseases. Results of trials also support the scientific promise of JAK1 and JAK2 inhibition in the treatment of myeloproliferative neoplasms as demonstrated in the clinical improvement that might have been achieved for the first time in treating myelofibrosis, as Paul A. Friedman, Chief Executive Officer and President of Incyte has stated.
For the victims of myelofibrosis and their doctors, they have yet to see whether the clinical improvements experienced would contribute to increased survivability, especially that the drug seems to have been unsuccessful in reversing the anemia, despite its success in shrinking the spleen size. Maybe red blood growth factor, or any other product added to INCB18424 would do the job remains to be seen. On the other hand, shrinking the enlarged spleen is, by itself, a great achievement. A tremendous increase in spleen size poses a huge discomfort and great negative impact on patients’ wellbeing. Adding the other clinical improvements to the effect of the drug on the spleen would motivate the FDA to approve the drug.
For investors, myelofibrosis has a minute market, but the drug promises to treat many diseases. Also, the firm’s technology and product pipeline are promising. Results of the drug on polycythemia vera are encouraging, which add to the market’s size in case of approval. We will be following up on this firm’s activities, focusing on its products’ early and late clinical trials. Added to the drug intended for myeloproliferative diseases, the addition of the firm’s molecules addressing inflammatory diseases could turn this small firm into a full-fledged drug developer.
---------
Muss man der Grechtigkeit halber anmerken, dass im Blog von Ohad Hammer sämtliche Hintergründe und die hervorragenden Perspektiven schon ab August 2009 dargestellt worden waren. Damals war INCY auch noch deutlich preiswerter zu haben, während ich jetzt der Meinung bin, dass 18424 schon weitgehend eingepreist ist. Aber INCY ist andererseits auch nicht nur 18424...
INCYTE’S (INCY) DRUG NBC 18424
NCB18424 is topping the news in the category of drugs that have chance of approval. The drug, which is partnered with Novartis is for the treatment of myeloproliferative diseases, which include myelofibrosis, polycythemia vera, and essential thrombocythemia. Recent promising results of phase 1/2 studies of NCB18424 on myelofibrosis were published in The New England Journal of Medicine. This rare, life-threatening hematologic disease is typified by malfunctioning fibrosed bone marrow that causes the formation of abnormally shaped red blood cells. The disease is characterized by bone marrow failure, anemia, enlarged spleen (splenomegaly), and debilitating symptoms, including extreme fatigue, poor quality of life, pancytopenia, weight loss and shortened survival, among other symptoms.
INCB18424 is a janus kinase (JAK) inhibitor. There are four Janus kinase families. JAK1 and JAK2 are involved in interferon-gamma signalling, whereas JAK1 and tyrosine kinase 2 (TYK2) are involved in type I interferon signaling. Mice that do not express TYK2 have defective natural cell killer function. Transgenic mice that do not express JAK1 have defective responses to some cytokines such as interferon gamma, which is critical for innate and adoptive immunity against viral and intracellular bacterial infections and for tumor control. Aberrant interferon gamma expression is associated with a number of autoinflammatory and autoimmune diseases. The cytokine inhibits viral replication directly, and has immunostimulatory and immunomodulatory effects. There is a strong association between abnormal JAK signaling and the development of myeloproliferative diseases.
Trial results of INCB18424 on myelofibrosis demonstrate that 70% to 82% of patients receiving oral INCB18424 twice daily had marked reduction i(25% or more) in palpable spleen size that was durable for more than a year of follow-up. More than half the subjects treated with an optimized dose regimen, which began with 15 mg twice daily, achieved at least a 50% reduction in palpable spleen size. This marked reduction was confirmed using objective measurements by MRI, where 48% of patients on optimized regimens had at least a 35% reduction in spleen volume. After one month of therapy, patients with symptoms including fatigue, night sweats and pruritus achieved more than 50% improvement in symptom scores.
Patients with prior weight loss, experienced a clinically meaningful gain in total body weight following treatment. The drug caused improvement in patients’ performance status, increased exercise capacity, reductions of circulating cytokines (inflammation-causing proteins in the blood, which are markedly elevated in patients with myelofibrosis.
Two Phase 3 clinical trials, COMFORT-I in the US, Canada and Australia, and COMFORT-II in Europe, have completed enrollment and are evaluating the benefits of treatment with INCB18424 compared to either placebo or best available treatment.
Conclusion: The discovery of JAK mutations common to myelofibrosis, polycythemia vera and essential thrombocythemia, has linked them on a molecular level. Together with the fact that the patients tend to have elevated inflammatory cytokines that signal through JAK1 and JAK2, led Incyte to the discovery and development of INCB18424, a potent, selective inhibitor of the JAK1 and JAK2 tyrosine kinases. So, there exist a solid rationale behind the development of the drug for the selected diseases. Results of trials also support the scientific promise of JAK1 and JAK2 inhibition in the treatment of myeloproliferative neoplasms as demonstrated in the clinical improvement that might have been achieved for the first time in treating myelofibrosis, as Paul A. Friedman, Chief Executive Officer and President of Incyte has stated.
For the victims of myelofibrosis and their doctors, they have yet to see whether the clinical improvements experienced would contribute to increased survivability, especially that the drug seems to have been unsuccessful in reversing the anemia, despite its success in shrinking the spleen size. Maybe red blood growth factor, or any other product added to INCB18424 would do the job remains to be seen. On the other hand, shrinking the enlarged spleen is, by itself, a great achievement. A tremendous increase in spleen size poses a huge discomfort and great negative impact on patients’ wellbeing. Adding the other clinical improvements to the effect of the drug on the spleen would motivate the FDA to approve the drug.
For investors, myelofibrosis has a minute market, but the drug promises to treat many diseases. Also, the firm’s technology and product pipeline are promising. Results of the drug on polycythemia vera are encouraging, which add to the market’s size in case of approval. We will be following up on this firm’s activities, focusing on its products’ early and late clinical trials. Added to the drug intended for myeloproliferative diseases, the addition of the firm’s molecules addressing inflammatory diseases could turn this small firm into a full-fledged drug developer.
---------
Muss man der Grechtigkeit halber anmerken, dass im Blog von Ohad Hammer sämtliche Hintergründe und die hervorragenden Perspektiven schon ab August 2009 dargestellt worden waren. Damals war INCY auch noch deutlich preiswerter zu haben, während ich jetzt der Meinung bin, dass 18424 schon weitgehend eingepreist ist. Aber INCY ist andererseits auch nicht nur 18424...
Aveo macht heute mit +9% richtig Spaß. Dabei las sich die folgende Meldung für mich eigentlich negativ. Fühlt sich jemand in der Lage, das etwas einzuordnen?
AVEO Pharmaceuticals Regains Worldwide Rights to Develop and Commercialize Anti-HGF Antibody Candidate AV-299
CAMBRIDGE, Mass., Sep 30, 2010 (BUSINESS WIRE) -- AVEO Pharmaceuticals, Inc. /quotes/comstock/15*!aveo/quotes/nls/aveo (AVEO 10.90, +0.91, +9.11%) , a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced that AVEO has regained worldwide rights from Merck (through its subsidiary, Schering Corporation) to develop and commercialize AV-299 (also known as SCH 900105), its anti-hepatocyte growth factor (HGF) antibody candidate.
AV-299 is a potent, functional anti-HGF antibody that was discovered by AVEO through its Human Response Platform(TM). Data from Phase 1 clinical trials of AV-299 indicate a favorable tolerability profile and good combinability with EGFR inhibitors, erlotinib (Tarceva(R)) and gefitinib (Iressa(R)). In June 2010, AVEO initiated a Phase 2 clinical trial evaluating AV-299 in combination with gefitinib versus gefitinib monotherapy in patients with non-small cell lung cancer (NSCLC). In conjunction with the Phase 2 trial initiation, AVEO received an $8.5 million milestone payment from Merck under the terms of the license agreement. Top-line efficacy data from the AV-299 Phase 2 trial are expected in late 2011.
"AVEO is very pleased to regain worldwide rights for the development and commercialization of AV-299," stated Tuan Ha-Ngoc, president and chief executive officer of AVEO Pharmaceuticals. "AVEO now holds significant commercialization rights to all oncology products in our pipeline, and we believe that we are well-positioned to move toward our goal of becoming a fully-integrated commercial organization. Our expected year-end 2010 cash balance remains unchanged, and we reaffirm that AVEO has sufficient capital to take us beyond data from TIVO-1, our ongoing Phase 3 clinical trial of tivozanib in patients with renal cell carcinoma. We look forward to sharing top-line TIVO-1 data in mid-2011."
"Merck is pleased with our history of collaborating with AVEO, and would welcome the opportunity to work with AVEO again in the future," said David Nicholson, Ph.D., senior vice president and head of worldwide licensing and knowledge management at Merck. "The decision to return this program to AVEO is a result of portfolio prioritization."
About AV-299 and the HGF/c-Met Pathway
The HGF/c-Met pathway is believed to play an important role in regulating tumor growth, invasion and metastasis, making it an exciting novel target in oncology. In addition, preclinical and clinical observations suggest that increased HGF and/or c-Met receptor amplification may confer resistance to EGFR inhibitors. Recently, encouraging Phase 2 clinical data were reported at the 2010 ASCO Annual Meeting with a small molecule c-Met inhibitor in combination with an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) in patients with advanced, refractory NSCLC. These data signal the potential patient benefit from combination therapy of an EGFR TKI and an inhibitor of the HGF/c-Met pathway.
AV-299 is a potent, functional anti-HGF antibody that was discovered by AVEO through its Human Response Platform(TM) (HRP). In AVEO's proprietary tumor models with elevated HGF/c-Met signaling, AV-299 exhibited strong additive anti-tumor effect when given in combination with other approved anti-cancer agents such as erlotinib (Tarceva(R)), cetuximab (Erbitux(R)) and temozolomide (Temodar(R)). In additional preclinical studies, AV-299 was more effective at inhibiting tumor growth (at the dose tested) than other anti-HGF antibodies currently in clinical development.
Data from a recently completed a Phase 1 clinical trial with AV-299 showed a favorable combinability and tolerability profile with no dose-limiting toxicities up to the highest dose tested (20 mg/kg, bi-weekly). Based on the promising preclinical data and Phase 1 results, in June 2010, AVEO initiated a Phase 2 clinical trial evaluating AV-299 in combination with gefitinib (Iressa(TM)) versus gefitinib monotherapy in patients with NSCLC. Top-line efficacy data from the ongoing Phase 2 trial are expected in late 2011.
AVEO Pharmaceuticals Regains Worldwide Rights to Develop and Commercialize Anti-HGF Antibody Candidate AV-299
CAMBRIDGE, Mass., Sep 30, 2010 (BUSINESS WIRE) -- AVEO Pharmaceuticals, Inc. /quotes/comstock/15*!aveo/quotes/nls/aveo (AVEO 10.90, +0.91, +9.11%) , a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced that AVEO has regained worldwide rights from Merck (through its subsidiary, Schering Corporation) to develop and commercialize AV-299 (also known as SCH 900105), its anti-hepatocyte growth factor (HGF) antibody candidate.
AV-299 is a potent, functional anti-HGF antibody that was discovered by AVEO through its Human Response Platform(TM). Data from Phase 1 clinical trials of AV-299 indicate a favorable tolerability profile and good combinability with EGFR inhibitors, erlotinib (Tarceva(R)) and gefitinib (Iressa(R)). In June 2010, AVEO initiated a Phase 2 clinical trial evaluating AV-299 in combination with gefitinib versus gefitinib monotherapy in patients with non-small cell lung cancer (NSCLC). In conjunction with the Phase 2 trial initiation, AVEO received an $8.5 million milestone payment from Merck under the terms of the license agreement. Top-line efficacy data from the AV-299 Phase 2 trial are expected in late 2011.
"AVEO is very pleased to regain worldwide rights for the development and commercialization of AV-299," stated Tuan Ha-Ngoc, president and chief executive officer of AVEO Pharmaceuticals. "AVEO now holds significant commercialization rights to all oncology products in our pipeline, and we believe that we are well-positioned to move toward our goal of becoming a fully-integrated commercial organization. Our expected year-end 2010 cash balance remains unchanged, and we reaffirm that AVEO has sufficient capital to take us beyond data from TIVO-1, our ongoing Phase 3 clinical trial of tivozanib in patients with renal cell carcinoma. We look forward to sharing top-line TIVO-1 data in mid-2011."
"Merck is pleased with our history of collaborating with AVEO, and would welcome the opportunity to work with AVEO again in the future," said David Nicholson, Ph.D., senior vice president and head of worldwide licensing and knowledge management at Merck. "The decision to return this program to AVEO is a result of portfolio prioritization."
About AV-299 and the HGF/c-Met Pathway
The HGF/c-Met pathway is believed to play an important role in regulating tumor growth, invasion and metastasis, making it an exciting novel target in oncology. In addition, preclinical and clinical observations suggest that increased HGF and/or c-Met receptor amplification may confer resistance to EGFR inhibitors. Recently, encouraging Phase 2 clinical data were reported at the 2010 ASCO Annual Meeting with a small molecule c-Met inhibitor in combination with an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) in patients with advanced, refractory NSCLC. These data signal the potential patient benefit from combination therapy of an EGFR TKI and an inhibitor of the HGF/c-Met pathway.
AV-299 is a potent, functional anti-HGF antibody that was discovered by AVEO through its Human Response Platform(TM) (HRP). In AVEO's proprietary tumor models with elevated HGF/c-Met signaling, AV-299 exhibited strong additive anti-tumor effect when given in combination with other approved anti-cancer agents such as erlotinib (Tarceva(R)), cetuximab (Erbitux(R)) and temozolomide (Temodar(R)). In additional preclinical studies, AV-299 was more effective at inhibiting tumor growth (at the dose tested) than other anti-HGF antibodies currently in clinical development.
Data from a recently completed a Phase 1 clinical trial with AV-299 showed a favorable combinability and tolerability profile with no dose-limiting toxicities up to the highest dose tested (20 mg/kg, bi-weekly). Based on the promising preclinical data and Phase 1 results, in June 2010, AVEO initiated a Phase 2 clinical trial evaluating AV-299 in combination with gefitinib (Iressa(TM)) versus gefitinib monotherapy in patients with NSCLC. Top-line efficacy data from the ongoing Phase 2 trial are expected in late 2011.
Aveo auch heute gut im Plus und wie gestern mit - für diese Aktie - hohen Umsätzen.
Diesmal gibts auch positive News, die indes eigentlich bestenfalls teilweise wirklich neu sind...
October 01, 2010 06:30 AM Eastern Daylight Time
AVEO Pharmaceuticals’ Tivozanib Featured at Ninth International Kidney Cancer Symposium
Tivozanib Clinical Data and Pipeline Overview Presented
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced that previously reported results from its Phase 2 study evaluating tivozanib for the treatment of advanced renal cell carcinoma (RCC) will be presented at the Ninth International Kidney Cancer Symposium (IKCS) in Chicago, Illinois, October 1-2, 2010. Additionally, data from an ongoing Phase 1b trial evaluating tivozanib in combination with temsirolimus (Torisel®), an approved mTOR inhibitor, in patients with RCC will be presented at the meeting.
“The Kidney Cancer Association has made tremendous strides in their effort to improve the treatment and care of patients living with kidney cancer, and we are honored to have the opportunity to share tivozanib clinical data with key constituents of the kidney cancer community,” said William Slichenmyer, M.D., Sc.M., chief medical officer at AVEO. “We believe that there is an urgent need for a differentiated kidney cancer therapy with strong efficacy and favorable tolerability, and we look forward to continued collaboration with the Kidney Cancer Association, clinicians and patients as we make efforts to advance tivozanib toward regulatory approval.”
The Phase 2 clinical trial evaluated tivozanib in 272 patients with advanced RCC. Data showed that the median PFS achieved by patients with advanced clear cell RCC who had undergone a prior nephrectomy was 14.8 months – comparing favorably to historical data from trials testing other currently approved multikinase inhibitors in RCC. Hypertension was the most commonly reported treatment-related adverse effect, and was observed in 50% of treated patients. Development of hypertension was directly associated with improved clinical outcomes among patients overall and in the subset of patients with clear cell RCC who had undergone a prior nephrectomy. Off-target toxicities commonly associated with other targeted therapies, such as mucositis, fatigue and hand-foot syndrome, were notably low in the tivozanib group, which AVEO believes underscores a potential tolerable safety profile and potential for combinability with other therapeutic agents.
“A growing body of evidence suggests that tivozanib is highly differentiated in its ability to potently and selectively target VEGF, which may result in favorable tolerability, increased combinability and extended median progression-free survival in patients,” said Thomas E. Hutson, D.O., Pharm.D., Baylor University Medical Center, Sammons Cancer Center, Dallas, and the co-primary investigator for TIVO-1, AVEO’s Phase 3 tivozanib trial in advanced kidney cancer. “TIVO-1, the first-ever Phase 3 study in RCC designed to evaluate superiority in a head-to-head comparison against a widely prescribed anti-angiogenesis therapy in first-line treatment, has been designed to equip oncologists with the information necessary to make the most appropriate treatment decisions for their patients battling kidney cancer.”
Results from a Phase 1 clinical study evaluating tivozanib in combination with the mTOR inhibitor temsirolimus (Torisel), a rapamycin pro-drug, in patients with advanced kidney cancer will be presented in an oral session by Fairooz Kabbinavar, M.D., professor of medicine and urologic oncology, Henry Alvin and Carrie L. Meinhardt chair in Kidney Cancer Research, director, Hematology-Oncology Fellowship Program, medical director, Thoracic Oncology and Kidney Cancer Programs, David Geffen School of Medicine, University of California, Los Angeles. Study results showed tumor shrinkage in 12 out of 16 patients evaluated, and two partial responses as assessed by RECIST criteria. The combination was well-tolerated with no dose limiting toxicities (DLT).
Murray Robinson, Ph.D., senior vice president, translational medicine at AVEO, will also be making a presentation titled, “Tivozanib: clinical progress and response biomarkers,” highlighting AVEO’s ongoing tivozanib clinical research efforts, and the company’s commitment to exploring how protein biomarker research may help clinicians determine optimal treatment combinations for patients battling kidney cancer and other tumor types.
“Understanding how predictive biomarkers fit into the kidney cancer landscape, and how they may help inform optimal treatment decisions for patients, is new and exciting ground for us in the oncology medical community,” added Dr. Hutson. “I applaud AVEO’s research efforts, and the company’s goal of further advancing our understanding of how biomarkers may ultimately improve the treatment and care of patients living with kidney cancer.”
About TIVO-1
AVEO recently completed patient enrollment in TIVO-1, a global Phase 3 clinical trial of tivozanib in patients with advanced RCC who have not received prior VEGF-targeted therapy. Initiated in February of this year, TIVO-1 successfully reached the target enrollment of 500 patients in August 2010, ahead of schedule.
TIVO-1 is evaluating the efficacy of tivozanib compared to Nexavar® (sorafenib), an FDA and EMA approved therapy for RCC. Tivozanib is the only therapy currently in late-stage development for first-line treatment of kidney cancer. The primary endpoint of the trial is to compare the progression-free survival (PFS) of tivozanib vs. Nexavar. Secondary endpoints include overall survival, objective response rate, safety and quality of life. Patients who demonstrate disease progression during treatment with Nexavar will have the opportunity to be treated with tivozanib by participating in a separate long-term treatment protocol. This trial is being led by Robert Motzer, M.D. from the Memorial Sloan-Kettering Cancer Center. Topline efficacy data are expected mid-2011.
Diesmal gibts auch positive News, die indes eigentlich bestenfalls teilweise wirklich neu sind...
October 01, 2010 06:30 AM Eastern Daylight Time
AVEO Pharmaceuticals’ Tivozanib Featured at Ninth International Kidney Cancer Symposium
Tivozanib Clinical Data and Pipeline Overview Presented
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced that previously reported results from its Phase 2 study evaluating tivozanib for the treatment of advanced renal cell carcinoma (RCC) will be presented at the Ninth International Kidney Cancer Symposium (IKCS) in Chicago, Illinois, October 1-2, 2010. Additionally, data from an ongoing Phase 1b trial evaluating tivozanib in combination with temsirolimus (Torisel®), an approved mTOR inhibitor, in patients with RCC will be presented at the meeting.
“The Kidney Cancer Association has made tremendous strides in their effort to improve the treatment and care of patients living with kidney cancer, and we are honored to have the opportunity to share tivozanib clinical data with key constituents of the kidney cancer community,” said William Slichenmyer, M.D., Sc.M., chief medical officer at AVEO. “We believe that there is an urgent need for a differentiated kidney cancer therapy with strong efficacy and favorable tolerability, and we look forward to continued collaboration with the Kidney Cancer Association, clinicians and patients as we make efforts to advance tivozanib toward regulatory approval.”
The Phase 2 clinical trial evaluated tivozanib in 272 patients with advanced RCC. Data showed that the median PFS achieved by patients with advanced clear cell RCC who had undergone a prior nephrectomy was 14.8 months – comparing favorably to historical data from trials testing other currently approved multikinase inhibitors in RCC. Hypertension was the most commonly reported treatment-related adverse effect, and was observed in 50% of treated patients. Development of hypertension was directly associated with improved clinical outcomes among patients overall and in the subset of patients with clear cell RCC who had undergone a prior nephrectomy. Off-target toxicities commonly associated with other targeted therapies, such as mucositis, fatigue and hand-foot syndrome, were notably low in the tivozanib group, which AVEO believes underscores a potential tolerable safety profile and potential for combinability with other therapeutic agents.
“A growing body of evidence suggests that tivozanib is highly differentiated in its ability to potently and selectively target VEGF, which may result in favorable tolerability, increased combinability and extended median progression-free survival in patients,” said Thomas E. Hutson, D.O., Pharm.D., Baylor University Medical Center, Sammons Cancer Center, Dallas, and the co-primary investigator for TIVO-1, AVEO’s Phase 3 tivozanib trial in advanced kidney cancer. “TIVO-1, the first-ever Phase 3 study in RCC designed to evaluate superiority in a head-to-head comparison against a widely prescribed anti-angiogenesis therapy in first-line treatment, has been designed to equip oncologists with the information necessary to make the most appropriate treatment decisions for their patients battling kidney cancer.”
Results from a Phase 1 clinical study evaluating tivozanib in combination with the mTOR inhibitor temsirolimus (Torisel), a rapamycin pro-drug, in patients with advanced kidney cancer will be presented in an oral session by Fairooz Kabbinavar, M.D., professor of medicine and urologic oncology, Henry Alvin and Carrie L. Meinhardt chair in Kidney Cancer Research, director, Hematology-Oncology Fellowship Program, medical director, Thoracic Oncology and Kidney Cancer Programs, David Geffen School of Medicine, University of California, Los Angeles. Study results showed tumor shrinkage in 12 out of 16 patients evaluated, and two partial responses as assessed by RECIST criteria. The combination was well-tolerated with no dose limiting toxicities (DLT).
Murray Robinson, Ph.D., senior vice president, translational medicine at AVEO, will also be making a presentation titled, “Tivozanib: clinical progress and response biomarkers,” highlighting AVEO’s ongoing tivozanib clinical research efforts, and the company’s commitment to exploring how protein biomarker research may help clinicians determine optimal treatment combinations for patients battling kidney cancer and other tumor types.
“Understanding how predictive biomarkers fit into the kidney cancer landscape, and how they may help inform optimal treatment decisions for patients, is new and exciting ground for us in the oncology medical community,” added Dr. Hutson. “I applaud AVEO’s research efforts, and the company’s goal of further advancing our understanding of how biomarkers may ultimately improve the treatment and care of patients living with kidney cancer.”
About TIVO-1
AVEO recently completed patient enrollment in TIVO-1, a global Phase 3 clinical trial of tivozanib in patients with advanced RCC who have not received prior VEGF-targeted therapy. Initiated in February of this year, TIVO-1 successfully reached the target enrollment of 500 patients in August 2010, ahead of schedule.
TIVO-1 is evaluating the efficacy of tivozanib compared to Nexavar® (sorafenib), an FDA and EMA approved therapy for RCC. Tivozanib is the only therapy currently in late-stage development for first-line treatment of kidney cancer. The primary endpoint of the trial is to compare the progression-free survival (PFS) of tivozanib vs. Nexavar. Secondary endpoints include overall survival, objective response rate, safety and quality of life. Patients who demonstrate disease progression during treatment with Nexavar will have the opportunity to be treated with tivozanib by participating in a separate long-term treatment protocol. This trial is being led by Robert Motzer, M.D. from the Memorial Sloan-Kettering Cancer Center. Topline efficacy data are expected mid-2011.
So, für die nächste Zeit die letzte Bemerkung zu AVEO:
Der Chart hat sich in den letzten zwei Tagen aber auch wirklich etwas putzig entwickelt:

Hier ein Artikel, der die Rückgabe von AV-299 durch Merck tatsächlich positiv einordnet. Na ja...
Merck's Loss Is Aveo's Gain
By Lisa LaMotta Oct 01, 2010 3:00 pm
The biotech will move into a strong position as its cancer holdings become more significant now that Merck has pulled out.
Aveo Pharmaceuticals (AVEO) announced on Thursday that its three-year deal with Big Pharma has come to a close. While this sort of news is usually bad news (it often happens when a drug doesn’t work), Aveo could be the one lucking out.
The Cambridge, Massachusetts-based biotech entered into the agreement in April 2007 with Schering-Plough -- now Merck (MRK) -- for the larger company to partner on its then preclinical cancer treatment. At the time, Aveo received a $7.5 million upfront payment, as well as $10 million in equity investments. The biotech was also eligible to receive another $460 million in milestone payments.
The deal was structured so that Aveo made out like a bandit and doesn’t really lose anything now that Merck has decided to end the collaboration. The only downside is that Schering/Merck was funding all of the research and development, but Aveo was able to get the drug into phase II trials on the Big Pharma’s dime (Merck will provide funding for the program through the end of the year). The biotech now has the option to find a new partner who can take over that funding.
"Aveo is very pleased to regain worldwide rights for the development and commercialization of AV-299," said Aveo CEO Tuan Ha-Ngoc. "Aveo now holds significant commercialization rights to all oncology products in our pipeline, and we believe that we are well-positioned to move toward our goal of becoming a fully-integrated commercial organization.”
Aveo presented positive phase I results for the drug at the 46th Annual Meeting of the American Society of Clinical Oncology (ASCO) held in Chicago earlier this year. The drug was shown to be effective in several different tumor types and was safe at all doses. The biotech moved into its first phase II study of the drug, AV-299, in patients with non-small cell lung cancer and results are expected towards the end of next year.
If the drug looks so promising then why is Merck pulling out? Simple; the Big Pharma has finally gotten around to cleaning out the excess it has in its pipeline since purchasing Schering-Plough. "Merck is pleased with our history of collaborating with AVEO, and would welcome the opportunity to work with AVEO again in the future," said David Nicholson, Ph.D., senior vice president and head of worldwide licensing and knowledge management at Merck. "The decision to return this program to AVEO is a result of portfolio prioritization."
Some critics might say that Merck dropped the drug because the phase II study wasn’t panning out as the pharmaceutical giant had expected, but this scenario is highly unlikely, largely due to the phase II testing being in such early stages of the program. Aveo is also handling all of the development of the drug, thus it’s unlikely that Merck has had a clear picture of what is really going on there.
“We do not believe Merck's decision was due to negative findings of the ongoing Phase II trial of AV-299 in lung cancer,” said Leerink Swann analyst Howard Liang in a note to investors. “In our opinion, regaining rights to this agent bolsters Aveo's pipeline and it now has two agents, one in Phase III and one in Phase II, to which it has broad commercial rights.”
Aveo’s stock is already up 10% on the news to trade around $12 per share on Friday afternoon. Expect the biotech to gain further exposure in mid-2011 when it announces late-stage trial data for its lead product, Tivozanib, and then again toward the end of the year when it announces the results of the phase II study for AV-299. Aveo could be pushed further into the limelight as its competitors announce data for their drugs that are in the same class as AV-299 – specifically ArQule’s (ARQL) ARQ-197.
http://www.minyanville.com/businessmarkets/articles/cancer-h…
Der Chart hat sich in den letzten zwei Tagen aber auch wirklich etwas putzig entwickelt:
Hier ein Artikel, der die Rückgabe von AV-299 durch Merck tatsächlich positiv einordnet. Na ja...
Merck's Loss Is Aveo's Gain
By Lisa LaMotta Oct 01, 2010 3:00 pm
The biotech will move into a strong position as its cancer holdings become more significant now that Merck has pulled out.
Aveo Pharmaceuticals (AVEO) announced on Thursday that its three-year deal with Big Pharma has come to a close. While this sort of news is usually bad news (it often happens when a drug doesn’t work), Aveo could be the one lucking out.
The Cambridge, Massachusetts-based biotech entered into the agreement in April 2007 with Schering-Plough -- now Merck (MRK) -- for the larger company to partner on its then preclinical cancer treatment. At the time, Aveo received a $7.5 million upfront payment, as well as $10 million in equity investments. The biotech was also eligible to receive another $460 million in milestone payments.
The deal was structured so that Aveo made out like a bandit and doesn’t really lose anything now that Merck has decided to end the collaboration. The only downside is that Schering/Merck was funding all of the research and development, but Aveo was able to get the drug into phase II trials on the Big Pharma’s dime (Merck will provide funding for the program through the end of the year). The biotech now has the option to find a new partner who can take over that funding.
"Aveo is very pleased to regain worldwide rights for the development and commercialization of AV-299," said Aveo CEO Tuan Ha-Ngoc. "Aveo now holds significant commercialization rights to all oncology products in our pipeline, and we believe that we are well-positioned to move toward our goal of becoming a fully-integrated commercial organization.”
Aveo presented positive phase I results for the drug at the 46th Annual Meeting of the American Society of Clinical Oncology (ASCO) held in Chicago earlier this year. The drug was shown to be effective in several different tumor types and was safe at all doses. The biotech moved into its first phase II study of the drug, AV-299, in patients with non-small cell lung cancer and results are expected towards the end of next year.
If the drug looks so promising then why is Merck pulling out? Simple; the Big Pharma has finally gotten around to cleaning out the excess it has in its pipeline since purchasing Schering-Plough. "Merck is pleased with our history of collaborating with AVEO, and would welcome the opportunity to work with AVEO again in the future," said David Nicholson, Ph.D., senior vice president and head of worldwide licensing and knowledge management at Merck. "The decision to return this program to AVEO is a result of portfolio prioritization."
Some critics might say that Merck dropped the drug because the phase II study wasn’t panning out as the pharmaceutical giant had expected, but this scenario is highly unlikely, largely due to the phase II testing being in such early stages of the program. Aveo is also handling all of the development of the drug, thus it’s unlikely that Merck has had a clear picture of what is really going on there.
“We do not believe Merck's decision was due to negative findings of the ongoing Phase II trial of AV-299 in lung cancer,” said Leerink Swann analyst Howard Liang in a note to investors. “In our opinion, regaining rights to this agent bolsters Aveo's pipeline and it now has two agents, one in Phase III and one in Phase II, to which it has broad commercial rights.”
Aveo’s stock is already up 10% on the news to trade around $12 per share on Friday afternoon. Expect the biotech to gain further exposure in mid-2011 when it announces late-stage trial data for its lead product, Tivozanib, and then again toward the end of the year when it announces the results of the phase II study for AV-299. Aveo could be pushed further into the limelight as its competitors announce data for their drugs that are in the same class as AV-299 – specifically ArQule’s (ARQL) ARQ-197.
http://www.minyanville.com/businessmarkets/articles/cancer-h…
Neuigkeiten bei Array, die nachbörslich gut aufgenommen worden sind (+ 16%):
(wobei diese nachbörslichen Kurse angesichts der lächerlichen Volumina nicht sehr ernst genommen werden dürfen)
October 01, 2010 05:30 PM Eastern Daylight Time
Array BioPharma Announces Positive Results for HER2 Inhibitor, ARRY-380, in Breast Cancer Patients
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (NASDAQ: ARRY) today announced positive interim results of its novel, oral HER2 (ErbB2) inhibitor, ARRY-380, in a Phase 1 trial in patients with advanced cancer. These results are being presented at the 2010 ASCO Breast Cancer Symposium in National Harbor, Maryland. The complete poster (#A7) entitled, “A Phase 1 Study to Assess the Safety, Tolerability and Pharmacokinetics of ARRY-380 - an Oral Inhibitor of HER2,” is available as a PDF on Array's website: http://www.arraybiopharma.com/PatentsPublications/Default.as…
Interim results were presented on 17 HER2 positive metastatic breast cancer patients treated with ARRY-380 at doses greater than or equal to 200 mg BID. All of these patients had been previously treated with Herceptin® (trastuzumab), and 81 percent were previously treated with Tykerb® (lapatinib). Twenty-nine percent of these patients had a partial response or stable disease for six months or longer. Thirteen of the 17 patients had measurable disease as defined by the Response Evaluation Criteria in Solid Tumors (RECIST); of these patients, seven (54 percent) had regressions in target lesions. Of the four patients with no measurable disease, three had regressions of non-target chest wall lesions as documented by photographic evaluation.
ARRY-380 has been well-tolerated; the predominant treatment-related adverse events have been Grade 1. Because ARRY-380 is selective for HER2 and does not inhibit EGFR, there was, as expected, a low incidence and severity of diarrhea, rash and fatigue. Additionally, there were no Grade 4 events or adverse cardiac events reported. The maximum tolerated dose of ARRY-380 established in this Phase 1 trial is 600 mg (twice daily). An expansion cohort in patients with HER2 positive metastatic breast cancer is ongoing to confirm safety and explore efficacy and pharmacodynamic markers.
“I am optimistic about this orally available, selective HER2 inhibitor, ARRY-380. To date, it has been well-tolerated and has shown activity in patients previously treated with trastuzumab and lapatinib,” said Stacy Moulder, M.D., Clinical Investigator, M.D. Anderson Cancer Center. “We remain enthusiastic about prospects for the drug based on this new data.”
“Maintaining tumor remission while improving breast cancer patients’ quality of life represents one of the largest commercial opportunities in cancer therapy,” said Kevin Koch, Ph.D., President and Chief Scientific Officer. “In this clinical trial, ARRY-380 has been well-tolerated and has shown signs of clinical activity, including the shrinking of visceral metastases.”
About HER2 and ARRY-380
HER2, also known as ErbB2, is a receptor tyrosine kinase that has been found to be over-expressed in breast cancer and other cancers such as gastric and ovarian. Herceptin® (trastuzumab), the intravenously-dosed protein inhibitor that modulates HER2, has been approved as an adjuvant to surgery in early stage breast cancer patients; it had already been approved for the treatment of HER2+ metastatic breast cancer. This new indication has significantly expanded the number of breast cancer patients eligible for treatment with an HER2 inhibitor.
ARRY-380 is a potent, orally active small molecule drug that has been well-tolerated to date and selectively inhibits tumor growth in preclinical models of HER2-dependent tumors. ARRY-380 also demonstrated significant dose-related tumor growth inhibition in preclinical models that was superior to Herceptin and Tykerb® (lapatinib) and was additive for tumor growth inhibition in preclinical models when dosed in combination with the standard of care therapeutics such as Herceptin or Taxotere® (docetaxel). Based on its preclinical activity, tolerability and pharmacokinetics, and the results of the Phase 1 study to evaluate the safety, tolerability and pharmacokinetic profile and to establish the maximum tolerated dose of ARRY-380, Array is conducting an expansion cohort in patients with HER2 positive metastatic breast cancer to confirm safety and explore efficacy and pharmacodynamic markers.
(wobei diese nachbörslichen Kurse angesichts der lächerlichen Volumina nicht sehr ernst genommen werden dürfen)
October 01, 2010 05:30 PM Eastern Daylight Time
Array BioPharma Announces Positive Results for HER2 Inhibitor, ARRY-380, in Breast Cancer Patients
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (NASDAQ: ARRY) today announced positive interim results of its novel, oral HER2 (ErbB2) inhibitor, ARRY-380, in a Phase 1 trial in patients with advanced cancer. These results are being presented at the 2010 ASCO Breast Cancer Symposium in National Harbor, Maryland. The complete poster (#A7) entitled, “A Phase 1 Study to Assess the Safety, Tolerability and Pharmacokinetics of ARRY-380 - an Oral Inhibitor of HER2,” is available as a PDF on Array's website: http://www.arraybiopharma.com/PatentsPublications/Default.as…
Interim results were presented on 17 HER2 positive metastatic breast cancer patients treated with ARRY-380 at doses greater than or equal to 200 mg BID. All of these patients had been previously treated with Herceptin® (trastuzumab), and 81 percent were previously treated with Tykerb® (lapatinib). Twenty-nine percent of these patients had a partial response or stable disease for six months or longer. Thirteen of the 17 patients had measurable disease as defined by the Response Evaluation Criteria in Solid Tumors (RECIST); of these patients, seven (54 percent) had regressions in target lesions. Of the four patients with no measurable disease, three had regressions of non-target chest wall lesions as documented by photographic evaluation.
ARRY-380 has been well-tolerated; the predominant treatment-related adverse events have been Grade 1. Because ARRY-380 is selective for HER2 and does not inhibit EGFR, there was, as expected, a low incidence and severity of diarrhea, rash and fatigue. Additionally, there were no Grade 4 events or adverse cardiac events reported. The maximum tolerated dose of ARRY-380 established in this Phase 1 trial is 600 mg (twice daily). An expansion cohort in patients with HER2 positive metastatic breast cancer is ongoing to confirm safety and explore efficacy and pharmacodynamic markers.
“I am optimistic about this orally available, selective HER2 inhibitor, ARRY-380. To date, it has been well-tolerated and has shown activity in patients previously treated with trastuzumab and lapatinib,” said Stacy Moulder, M.D., Clinical Investigator, M.D. Anderson Cancer Center. “We remain enthusiastic about prospects for the drug based on this new data.”
“Maintaining tumor remission while improving breast cancer patients’ quality of life represents one of the largest commercial opportunities in cancer therapy,” said Kevin Koch, Ph.D., President and Chief Scientific Officer. “In this clinical trial, ARRY-380 has been well-tolerated and has shown signs of clinical activity, including the shrinking of visceral metastases.”
About HER2 and ARRY-380
HER2, also known as ErbB2, is a receptor tyrosine kinase that has been found to be over-expressed in breast cancer and other cancers such as gastric and ovarian. Herceptin® (trastuzumab), the intravenously-dosed protein inhibitor that modulates HER2, has been approved as an adjuvant to surgery in early stage breast cancer patients; it had already been approved for the treatment of HER2+ metastatic breast cancer. This new indication has significantly expanded the number of breast cancer patients eligible for treatment with an HER2 inhibitor.
ARRY-380 is a potent, orally active small molecule drug that has been well-tolerated to date and selectively inhibits tumor growth in preclinical models of HER2-dependent tumors. ARRY-380 also demonstrated significant dose-related tumor growth inhibition in preclinical models that was superior to Herceptin and Tykerb® (lapatinib) and was additive for tumor growth inhibition in preclinical models when dosed in combination with the standard of care therapeutics such as Herceptin or Taxotere® (docetaxel). Based on its preclinical activity, tolerability and pharmacokinetics, and the results of the Phase 1 study to evaluate the safety, tolerability and pharmacokinetic profile and to establish the maximum tolerated dose of ARRY-380, Array is conducting an expansion cohort in patients with HER2 positive metastatic breast cancer to confirm safety and explore efficacy and pharmacodynamic markers.
Antwort auf Beitrag Nr.: 40.254.978 von SLGramann am 03.10.10 09:28:59
Hammer hat das kurz so eingeordnet:
The 380 data looks interesting but very early. The value of the drug is in combination with Herceptin and head to head vs. Tykerb. In theory this drug could be very meaningful as the only selective HER2 inhibitor (selective compounds are all the range right now…)but there is not enough data. The p1 results cetainly merit a phase II study.
Er will demnächst einen größeren Artikel über Array schreiben und wird seine Position aufstocken (und zwar nicht (nur) wegen dieser Medlung).
Hammer hat das kurz so eingeordnet:
The 380 data looks interesting but very early. The value of the drug is in combination with Herceptin and head to head vs. Tykerb. In theory this drug could be very meaningful as the only selective HER2 inhibitor (selective compounds are all the range right now…)but there is not enough data. The p1 results cetainly merit a phase II study.
Er will demnächst einen größeren Artikel über Array schreiben und wird seine Position aufstocken (und zwar nicht (nur) wegen dieser Medlung).
Antwort auf Beitrag Nr.: 40.255.797 von SLGramann am 03.10.10 15:32:31
Ohad Hammer mit einem laaaangen Artikel zu Array (so schnell ging das jetzt):
Array’s (ARRY) shares keep on fluctuating in the $2.5-$3.5 range, relatively unchanged from the beginning of 2010. It seems that the market is having trouble assessing the real value of the company and its pipeline, which includes 13 (!) drugs in clinical trials. With a market cap of ~$170M, the market puts an average price tag of $13M per asset, a ridiculously low valuation (assuming no value is assigned to the company’s discovery platform). The company’s long term debt (due in 2014) could be partially blamed for this anomaly, but the problem seems to be more related to the company’s business model. The good news is that during the next year the company is looking at multiple events that might change the way Wall Street views Array.
...
hier gehts weiter:
http://www.hammerstockblog.com/array-biopharma%E2%80%93-a-wa…
Ohad Hammer mit einem laaaangen Artikel zu Array (so schnell ging das jetzt):
Array’s (ARRY) shares keep on fluctuating in the $2.5-$3.5 range, relatively unchanged from the beginning of 2010. It seems that the market is having trouble assessing the real value of the company and its pipeline, which includes 13 (!) drugs in clinical trials. With a market cap of ~$170M, the market puts an average price tag of $13M per asset, a ridiculously low valuation (assuming no value is assigned to the company’s discovery platform). The company’s long term debt (due in 2014) could be partially blamed for this anomaly, but the problem seems to be more related to the company’s business model. The good news is that during the next year the company is looking at multiple events that might change the way Wall Street views Array.
...
hier gehts weiter:
http://www.hammerstockblog.com/array-biopharma%E2%80%93-a-wa…
Jason Chew hat die Entwicklung bei Incyte betrachtet. Nichts wirklich neues, aber dennoch eine interessante Zusammenfassung:
Monday, October 4, 2010
Incyte Therapeutics’ JAK: Just Another Kinase?
Jason Chew
With a nearly $2 billion market cap, Incyte is currently the most valuable biotech stock with no marketed product. Is it justified? Practically all of the company’s value is wrapped up in two related Janus Kinase (JAK) inhibitors, both of which are partnered. INCB18424 for oncology is partnered with Novartis and the related compound, INCB28050, for inflammation is partnered with Eli Lilly.
There is a tremendous amount of interest in the JAK inhibitor space. At least six other companies, including such giants as Pfizer, AstraZeneca, and Sanofi Aventis have compounds in mid to late stage development. Incyte, though, may be first to market, with final data in Myelofibrosis (MF) due end of this year and a regulatory filing expected the first half of next. MF is one of a closely related group of hematological malignancies known as Myeloproliferative disorders (MPDs) in which the bone marrow cells that produce the body's blood cells develop and function abnormally. According to the CEO, INCB18424 has an 18 to 24 month lead on its closes competition.
INCB18424 is also beginning a Phase III trial for a second MPD, Polycythaemia Vera (PV) this year. In this indication, the company feels it has an even larger head start on its competitors than in MF. As with MF, investors have high hopes for success in this trial due to exceptional early data. In addition, the compound is in Phase I/II trials for ALL and Phase II trials for AML, two blood cancer indications.
Focusing only on MF and PV, where INCB18424 is closest to market, Incyte looks well positioned to join the ranks of profitable biotechs. According to the MPD foundation, there are 30,000 patients in the US living with MF and 71,000 with PV. A recent Incyte presentation pegged the number of MF patients at 16,000-18,500 and PV at 95,000. INCB18424 will be priced on par with other new cancer medicines, somewhere between $40k and $60k. Penetration in the MF market is expected to be thorough with high levels of usage due to a lack of alternatives. In PV, the target population is the 20-25% of patients intolerant to hydroxyurea therapy. Usage in this population is also expected to be quite thorough.
Based on these assumptions and estimates, INCB18424 can conceivably achieve blockbuster sales in these two indications in the US alone. It has the potential to be a first in class drug, serving multiple unmet needs.
Incyte’s second JAK inhibitor, INCB28050, is only in mid-stage trials for rheumatoid arthritis (RA) so far. Partial data from an ongoing Phase IIa trial showed the compound to be comparable to Pfizer’s competitor JAK inhibitor, CP690550. Pfizer’s compound is further ahead in the clinic; Phase III results are expected later this year, with a possible launch in 2013. Incyte is to begin Phase IIb trials for its compounds later this year and is about two years behind Pfizer.
INCB28050, CP690550, together with Syk kinase inhibitor, R788, from Rigel/AstraZeneca make up the trifecta of oral compounds knocking on the door of the rheumatoid arthritis/autoimmune disorder market- one currently held firm by anti-TNF therapies. Anti-TNF drugs, including Humira, Remicade, Enbrel, and Cimzia, had sales of $16 billion in 2008. Sales of these biologics are projected to grow to $25 billion by 2014. The top selling drug, Humira, priced at $13,000 during its launch in 2003, is projected to reach sales of $13.7 billion in 2016, placing it solidy as the world’s best selling drug. One reason for its success- of all the anti-TNF drugs, it is the only one that can be self-administered.
This bodes well for the oral compounds. Based on early data, INCB28050 certainly appears efficacious. It also has the convenience of oral administration- no more needles. Another advantage: the compound appears to be active as a monotherapy. Compare this to current anti-TNF therapies, which must be taken concurrently with a methotrexate background treatment.
The biggest hurdle to approval remains safety. There is little safety data available for INCB28050 due to its relatively early stage in clinical development. So far, management has painted a picture of a compound with a very clean profile, “adverse events were mild to moderate in intensity with frequency similar to that observed in patients treated with placebo” is how they described results released this May from their ongoing Phase IIa trial. We’ll find out more details at the American College of Rheumatology (ACR) meeting come November. But already, management is on the lookout for total cholesterol increases and liver enzyme elevations- two events seen with Pfizer’s JAK inhibitor.
If these early results are maintained in late stage trials and the compound isn’t derailed by a previously undetected adverse effect, INCB28050 has huge potential. Both management and rheumatologists believe oral therapies will eventually become the standard of care for RA treatment before the use of anti-TNF and other biologics. If Incyte’s compound is able to penetrate just 5% of the anti-TNF market, it will be a blockbuster (I certainly see significantly higher penetration levels). It won’t all flow to Incyte. Worldwide rights have been licensed to Eli Lilly, Incyte has opted in to co-develop the compound and will receive somewhere between 20-30% in royalties. Not too shabby.
So while analysts are projecting Humira to be the world’s top selling drug in 2016, note that it will likely have three novel competitors by then. Don’t expect its reign to last.
Author is Long INCY, ABT
http://biopharmareport.blogspot.com/2010/10/incyte-therapeut…
Monday, October 4, 2010
Incyte Therapeutics’ JAK: Just Another Kinase?
Jason Chew
With a nearly $2 billion market cap, Incyte is currently the most valuable biotech stock with no marketed product. Is it justified? Practically all of the company’s value is wrapped up in two related Janus Kinase (JAK) inhibitors, both of which are partnered. INCB18424 for oncology is partnered with Novartis and the related compound, INCB28050, for inflammation is partnered with Eli Lilly.
There is a tremendous amount of interest in the JAK inhibitor space. At least six other companies, including such giants as Pfizer, AstraZeneca, and Sanofi Aventis have compounds in mid to late stage development. Incyte, though, may be first to market, with final data in Myelofibrosis (MF) due end of this year and a regulatory filing expected the first half of next. MF is one of a closely related group of hematological malignancies known as Myeloproliferative disorders (MPDs) in which the bone marrow cells that produce the body's blood cells develop and function abnormally. According to the CEO, INCB18424 has an 18 to 24 month lead on its closes competition.
INCB18424 is also beginning a Phase III trial for a second MPD, Polycythaemia Vera (PV) this year. In this indication, the company feels it has an even larger head start on its competitors than in MF. As with MF, investors have high hopes for success in this trial due to exceptional early data. In addition, the compound is in Phase I/II trials for ALL and Phase II trials for AML, two blood cancer indications.
Focusing only on MF and PV, where INCB18424 is closest to market, Incyte looks well positioned to join the ranks of profitable biotechs. According to the MPD foundation, there are 30,000 patients in the US living with MF and 71,000 with PV. A recent Incyte presentation pegged the number of MF patients at 16,000-18,500 and PV at 95,000. INCB18424 will be priced on par with other new cancer medicines, somewhere between $40k and $60k. Penetration in the MF market is expected to be thorough with high levels of usage due to a lack of alternatives. In PV, the target population is the 20-25% of patients intolerant to hydroxyurea therapy. Usage in this population is also expected to be quite thorough.
Based on these assumptions and estimates, INCB18424 can conceivably achieve blockbuster sales in these two indications in the US alone. It has the potential to be a first in class drug, serving multiple unmet needs.
Incyte’s second JAK inhibitor, INCB28050, is only in mid-stage trials for rheumatoid arthritis (RA) so far. Partial data from an ongoing Phase IIa trial showed the compound to be comparable to Pfizer’s competitor JAK inhibitor, CP690550. Pfizer’s compound is further ahead in the clinic; Phase III results are expected later this year, with a possible launch in 2013. Incyte is to begin Phase IIb trials for its compounds later this year and is about two years behind Pfizer.
INCB28050, CP690550, together with Syk kinase inhibitor, R788, from Rigel/AstraZeneca make up the trifecta of oral compounds knocking on the door of the rheumatoid arthritis/autoimmune disorder market- one currently held firm by anti-TNF therapies. Anti-TNF drugs, including Humira, Remicade, Enbrel, and Cimzia, had sales of $16 billion in 2008. Sales of these biologics are projected to grow to $25 billion by 2014. The top selling drug, Humira, priced at $13,000 during its launch in 2003, is projected to reach sales of $13.7 billion in 2016, placing it solidy as the world’s best selling drug. One reason for its success- of all the anti-TNF drugs, it is the only one that can be self-administered.
This bodes well for the oral compounds. Based on early data, INCB28050 certainly appears efficacious. It also has the convenience of oral administration- no more needles. Another advantage: the compound appears to be active as a monotherapy. Compare this to current anti-TNF therapies, which must be taken concurrently with a methotrexate background treatment.
The biggest hurdle to approval remains safety. There is little safety data available for INCB28050 due to its relatively early stage in clinical development. So far, management has painted a picture of a compound with a very clean profile, “adverse events were mild to moderate in intensity with frequency similar to that observed in patients treated with placebo” is how they described results released this May from their ongoing Phase IIa trial. We’ll find out more details at the American College of Rheumatology (ACR) meeting come November. But already, management is on the lookout for total cholesterol increases and liver enzyme elevations- two events seen with Pfizer’s JAK inhibitor.
If these early results are maintained in late stage trials and the compound isn’t derailed by a previously undetected adverse effect, INCB28050 has huge potential. Both management and rheumatologists believe oral therapies will eventually become the standard of care for RA treatment before the use of anti-TNF and other biologics. If Incyte’s compound is able to penetrate just 5% of the anti-TNF market, it will be a blockbuster (I certainly see significantly higher penetration levels). It won’t all flow to Incyte. Worldwide rights have been licensed to Eli Lilly, Incyte has opted in to co-develop the compound and will receive somewhere between 20-30% in royalties. Not too shabby.
So while analysts are projecting Humira to be the world’s top selling drug in 2016, note that it will likely have three novel competitors by then. Don’t expect its reign to last.
Author is Long INCY, ABT
http://biopharmareport.blogspot.com/2010/10/incyte-therapeut…
Hi ipollit,
neue Daten zu T-DM1. Was hältst Du davon? Meiner Meinung nach ist T-DM1 weiterhin auf dem Weg, eines Tages Herceptin mehr oder weniger komplett zu ersetzen. Siehst Du das auch so oder übertreibe ich?
WALTHAM, Mass., Oct 08, 2010 (BUSINESS WIRE) -- ImmunoGen, Inc. / , a biotechnology company that develops targeted antibody-based anticancer products, today announced the reporting of positive interim clinical data with trastuzumab-DM1 (T-DM1) for first-line treatment of HER2+ metastatic breast cancer in a press release issued by the European Society for Medical Oncology (ESMO). T-DM1 consists of ImmunoGen's DM1 cancer cell-killing agent attached to the HER2-targeting antibody, trastuzumab, developed by Genentech, a member of the Roche Group. The compound is in global development by Roche under a collaboration agreement between ImmunoGen and Genentech.
The ESMO press release reports on data to be presented on Monday, October 11 in its Presidential Symposium. These data are initial findings from a 137-patient randomized Phase II clinical trial comparing T-DM1 versus Herceptin(R) (trastuzumab) plus chemotherapy (docetaxel) for the treatment of HER2+ metastatic breast cancer that has not previously been treated with chemotherapy. Herceptin plus chemotherapy is current standard first-line therapy for this cancer.
The objective response rate for patients treated with T-DM1 was 47.8%, compared to 41.4% for patients treated with Herceptin plus chemotherapy, with a minimum of 5.9 months of patient follow-up. Longer follow-up is required for analysis of progression-free survival (PFS) -- the primary endpoint of the study -- and for final, mature objective response rate data. Final PFS data are expected to be available in 2Q 2011.
Of particular note, the incidence of more serious side effects (Grade 3 and higher) reported with T-DM1 was approximately half of that reported with trastuzumab plus docetaxel (37% vs. 75%, respectively), with no increased risk of cardiotoxicity. There were no new safety-signals reported with T-DM1 in this trial compared with earlier T-DM1 clinical studies.
The most common adverse events (all grades) in the Herceptin plus docetaxel arm of the study were alopecia (66.2% compared to 1.5% for the T-DM1 arm), neutropenia (57.4% vs. 7.5%) and diarrhea (45.6% vs. 10.4%). The most common adverse events in the T-DM1 arm of the study were nausea (47.8% vs. 39.7% for the Herceptin plus docetaxel arm), fatigue (46.3% vs. 46.2%) and temperature increase (35.8% vs. 20.6%).
This early analysis supports further investigation of T-DM1 for first-line treatment of HER2+ metastatic breast cancer. In July 2010, Roche initiated a Phase III trial, MARIANNE, to assess T-DM1 further for this use. MARIANNE compares both T-DM1 alone and T-DM1 used in combination with pertuzumab to Herceptin used in combination with a taxane in patients with advanced HER2+ breast cancer not previously treated for advanced disease.
neue Daten zu T-DM1. Was hältst Du davon? Meiner Meinung nach ist T-DM1 weiterhin auf dem Weg, eines Tages Herceptin mehr oder weniger komplett zu ersetzen. Siehst Du das auch so oder übertreibe ich?
WALTHAM, Mass., Oct 08, 2010 (BUSINESS WIRE) -- ImmunoGen, Inc. / , a biotechnology company that develops targeted antibody-based anticancer products, today announced the reporting of positive interim clinical data with trastuzumab-DM1 (T-DM1) for first-line treatment of HER2+ metastatic breast cancer in a press release issued by the European Society for Medical Oncology (ESMO). T-DM1 consists of ImmunoGen's DM1 cancer cell-killing agent attached to the HER2-targeting antibody, trastuzumab, developed by Genentech, a member of the Roche Group. The compound is in global development by Roche under a collaboration agreement between ImmunoGen and Genentech.
The ESMO press release reports on data to be presented on Monday, October 11 in its Presidential Symposium. These data are initial findings from a 137-patient randomized Phase II clinical trial comparing T-DM1 versus Herceptin(R) (trastuzumab) plus chemotherapy (docetaxel) for the treatment of HER2+ metastatic breast cancer that has not previously been treated with chemotherapy. Herceptin plus chemotherapy is current standard first-line therapy for this cancer.
The objective response rate for patients treated with T-DM1 was 47.8%, compared to 41.4% for patients treated with Herceptin plus chemotherapy, with a minimum of 5.9 months of patient follow-up. Longer follow-up is required for analysis of progression-free survival (PFS) -- the primary endpoint of the study -- and for final, mature objective response rate data. Final PFS data are expected to be available in 2Q 2011.
Of particular note, the incidence of more serious side effects (Grade 3 and higher) reported with T-DM1 was approximately half of that reported with trastuzumab plus docetaxel (37% vs. 75%, respectively), with no increased risk of cardiotoxicity. There were no new safety-signals reported with T-DM1 in this trial compared with earlier T-DM1 clinical studies.
The most common adverse events (all grades) in the Herceptin plus docetaxel arm of the study were alopecia (66.2% compared to 1.5% for the T-DM1 arm), neutropenia (57.4% vs. 7.5%) and diarrhea (45.6% vs. 10.4%). The most common adverse events in the T-DM1 arm of the study were nausea (47.8% vs. 39.7% for the Herceptin plus docetaxel arm), fatigue (46.3% vs. 46.2%) and temperature increase (35.8% vs. 20.6%).
This early analysis supports further investigation of T-DM1 for first-line treatment of HER2+ metastatic breast cancer. In July 2010, Roche initiated a Phase III trial, MARIANNE, to assess T-DM1 further for this use. MARIANNE compares both T-DM1 alone and T-DM1 used in combination with pertuzumab to Herceptin used in combination with a taxane in patients with advanced HER2+ breast cancer not previously treated for advanced disease.
Antwort auf Beitrag Nr.: 40.292.430 von SLGramann am 08.10.10 21:25:24hi SLGramann!
für mich hört es sich auch positiv an. Die Idee, damit die Chemo gezielter einzusetzen, scheint ja zu funktionieren. Halb soviele Nebenwirkungen bei einer vielleicht sogar etwas höheren Wirkung wäre ja schon eine deutliche Verbesserung. Sehr deutlich sieht man das auch beim Haarausfall, der bei Taxotere mit 66% sehr häufig und bei T-DM1 fast garnicht (1,5%) auftritt. Mal schauen wie am Montag weitere Details aussehen. Zudem sind es ja erst die vorläufigen Daten... vielleicht sieht es bei PFS oder Überlebensraten am Ende noch besser aus. Inzwischen habe ich auch ein paar IMGN... dafür kam mir der Rückschlag zuletzt ganz gelegen.
mfg ipollit
für mich hört es sich auch positiv an. Die Idee, damit die Chemo gezielter einzusetzen, scheint ja zu funktionieren. Halb soviele Nebenwirkungen bei einer vielleicht sogar etwas höheren Wirkung wäre ja schon eine deutliche Verbesserung. Sehr deutlich sieht man das auch beim Haarausfall, der bei Taxotere mit 66% sehr häufig und bei T-DM1 fast garnicht (1,5%) auftritt. Mal schauen wie am Montag weitere Details aussehen. Zudem sind es ja erst die vorläufigen Daten... vielleicht sieht es bei PFS oder Überlebensraten am Ende noch besser aus. Inzwischen habe ich auch ein paar IMGN... dafür kam mir der Rückschlag zuletzt ganz gelegen.
mfg ipollit
ONXX... die Beantragung der Martkzulassung von Carfilzomib verschiebt sich um ein halbes Jahr, da noch Fragen zum Herstellungsprozess bestehen.
http://www.fool.com/investing/high-growth/2010/10/08/onyx-de…
Application As FDA Asks for More Data
By Luke Timmerman
October 8, 2010
Some minor bad news came out Thursday afternoon from Onyx Pharmaceuticals (Nasdaq: ONXX) and its lead drug candidate for patients with multiple myeloma.
The Emeryville, CA-based drug developer said it is delaying the new drug application for carfilzomib after FDA officials asked for more data to show it can manufacture larger commercial batches that are consistent with the molecules it produced and tested in clinical trials. Onyx had previously forecasted it would turn in the application by the end of 2010, and now says it will come " as early as mid-year 2011."
" This is a timing issue," Onyx CEO Tony Coles said on a conference call with analysts. " We believe the fundamental value proposition for carfilzomib is unchanged and the commercial opportunity is significant."
The FDA's request for more data came after the company made some modifications to its manufacturing process as it attempts to start scaling up for commercial demand. The company saw some " minor variations" that are thought to be related to minor changes in equipment temperatures at the larger scale. Onyx has generated some data already, and is working to produce more, to show its larger-scale process is reproducible and creates a product with the same properties that were observed in the clinical trials.
The new drug is vitally important to Onyx's growth prospects, as it seeks to diversify beyond its lone marketed product at the moment, sorafenib (Nexavar) for kidney and liver cancer. Onyx agreed to pay $276 million upfront, plus another potential $535 million in milestone payments, to obtain carfilzomib last year through the acquisition of South San Francisco-based Proteolix. A few months later, in July, Onyx reported that the new drug helped shrink tumors in about one-fourth of patients who had relapsed after getting prior rounds of treatment. The results were compelling enough to seek FDA approval, even though the drug hasn't yet completed the third and final stage of clinical trials usually required before a drug can be sold in the U.S.
If the drug is approved, it would be a new treatment option for a disease that hits about 20,000 people each year in the U.S., and kills about half that many every year, according to the American Cancer Society.
Onyx shares fell about 7% in after-hours trading following news of the delay.

mfg ipollit
http://www.fool.com/investing/high-growth/2010/10/08/onyx-de…
Application As FDA Asks for More Data
By Luke Timmerman
October 8, 2010
Some minor bad news came out Thursday afternoon from Onyx Pharmaceuticals (Nasdaq: ONXX) and its lead drug candidate for patients with multiple myeloma.
The Emeryville, CA-based drug developer said it is delaying the new drug application for carfilzomib after FDA officials asked for more data to show it can manufacture larger commercial batches that are consistent with the molecules it produced and tested in clinical trials. Onyx had previously forecasted it would turn in the application by the end of 2010, and now says it will come " as early as mid-year 2011."
" This is a timing issue," Onyx CEO Tony Coles said on a conference call with analysts. " We believe the fundamental value proposition for carfilzomib is unchanged and the commercial opportunity is significant."
The FDA's request for more data came after the company made some modifications to its manufacturing process as it attempts to start scaling up for commercial demand. The company saw some " minor variations" that are thought to be related to minor changes in equipment temperatures at the larger scale. Onyx has generated some data already, and is working to produce more, to show its larger-scale process is reproducible and creates a product with the same properties that were observed in the clinical trials.
The new drug is vitally important to Onyx's growth prospects, as it seeks to diversify beyond its lone marketed product at the moment, sorafenib (Nexavar) for kidney and liver cancer. Onyx agreed to pay $276 million upfront, plus another potential $535 million in milestone payments, to obtain carfilzomib last year through the acquisition of South San Francisco-based Proteolix. A few months later, in July, Onyx reported that the new drug helped shrink tumors in about one-fourth of patients who had relapsed after getting prior rounds of treatment. The results were compelling enough to seek FDA approval, even though the drug hasn't yet completed the third and final stage of clinical trials usually required before a drug can be sold in the U.S.
If the drug is approved, it would be a new treatment option for a disease that hits about 20,000 people each year in the U.S., and kills about half that many every year, according to the American Cancer Society.
Onyx shares fell about 7% in after-hours trading following news of the delay.
mfg ipollit
Antwort auf Beitrag Nr.: 39.734.598 von ipollit am 25.06.10 00:23:36REGN - Im Juni kam von Pfizer die Meldung, dass Studien zum Schmerzmittel Tanezumab wegen unerwarteter Nebenwirkungen abgebrochen werden mussten, da es bei Patienten mit Osteoarthritis zu Gelenkschäden kam. Tanezumab ist als NGF-AK sehr ähnlich zu Sanofi/REGN's REGN475, das sich gerade in PII befindet.
Nun hat sich herausgestellt, dass es sich bei dieser "Nebenwirkung" eigentlich um eine zu gute Wirkung von Tanezumab handelt. Die Schmerzen werden derart wirksam unterdrückt, dass die Patienten ohne Schmerzen ihre Gelenke zu sehr belasten. Mal sehen wie man bei Regeneron damit umgeht REGN475... zumindest die Wirksamkeit von REGN475 scheint nicht in Frage zu stehen.
http://abcnews.go.com/Health/PainArthritis/pfizer-arthritis-…
'Game-Changing' Arthritis Drug Blocks Pain Too Well in Some
Tanezumab Blunted Pain So Effectively in Test That Patients Never Felt Warning Signs of Injury
By JANE E. ALLEN, ABC News Medical Unit
Sept. 30, 2010
An experimental pain-relieving drug has yielded an unexpected finding for patients with osteoarthritis.
It's not that the drug doesn't work; indeed, it appears to be remarkably effective against pain in many with arthritic knees. But in some patients, the drug blunted joint pain so powerfully they never felt the warning signs or twinges that they were overdoing it -- and suffered joint destruction as a result.
In the quest for new pain relievers with minimal side effects, researchers have been focusing on a chemical known as nerve growth factor, which has been associated with increased pain from a variety of injuries and inflammatory conditions.
The experimental drug in this study aims to inhibit nerve growth factor. Its effect is significant, especially in light of the prevalence of osteoarthritis, a common result of excessive wear-and-tear on the joints, which plagues an estimated 27 million American adults. Many sufferers seek pain relief from non-narcotic medications.
"This is a radical notion for most people: that pain can be protective, but if you think about it, without pain signals, we would injure ourselves all the time" said Dr. Jack Choueka, chairman of orthopedics at Maimonides Medical Center in Brooklyn, N.Y., who was not involved in the study.
Doctors strive to reduce chronic pain, but they need to preserve at least some of it. It is the body's way of putting up a red flag warning about imminent tissue damage, Choueka said. "So it's important for doctors to help patients cope with pain, but not to the point where their ability to feel pain is impaired and places them in danger. Ergo: a little pain is a good thing."
The drug that worked "too well," tanezumab, is among a class of targeted treatments using monoclonal antibodies that latch onto a specific target, in this case nerve growth factor, and neutralize it.
In a clinical trial, reported in this week's New England Journal of Medicine, researchers assessed the safety and pain-relieving effect of the Pfizer drug on patients who suffered severe knee pain while walking on a flat surface. All 450 men and women were either unwilling to take non-narcotic pain relievers, hadn't gotten relief from such medications, or were facing knee surgery or injections.
The researchers, led by Dr. Nancy E. Lane, director of the Center for Healthy Aging at the University of California, Davis, randomly assigned them to receive one of five different doses of tanezumab intravenously -- on the first and 56th day of the study -- or a placebo.
Drug 'Like Novocain' Blocks Even Useful Pain in Some
Indisputably, tanezumab did a good job. Patients reported it reduced their knee pain anywhere from 45 percent to 62 percent on average, compared with a 22 percent average reduction for those getting a placebo. The drug often achieved this in the first couple of days after treatment, according to patient diary entries, Lane said. That surpassed the 30 percent reduction in pain intensity often cited as a threshold for meaningful pain relief.
The effect persisted through four months of treatment, allowing some patients to do previously unheard-of things like dance again, Lane said. The medication also reduced stiffness and improved physical function more than the placebo, with only mild to moderate side effects, which generally dissipated in a few days. The most common were headache, upper respiratory infection and pricking or tingling sensations in the hands and feet.
But tanezumab's potential as an arthritis pain reliever has been tempered by emerging evidence that use can lead to joint failure, said John N. Wood of the Wolfson Institute for Biomedical Research at University College, London, in an accompanying editorial in the New England Journal. Since the UC Davis-led study ended on May 24, 16 patients in a Phase III study of the drug in knee and hip osteoarthritis suffered joint breakdown that required total joint replacements.
At the FDA's request, Pfizer in June suspended studies of the drug for osteoarthritis. In July, it also suspended tanezumab studies for lower back pain and diabetic nerve damage, but said it would continue investigating its potential for other conditions, including cancer pain.
In an interview Wednesday, Lane called the tanezumab "a game-changing medication. We've never had an agent this potent. You get something that takes the pain away, you're bound to accelerate the disease."
She described researchers as so dazzled by tanezumab's powerful analgesic effect that they didn't warn their patients to take it easy.
"We've never had anything like this. We didn't know to counsel our patients, to say, 'This is like Novocain that the dentist gives. You're not going to feel anything...
"' Everyone got so excited by the findings, they didn't realize that everyone who has pain -- they aren't necessarily going to sit in a chair all day."
Experimental Drug May Have Advantages Over Current Treatment
Of those patients, she said: "We didn't monitor activity ... or how hard they were pushing, loading their joints. We just let them go to town. And they did."
Lane said the drug has multiple advantages over current analgesics: "It's not a narcotic, so it doesn't mess with your brain and intestines. It doesn't hurt your stomach. It has no effect at all on the kidneys and liver." With so many factors in its favor, she said it's up to the medical research community "to figure out how to use it so patients can benefit. We want to maximize the benefit and lower the risk."
Lane said she was grateful that the destructive side effects appeared while the drug was still experimental, rather than "when the drug was out. Now we can counsel patients, rather than have the drug withdrawn like the COX-2 inhibitors," which were taken off the market when they caused heart problems. "I think this is responsible drug development. Thank goodness it happened now."
While studies of the drug remain suspended at the request of the FDA, she said, "there are there other companies that have this type of a medication in development right now." Those companies have told her the FDA advised them to "go ahead cautiously. I think we will see a nerve growth factor inhibitor on the market -- whether it will be Pfizer's, I can't say."
Dr. Alton J. Stubbs, an assistant professor of orthopedic surgery at Wake Forest University Baptist Medical Center Winston-Salem, N.C., called the study "very important" and said that scientists in the last 15 to 20 years have "become very good at targeting specific proteins in the body that may be associated with chronic disease."
He, too, wasn't sure if tenazumab would ultimately win FDA approval, but said the take-home message of Lane's study was "that the science is advancing, there are some positive therapies on the horizon, and that patients should have a lot to look forward to in the next 5-10 years."
The study has several reported conflicts of interest. Rinat Neuroscience, now a subsidiary of pharmacy giant Pfizer, designed and coordinated the study. Lane, who holds an endowed chair in medicine and rheumatology, has been a paid consultant to Pfizer as well as Amgen, Merck, Aventis and NicOx.
mfg ipollit
Nun hat sich herausgestellt, dass es sich bei dieser "Nebenwirkung" eigentlich um eine zu gute Wirkung von Tanezumab handelt. Die Schmerzen werden derart wirksam unterdrückt, dass die Patienten ohne Schmerzen ihre Gelenke zu sehr belasten. Mal sehen wie man bei Regeneron damit umgeht REGN475... zumindest die Wirksamkeit von REGN475 scheint nicht in Frage zu stehen.
http://abcnews.go.com/Health/PainArthritis/pfizer-arthritis-…
'Game-Changing' Arthritis Drug Blocks Pain Too Well in Some
Tanezumab Blunted Pain So Effectively in Test That Patients Never Felt Warning Signs of Injury
By JANE E. ALLEN, ABC News Medical Unit
Sept. 30, 2010
An experimental pain-relieving drug has yielded an unexpected finding for patients with osteoarthritis.
It's not that the drug doesn't work; indeed, it appears to be remarkably effective against pain in many with arthritic knees. But in some patients, the drug blunted joint pain so powerfully they never felt the warning signs or twinges that they were overdoing it -- and suffered joint destruction as a result.
In the quest for new pain relievers with minimal side effects, researchers have been focusing on a chemical known as nerve growth factor, which has been associated with increased pain from a variety of injuries and inflammatory conditions.
The experimental drug in this study aims to inhibit nerve growth factor. Its effect is significant, especially in light of the prevalence of osteoarthritis, a common result of excessive wear-and-tear on the joints, which plagues an estimated 27 million American adults. Many sufferers seek pain relief from non-narcotic medications.
"This is a radical notion for most people: that pain can be protective, but if you think about it, without pain signals, we would injure ourselves all the time" said Dr. Jack Choueka, chairman of orthopedics at Maimonides Medical Center in Brooklyn, N.Y., who was not involved in the study.
Doctors strive to reduce chronic pain, but they need to preserve at least some of it. It is the body's way of putting up a red flag warning about imminent tissue damage, Choueka said. "So it's important for doctors to help patients cope with pain, but not to the point where their ability to feel pain is impaired and places them in danger. Ergo: a little pain is a good thing."
The drug that worked "too well," tanezumab, is among a class of targeted treatments using monoclonal antibodies that latch onto a specific target, in this case nerve growth factor, and neutralize it.
In a clinical trial, reported in this week's New England Journal of Medicine, researchers assessed the safety and pain-relieving effect of the Pfizer drug on patients who suffered severe knee pain while walking on a flat surface. All 450 men and women were either unwilling to take non-narcotic pain relievers, hadn't gotten relief from such medications, or were facing knee surgery or injections.
The researchers, led by Dr. Nancy E. Lane, director of the Center for Healthy Aging at the University of California, Davis, randomly assigned them to receive one of five different doses of tanezumab intravenously -- on the first and 56th day of the study -- or a placebo.
Drug 'Like Novocain' Blocks Even Useful Pain in Some
Indisputably, tanezumab did a good job. Patients reported it reduced their knee pain anywhere from 45 percent to 62 percent on average, compared with a 22 percent average reduction for those getting a placebo. The drug often achieved this in the first couple of days after treatment, according to patient diary entries, Lane said. That surpassed the 30 percent reduction in pain intensity often cited as a threshold for meaningful pain relief.
The effect persisted through four months of treatment, allowing some patients to do previously unheard-of things like dance again, Lane said. The medication also reduced stiffness and improved physical function more than the placebo, with only mild to moderate side effects, which generally dissipated in a few days. The most common were headache, upper respiratory infection and pricking or tingling sensations in the hands and feet.
But tanezumab's potential as an arthritis pain reliever has been tempered by emerging evidence that use can lead to joint failure, said John N. Wood of the Wolfson Institute for Biomedical Research at University College, London, in an accompanying editorial in the New England Journal. Since the UC Davis-led study ended on May 24, 16 patients in a Phase III study of the drug in knee and hip osteoarthritis suffered joint breakdown that required total joint replacements.
At the FDA's request, Pfizer in June suspended studies of the drug for osteoarthritis. In July, it also suspended tanezumab studies for lower back pain and diabetic nerve damage, but said it would continue investigating its potential for other conditions, including cancer pain.
In an interview Wednesday, Lane called the tanezumab "a game-changing medication. We've never had an agent this potent. You get something that takes the pain away, you're bound to accelerate the disease."
She described researchers as so dazzled by tanezumab's powerful analgesic effect that they didn't warn their patients to take it easy.
"We've never had anything like this. We didn't know to counsel our patients, to say, 'This is like Novocain that the dentist gives. You're not going to feel anything...
"' Everyone got so excited by the findings, they didn't realize that everyone who has pain -- they aren't necessarily going to sit in a chair all day."
Experimental Drug May Have Advantages Over Current Treatment
Of those patients, she said: "We didn't monitor activity ... or how hard they were pushing, loading their joints. We just let them go to town. And they did."
Lane said the drug has multiple advantages over current analgesics: "It's not a narcotic, so it doesn't mess with your brain and intestines. It doesn't hurt your stomach. It has no effect at all on the kidneys and liver." With so many factors in its favor, she said it's up to the medical research community "to figure out how to use it so patients can benefit. We want to maximize the benefit and lower the risk."
Lane said she was grateful that the destructive side effects appeared while the drug was still experimental, rather than "when the drug was out. Now we can counsel patients, rather than have the drug withdrawn like the COX-2 inhibitors," which were taken off the market when they caused heart problems. "I think this is responsible drug development. Thank goodness it happened now."
While studies of the drug remain suspended at the request of the FDA, she said, "there are there other companies that have this type of a medication in development right now." Those companies have told her the FDA advised them to "go ahead cautiously. I think we will see a nerve growth factor inhibitor on the market -- whether it will be Pfizer's, I can't say."
Dr. Alton J. Stubbs, an assistant professor of orthopedic surgery at Wake Forest University Baptist Medical Center Winston-Salem, N.C., called the study "very important" and said that scientists in the last 15 to 20 years have "become very good at targeting specific proteins in the body that may be associated with chronic disease."
He, too, wasn't sure if tenazumab would ultimately win FDA approval, but said the take-home message of Lane's study was "that the science is advancing, there are some positive therapies on the horizon, and that patients should have a lot to look forward to in the next 5-10 years."
The study has several reported conflicts of interest. Rinat Neuroscience, now a subsidiary of pharmacy giant Pfizer, designed and coordinated the study. Lane, who holds an endowed chair in medicine and rheumatology, has been a paid consultant to Pfizer as well as Amgen, Merck, Aventis and NicOx.
mfg ipollit
REGN - eine KE soll 150 Mio USD Cash bringen
Regeneron Pharmaceuticals boosts stock offering to $154 million
On Friday October 8, 2010, 10:31 am EDT
NEW YORK (AP) -- Regeneron Pharmaceuticals Inc. said Friday it is increasing the size of its public offering of common stock to $154 million from $126 million.
The company said it is offering 5.5 million shares at $28 apiece. The stock closed at $27.98 on Thursday.
Citi is serving as the sole underwriter and has a 30-day option to buy up to 825,000 shares to cover overallotments.
Late Wednesday, the company announced an offering for 4.5 million shares at $28 apiece, giving underwriters an overallotment option to buy up to 675,000 shares.
The company plans to use proceeds for general corporate purposes. It has about 79.9 million share of common stock outstanding.
The offering is expected to close on or about Oct. 13

mfg ipollit
Regeneron Pharmaceuticals boosts stock offering to $154 million
On Friday October 8, 2010, 10:31 am EDT
NEW YORK (AP) -- Regeneron Pharmaceuticals Inc. said Friday it is increasing the size of its public offering of common stock to $154 million from $126 million.
The company said it is offering 5.5 million shares at $28 apiece. The stock closed at $27.98 on Thursday.
Citi is serving as the sole underwriter and has a 30-day option to buy up to 825,000 shares to cover overallotments.
Late Wednesday, the company announced an offering for 4.5 million shares at $28 apiece, giving underwriters an overallotment option to buy up to 675,000 shares.
The company plans to use proceeds for general corporate purposes. It has about 79.9 million share of common stock outstanding.
The offering is expected to close on or about Oct. 13
mfg ipollit
Antwort auf Beitrag Nr.: 40.295.573 von ipollit am 10.10.10 15:08:55
Verstehe nicht recht, was die KE jetzt soll. REGN ist doch nicht arm an Cash und im Dezember kommen die Daten für Trap-Eye. Will man da vorbauen?
----------
Es gibt massenweise (gute) Daten und Nachrichten von der ESMO. Aufgefallen sind mir ArQule, Exelixis, Immunogen, Micromet und Seattle Genetics. Muss man mal demnächst etwas sortieren...
Verstehe nicht recht, was die KE jetzt soll. REGN ist doch nicht arm an Cash und im Dezember kommen die Daten für Trap-Eye. Will man da vorbauen?
----------
Es gibt massenweise (gute) Daten und Nachrichten von der ESMO. Aufgefallen sind mir ArQule, Exelixis, Immunogen, Micromet und Seattle Genetics. Muss man mal demnächst etwas sortieren...
Antwort auf Beitrag Nr.: 40.302.242 von SLGramann am 11.10.10 23:45:34
Fangen wir mal mit Arqule an:
a) Das SPA für die P III für ARQ 197 wurde nunmehr vereinbart. Die P III stratet noch in diesem Jahr:
ArQule and Daiichi Sankyo Reach Agreement with FDA on Special Protocol Assessment
WOBURN, Mass., Oct 11, 2010 (BUSINESS WIRE) -- ArQule, Inc. (Nasdaq: ARQL) today announced a Special Protocol Assessment (SPA) agreement with the U.S. Food and Drug Administration (FDA) for the design of a Phase 3 trial of ARQ 197 in patients with non-small cell lung cancer (NSCLC) of non-squamous histology.
Daiichi Sankyo Co., Ltd., the holder of the Investigational New Drug application for ARQ 197 and ArQule's partner for the development of the compound, will conduct the Phase 3 trial, which is planned for initiation later this year.
http://investors.arqule.com/releasedetail.cfm?ReleaseID=5169…
b) Die Kooperation mit Daiichi wird ausgeweitet:
ArQule and Daiichi Sankyo Expand Drug Discovery Collaboration in Oncology
WOBURN, Massachusetts, October 12, 2010 /PRNewswire/ -- ArQule, Inc. (NASDAQ: ARQL) and Daiichi Sankyo Co., Ltd. (TSE 4568) today announced the expansion of their research, development and license agreement for the discovery of novel kinase inhibitors in the field of oncology. This expanded agreement establishes a third therapeutic target, with an option for a fourth, in the field of oncology, and it includes a two-year extension based on the application of the proprietary ArQule Kinase Inhibitor Platform (AKIP(TM)) technology.
http://www.prnewswire.co.uk/cgi/news/release?id=300346
Keine weltumstürzenden News, aber insofern wichtig, als dass sie zeigen, dass bei Arqule weiterhin alles auf dem richtigen Weg ist.
Fangen wir mal mit Arqule an:
a) Das SPA für die P III für ARQ 197 wurde nunmehr vereinbart. Die P III stratet noch in diesem Jahr:
ArQule and Daiichi Sankyo Reach Agreement with FDA on Special Protocol Assessment
WOBURN, Mass., Oct 11, 2010 (BUSINESS WIRE) -- ArQule, Inc. (Nasdaq: ARQL) today announced a Special Protocol Assessment (SPA) agreement with the U.S. Food and Drug Administration (FDA) for the design of a Phase 3 trial of ARQ 197 in patients with non-small cell lung cancer (NSCLC) of non-squamous histology.
Daiichi Sankyo Co., Ltd., the holder of the Investigational New Drug application for ARQ 197 and ArQule's partner for the development of the compound, will conduct the Phase 3 trial, which is planned for initiation later this year.
http://investors.arqule.com/releasedetail.cfm?ReleaseID=5169…
b) Die Kooperation mit Daiichi wird ausgeweitet:
ArQule and Daiichi Sankyo Expand Drug Discovery Collaboration in Oncology
WOBURN, Massachusetts, October 12, 2010 /PRNewswire/ -- ArQule, Inc. (NASDAQ: ARQL) and Daiichi Sankyo Co., Ltd. (TSE 4568) today announced the expansion of their research, development and license agreement for the discovery of novel kinase inhibitors in the field of oncology. This expanded agreement establishes a third therapeutic target, with an option for a fourth, in the field of oncology, and it includes a two-year extension based on the application of the proprietary ArQule Kinase Inhibitor Platform (AKIP(TM)) technology.
http://www.prnewswire.co.uk/cgi/news/release?id=300346
Keine weltumstürzenden News, aber insofern wichtig, als dass sie zeigen, dass bei Arqule weiterhin alles auf dem richtigen Weg ist.
Antwort auf Beitrag Nr.: 40.302.242 von SLGramann am 11.10.10 23:45:34
Exelixis hat mal wieder einen Erfolg beim Auslizensieren - ausgerechnet bei BMY :
LOS ANGELES | Mon Oct 11, 2010 12:55pm EDT
LOS ANGELES (Reuters) - Exelixis Inc has licensed two more experimental drug programs to Bristol-Myers Squibb Co for $60 million upfront, and potential milestone payments of up to $505 million, sending shares of the biotech company up more than 3 percent
http://www.reuters.com/article/idUSTRE69A3K620101011
Exelixis hat mal wieder einen Erfolg beim Auslizensieren - ausgerechnet bei BMY
:LOS ANGELES | Mon Oct 11, 2010 12:55pm EDT
LOS ANGELES (Reuters) - Exelixis Inc has licensed two more experimental drug programs to Bristol-Myers Squibb Co for $60 million upfront, and potential milestone payments of up to $505 million, sending shares of the biotech company up more than 3 percent
http://www.reuters.com/article/idUSTRE69A3K620101011
Antwort auf Beitrag Nr.: 40.302.242 von SLGramann am 11.10.10 23:45:34
Micromet mit einem BiTE-IND:
Micromet Announces FDA Acceptance of Investigational New Drug Application for BiTE Antibody MT111/MEDI-565
-Phase 1 Clinical Trial Expected to Begin in First Half 2011 in Patients with Gastrointestinal Cancer-
BETHESDA, Md., Oct 11, 2010 (BUSINESS WIRE) -- Micromet, Inc. (NASDAQ: MITI) today announced that its licensee for MT111, MedImmune, plans to initiate a Phase 1 trial in patients with advanced gastrointestinal cancers based on an investigational new drug (IND) application recently accepted by the U.S. Food and Drug Administration (FDA). MT111, also known as MEDI-565, is a BiTE(R) antibody designed to direct a patient's T cells, the body's most potent killer cells, against cancer cells that express carcinoembryonic antigen (CEA). CEA is a protein found on the surface of a number of gastrointestinal cancers, including colorectal, esophageal and gastric.
http://phx.corporate-ir.net/phoenix.zhtml?c=197259&p=irol-ne…
Micromet mit einem BiTE-IND:
Micromet Announces FDA Acceptance of Investigational New Drug Application for BiTE Antibody MT111/MEDI-565
-Phase 1 Clinical Trial Expected to Begin in First Half 2011 in Patients with Gastrointestinal Cancer-
BETHESDA, Md., Oct 11, 2010 (BUSINESS WIRE) -- Micromet, Inc. (NASDAQ: MITI) today announced that its licensee for MT111, MedImmune, plans to initiate a Phase 1 trial in patients with advanced gastrointestinal cancers based on an investigational new drug (IND) application recently accepted by the U.S. Food and Drug Administration (FDA). MT111, also known as MEDI-565, is a BiTE(R) antibody designed to direct a patient's T cells, the body's most potent killer cells, against cancer cells that express carcinoembryonic antigen (CEA). CEA is a protein found on the surface of a number of gastrointestinal cancers, including colorectal, esophageal and gastric.
http://phx.corporate-ir.net/phoenix.zhtml?c=197259&p=irol-ne…
Antwort auf Beitrag Nr.: 40.302.242 von SLGramann am 11.10.10 23:45:34
Seattle Genetics bestätigt den durchschlagenden Erfolg von Brentuximab Vedotin nun auch in der Indikation systemic anaplastic large cell lymphoma (ALCL):
Seattle Genetics and Millennium Announce Positive Top-Line Brentuximab Vedotin (SGN-35) Data from Phase II Trial in Relapsed or Refractory ALCL
-Objective Responses Achieved in 86 Percent of Patients-
- Phase II ALCL and Pivotal Hodgkin Lymphoma Brentuximab Vedotin Oral Presentations
Scheduled at ASH Annual Meeting in December-
BOTHELL, Wash. & CAMBRIDGE, Mass., Oct 11, 2010 (BUSINESS WIRE) --
Seattle Genetics, Inc. (Nasdaq: SGEN) and Millennium: The Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited (TSE:4502), today announced positive top-line results from a phase II clinical trial of single-agent brentuximab vedotin (SGN-35), an antibody-drug conjugate (ADC) targeted to CD30. The trial was conducted in 58 relapsed or refractory systemic anaplastic large cell lymphoma (ALCL) patients.
http://investor.seagen.com/phoenix.zhtml?c=124860&p=irol-new…
außerdem gibt erste klinische Daten zu SGN 75:
Best clinical response among non-Hodgkin lymphoma patients included one patient with a complete remission, four patients with stable disease, one patient with progressive disease and one patient who was not evaluable. Best clinical response among RCC patients included one patient with a partial response, three patients with stable disease and five patients with progressive disease. The most common adverse events were fatigue, nausea and peripheral edema (swelling). A maximum tolerated dose has not been established with either dosing schedule and dose escalation continues.
http://investor.seagen.com/phoenix.zhtml?c=124860&p=irol-new…
Beurteilen müssen diese Daten andere. Aber bei insgesamt 16 Patienten 2 ORs und 7 x stable disease, ohne dass die MTD erreicht wurde, klingt aus Laiensicht hoffnungsvoll.
Seattle Genetics bestätigt den durchschlagenden Erfolg von Brentuximab Vedotin nun auch in der Indikation systemic anaplastic large cell lymphoma (ALCL):
Seattle Genetics and Millennium Announce Positive Top-Line Brentuximab Vedotin (SGN-35) Data from Phase II Trial in Relapsed or Refractory ALCL
-Objective Responses Achieved in 86 Percent of Patients-
- Phase II ALCL and Pivotal Hodgkin Lymphoma Brentuximab Vedotin Oral Presentations
Scheduled at ASH Annual Meeting in December-
BOTHELL, Wash. & CAMBRIDGE, Mass., Oct 11, 2010 (BUSINESS WIRE) --
Seattle Genetics, Inc. (Nasdaq: SGEN) and Millennium: The Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited (TSE:4502), today announced positive top-line results from a phase II clinical trial of single-agent brentuximab vedotin (SGN-35), an antibody-drug conjugate (ADC) targeted to CD30. The trial was conducted in 58 relapsed or refractory systemic anaplastic large cell lymphoma (ALCL) patients.
http://investor.seagen.com/phoenix.zhtml?c=124860&p=irol-new…
außerdem gibt erste klinische Daten zu SGN 75:
Best clinical response among non-Hodgkin lymphoma patients included one patient with a complete remission, four patients with stable disease, one patient with progressive disease and one patient who was not evaluable. Best clinical response among RCC patients included one patient with a partial response, three patients with stable disease and five patients with progressive disease. The most common adverse events were fatigue, nausea and peripheral edema (swelling). A maximum tolerated dose has not been established with either dosing schedule and dose escalation continues.
http://investor.seagen.com/phoenix.zhtml?c=124860&p=irol-new…
Beurteilen müssen diese Daten andere. Aber bei insgesamt 16 Patienten 2 ORs und 7 x stable disease, ohne dass die MTD erreicht wurde, klingt aus Laiensicht hoffnungsvoll.
Hammer beschäftigt sich in einem neuen Beitrag u.a. mit den ESMO-Daten von SGEN und ArQule.
Bei SGEN wird SGN 35 jetzt als faktisch "durch die Ziellinie" angesehen, während die Spannung in Zukunft von SGN 75 kommen könnte (obwohl hier die Datenlage natürlich noch sehr dünn ist).
Bei ArQule gehts natürlich ausschließlich um ARQ 197. Dieses Programm macht mir persönlich Kopfschmerzen. Es ist eine Riesenchance. Bei einem Erfolg kann ich mir für ArQule eine Marktkapitalisierung von um die 1 Milliarde Dollar gut vorstellen. Bei einem Fehlschlag kann es gut 40 bis 50% runtergehen. Die bisherigen Daten sind statistisch nicht signifikant, die P II ist eigentlich gescheitert. Aber allgemein ist man der Meinung, dass dies doch kein Scheitern war und der Kurs hat damals an einem einzigen Tag 80% zugelegt.
Ich schwanke sehr stark, ob ich mir das Ergebnis der P III von ARQ 197 als ArQule-Aktionär ansehen sollte oder neutral von der Seitenlinie.
Bis dahin ist ja noch einiges an Zeit...
ipollit, welches Gefühl hast Du bei ArQule? Weißt Du schon, ob die im Depot 2011 auftauchen werden? Vertraust Du ARQ 197?
Winners of ESMO 2010
The ESMO meeting is the most important oncology conference in Europe. This year in particular, it included very interesting data that affected the sentiment towards many biotech companies. Here, I intend to focus on what I view as three clear winners from the conference: Seattle Genetics (SGEN), Arqule (ARQL) and Dendreon (DNDN).
Seattle Genetics
Seattle Genetics is about to conclude the best year in its history, since it was incorporated 13 years ago. The company’s lead agent, SGN-35 (aka Brentuximab Vedotin), generated astonishing results in two types of blood cancers earlier this year. Based on the results in Hodkin’s Lymphoma, SGN-35’s approval seems inevitable, even though results are not from large randomized studies. Unlike T-DM1’s case, Seattle Genetics negotiated a special protocol assessment (SPA) with the FDA, implying that the trial design and endpoints are acceptable by the FDA.
Although initial market potential is limited ($250-$300M), this amount is still substantial for a company with a market cap of ~$1.7B, assuming a substantial portion of revenues will come from the US, where Seattle Genetics has exclusive rights. The approval will also serve as a final validation for Seattle Genetics’ technology and will position it as the leading antibody drug conjugate (ADC) company with the only approved ADC in the market (Mylotarg, the first ADC to get approved was recently withdrawn due to failed post-approval studies).
At ESMO, Seattle Genetics gave investors another reason for optimism, this time following positive data for another wholly owned agent, SGN-75. SGN-75 is an ADC for the treatment of NHL and renal cancer, two indications that represent a multi-billion dollar opportunity. Data at ESMO included preliminary findings implying that SGN-75 is active in both indications. The trial included only patients whose tumors express CD70, SGN-75’s target.
Seattle Genetics reported a complete response in one patient with NHL (out of 7 patients) and one partial response in a renal cancer patient (out of ~10 patients). As of data presentation the maximal dose was not reached, so the drug’s real activity could be substantially better. This is particularly relevant for solid cancers such as renal cancer, which typically require higher exposure compared to blood cancers. So far, SGN-75’s exposure was significantly lower than that achieved by other ADCs for solid tumors such as T-DM1, so the main challenge now is increasing the dose without causing too many side effects.
Although NHL is a larger market than Renal cancer, the latter should be viewed as more attractive for SGN-75 since it represents a higher unmet need with less competition, especially for antibodies. Until now, the market for renal cancer has been dominated by kinase inhibitors such as Pfizer’s (PFE) Sutent, Onyx’s (ONXX) Nexavar and Novartis’ (NVS) Afinitor. While these agents disrupt signals, ADCs such as SGN-75 deliver a toxic payload to tumor cells and can therefore be viewed as complimentary.
As with any early stage drug candidate, more patients are needed in order to better assess SGN-75’s potential. It remains to be seen whether higher doses lead to a better response rate. Another important issue is the ability to identify tumors which are more sensitive to SGN-75 based on the amount and pattern of CD70 expression. Preclinical data published by Seattle Genetics show good correlation between expression level s of CD70 and sensitivity to SGN-75. As CD70 is found in many other cancers but with less prevalence than in kidney cancer, there is potential opportunity for label expansion in lung, ovarian and pancreatic cancer, as shown by the elegant paper.
In summary, SGN-75 could become the next big ADC, as it contains all the right ingredients: New target, unmet need, potential utility in several solid tumor indications, good safety profile and clear efficacy. Based on the rapid recruitment, investors should have a sense of where this agent stands by next ASCO in June 2011.
Most importantly, SGN-75 is still unpartnered so positive results next year should have a huge impact on the compamy’s valuation. The antibody moiety in SGN-75 was part of a 2001 licensing deal with a research institute in the Netherlands. This implies that Seattle Genetics will probably pay low royalties on sales of SGN-75, but this should not prevent it from getting a lucrative licensing deal down the road.
Seattle Genetics is not the only one with a CD70 program. Medarex also has a CD70 ADC (MDX-1203) in clinical testing, based on its proprietary ADC technology, which is not as validated as that of Seattle Genetics. The two companies were running neck-to-neck with their CD70 programs during the past several years. Medarex beat Seattle Genetics in starting a phase I, probably due to the fact Seattle Genetics was developing a method for selecting patients based on CD70 expression. In contrast, Medarex is not selecting patients based on molecular markers but simply limit accrual to renal cancer and NHL. It will be intriguing to see first data for MDX-1203, which should give investors more perspective on its ADC technology compared to that of Seattle Genetics.
It is also very likely to see a CD70 ADC powered by Immunogen’s (IMGN) technology. Even if Immunogen or its partners are not working on the target, the recent data for SGN-75 clearly marks CD70 as the next hot target. Extrapolating from published work by Genentech, one can assume that Immunogen can use exactly the same non-cleavable linker currently used with T-DM1, which is much more validated than the rest of its linkers. With the exception of T-DM1, all of Immunogen’s compounds in the clinic are based on cleaveable linkers, which are viewed by some as less effective (even though some targets are not suitable for uncleaveable linker).
Arqule
Arqule and its partners presented results from several studies for the company’s lead agent, ARQ-197. The most important presentation was obviously final results from the randomized phase II in lung cancer, which laid the foundation for an upcoming phase III trial. The other trials included earlier data in additional indications that continued to show ARQ-197’s excellent safety profile even though the drug’s efficacy in these settings should be determined in more advanced studies.
The phase II lung cancer trial evaluated the addition of ARQ-197 to Tarceva vs. Tarceva alone in 2nd/3rd NSCLC patients. Technically, the trial did not meet the primary endpoint, which was showing a statistically significant improvement in progression-free survival (PFS, the time patients live without disease progression). However, as I previously discussed, the study generated a clear efficacy signal using two predefined and acceptable subset analyses.
The first analysis took into considerations imbalances in the genetic profile of patients between the two arms. Although usually, such retrospective analyses should be approached with skepticism, the two markers used for stratifying patients (EGFR and KRAS mutation) are well validated for predicting response to Tarceva. Therefore, the imbalances created an built-in bias in favor of the control arm. The second analysis, which was planned in advance, distinguished between two subtypes of NSCLC and found that in patients with non-squamous tumors the combination led to a statistically significant difference. This stratification is also widely acceptable, as the two subtypes are already known to differ in terms of response to certain agents, particularly Alimta.
The updated results included some interesting points that further establish ARQ-197 as one of the most promising agents for lung cancer. There was a clear trend towards better overall survival in non-squamous patients, although the trial was not powered to show a statistically significant difference. In addition, investigators presented interesting findings on the drug’s ability to delay the formation of new metastases. This property could be a huge advantage in earlier treatment lines, but it will have to be proven prospectively in larger studies. Lastly, in patients from the Tarceva arm who switched to the combination arm after progressing on Tarceva alone, there was a clear signal including 2 partial responses and several cases of prolonged disease stabilization. Interestingly, the two partial responses were in patients with high MET expression. All the findings above are retrospective or anecdotal, and by themselves cannot justify moving into phase III, but they provide supportive evidence that is in line with the known biologic function of MET.
The efficacy signal, coupled with the excellent safety profile led Arqule’s partner, Daiichi Sankyo, to launch a pivotal phase III trial in lung cancer. The study was designed based on the efficacy signal from the phase II, as it will enroll only non-squamous patients. The primary endpoint for the study is overall survival, which is harder to achieve but it is still considered the most relevant endpoint. In order to avoid the imbalances of the phase II, the mutation status of patients must be known at the point of randomization. The trial will be conducted under SPA, which significantly increases chances of approval if the trial meets the primary endpoint.
Metmab as a competitor
The session in which ARQ-197 was presented at ESMO included also results for Roche/Genentech’s Metmab in a very similar patient population but with many differences in terms of trial design and subset analysis. Like ARQ-197, MetMab targets MET but in a totally different approach. It is a monoclonal antibody that binds the external side of the receptor on cells, as opposed to ARQ-197 that inhibits signaling in the internal side of the target.
In the overall population, there was no difference between the two arms, which were balanced with respect to EGFR and KRAS mutations. This is in contrast to the signal ARQ-197 generated adjusting mutation imbalances (though retrospectively). Nevertheless, MetMab generated a clear efficacy signal in a subset of patients whose tumors expressed high levels of MET, even though this signal was just almost statistically significant. This is also in contrast to Arqule’s subset analysis which managed to achieve statistical significance based on a totally different stratification.
Patients with high MET expression had a PFS of 12.4 weeks vs. 6.4 weeks in the Tarceva arm. In terms of overall survival, the addition of MetMab in these patients led to a marginal improvement from 7.4 months to 7.7 months. Looking at the data, it seems that this number does not do justice to MetMab, as the curves look fairly separated. This kind of inconsistency might be explained by the relatively low sample size (65 MET positive patients in total). The most striking observation was that patients with low MET expression seemed to do worse on the MeMab+ Tarceva combination both in terms of PFS and overall survival.
MetMab’s data set further reinforces MET as a viable target, even though there are many open questions that arise from both studies. It is also very hard to compare ARQ-197 and MetMab, due to the substantial differences between the two trials. To begin with, investigators in each study used a totally different approach for distinguishing between MET positive and negative tumors. Roche/Genentech conducted its subset analysis based on MET expression, whereas Arqule used plain histology for identifying the right patients. Unlike ARQ-197, MetMab did not have superior activity in non-squamous patients, whereas ARQ-197 activity seemed to correlate with very high MET expression (although the data in the presentation is limited). In addition, the MetMab study did not include crossover, and therefore could not generate the anecdotal signs of activity like in the ARQ-197 study.
Due to all those differences and the fact that ARQ-197 and MetMab have never been compared directly, it is very hard to compare the two. MetMab’s results are more positive than negative for ARQ-197 since they provide another validation for MET, without showing clear superiority over ARQ-197. The apparent differences in activity could beeventually explained by the distinct modes of action the two compounds have.
The only troubling sign for Arqule is MetMab’s potential deleterious effect on MET negative/low tumors. The ARQ-197 presentation does not include this analysis, unfortunately, but one can assume that had such a signal been obtained, patients with MET negative tumors would have been excluded from the current ARQ-197 phase III trial. There is always the risk that Roche’s approach for quantifying MET expression is more reliable than that used with ARQ-197.
Another interesting trial that could shed more light on MET as a target for lung cancer is that of Aveo’s AV-299 (AVEO), currently in phase II. AV-299 targets the MET pathway using a third approach as it targets the ligand that activates MET as opposed to the receptor itself. Until recently, AV-299 was partnered with Merck, which gave back rights for the products to Aveo last month. The news sent Aveo’s shares higher, which is very rare in cases of termination by a large partner. Arqule’s and Roche’s data could explain the market’s paradoxical reaction.
http://www.hammerstockblog.com/winners-of-esmo-2010/
Bei SGEN wird SGN 35 jetzt als faktisch "durch die Ziellinie" angesehen, während die Spannung in Zukunft von SGN 75 kommen könnte (obwohl hier die Datenlage natürlich noch sehr dünn ist).
Bei ArQule gehts natürlich ausschließlich um ARQ 197. Dieses Programm macht mir persönlich Kopfschmerzen. Es ist eine Riesenchance. Bei einem Erfolg kann ich mir für ArQule eine Marktkapitalisierung von um die 1 Milliarde Dollar gut vorstellen. Bei einem Fehlschlag kann es gut 40 bis 50% runtergehen. Die bisherigen Daten sind statistisch nicht signifikant, die P II ist eigentlich gescheitert. Aber allgemein ist man der Meinung, dass dies doch kein Scheitern war und der Kurs hat damals an einem einzigen Tag 80% zugelegt.
Ich schwanke sehr stark, ob ich mir das Ergebnis der P III von ARQ 197 als ArQule-Aktionär ansehen sollte oder neutral von der Seitenlinie.
Bis dahin ist ja noch einiges an Zeit...
ipollit, welches Gefühl hast Du bei ArQule? Weißt Du schon, ob die im Depot 2011 auftauchen werden? Vertraust Du ARQ 197?
Winners of ESMO 2010
The ESMO meeting is the most important oncology conference in Europe. This year in particular, it included very interesting data that affected the sentiment towards many biotech companies. Here, I intend to focus on what I view as three clear winners from the conference: Seattle Genetics (SGEN), Arqule (ARQL) and Dendreon (DNDN).
Seattle Genetics
Seattle Genetics is about to conclude the best year in its history, since it was incorporated 13 years ago. The company’s lead agent, SGN-35 (aka Brentuximab Vedotin), generated astonishing results in two types of blood cancers earlier this year. Based on the results in Hodkin’s Lymphoma, SGN-35’s approval seems inevitable, even though results are not from large randomized studies. Unlike T-DM1’s case, Seattle Genetics negotiated a special protocol assessment (SPA) with the FDA, implying that the trial design and endpoints are acceptable by the FDA.
Although initial market potential is limited ($250-$300M), this amount is still substantial for a company with a market cap of ~$1.7B, assuming a substantial portion of revenues will come from the US, where Seattle Genetics has exclusive rights. The approval will also serve as a final validation for Seattle Genetics’ technology and will position it as the leading antibody drug conjugate (ADC) company with the only approved ADC in the market (Mylotarg, the first ADC to get approved was recently withdrawn due to failed post-approval studies).
At ESMO, Seattle Genetics gave investors another reason for optimism, this time following positive data for another wholly owned agent, SGN-75. SGN-75 is an ADC for the treatment of NHL and renal cancer, two indications that represent a multi-billion dollar opportunity. Data at ESMO included preliminary findings implying that SGN-75 is active in both indications. The trial included only patients whose tumors express CD70, SGN-75’s target.
Seattle Genetics reported a complete response in one patient with NHL (out of 7 patients) and one partial response in a renal cancer patient (out of ~10 patients). As of data presentation the maximal dose was not reached, so the drug’s real activity could be substantially better. This is particularly relevant for solid cancers such as renal cancer, which typically require higher exposure compared to blood cancers. So far, SGN-75’s exposure was significantly lower than that achieved by other ADCs for solid tumors such as T-DM1, so the main challenge now is increasing the dose without causing too many side effects.
Although NHL is a larger market than Renal cancer, the latter should be viewed as more attractive for SGN-75 since it represents a higher unmet need with less competition, especially for antibodies. Until now, the market for renal cancer has been dominated by kinase inhibitors such as Pfizer’s (PFE) Sutent, Onyx’s (ONXX) Nexavar and Novartis’ (NVS) Afinitor. While these agents disrupt signals, ADCs such as SGN-75 deliver a toxic payload to tumor cells and can therefore be viewed as complimentary.
As with any early stage drug candidate, more patients are needed in order to better assess SGN-75’s potential. It remains to be seen whether higher doses lead to a better response rate. Another important issue is the ability to identify tumors which are more sensitive to SGN-75 based on the amount and pattern of CD70 expression. Preclinical data published by Seattle Genetics show good correlation between expression level s of CD70 and sensitivity to SGN-75. As CD70 is found in many other cancers but with less prevalence than in kidney cancer, there is potential opportunity for label expansion in lung, ovarian and pancreatic cancer, as shown by the elegant paper.
In summary, SGN-75 could become the next big ADC, as it contains all the right ingredients: New target, unmet need, potential utility in several solid tumor indications, good safety profile and clear efficacy. Based on the rapid recruitment, investors should have a sense of where this agent stands by next ASCO in June 2011.
Most importantly, SGN-75 is still unpartnered so positive results next year should have a huge impact on the compamy’s valuation. The antibody moiety in SGN-75 was part of a 2001 licensing deal with a research institute in the Netherlands. This implies that Seattle Genetics will probably pay low royalties on sales of SGN-75, but this should not prevent it from getting a lucrative licensing deal down the road.
Seattle Genetics is not the only one with a CD70 program. Medarex also has a CD70 ADC (MDX-1203) in clinical testing, based on its proprietary ADC technology, which is not as validated as that of Seattle Genetics. The two companies were running neck-to-neck with their CD70 programs during the past several years. Medarex beat Seattle Genetics in starting a phase I, probably due to the fact Seattle Genetics was developing a method for selecting patients based on CD70 expression. In contrast, Medarex is not selecting patients based on molecular markers but simply limit accrual to renal cancer and NHL. It will be intriguing to see first data for MDX-1203, which should give investors more perspective on its ADC technology compared to that of Seattle Genetics.
It is also very likely to see a CD70 ADC powered by Immunogen’s (IMGN) technology. Even if Immunogen or its partners are not working on the target, the recent data for SGN-75 clearly marks CD70 as the next hot target. Extrapolating from published work by Genentech, one can assume that Immunogen can use exactly the same non-cleavable linker currently used with T-DM1, which is much more validated than the rest of its linkers. With the exception of T-DM1, all of Immunogen’s compounds in the clinic are based on cleaveable linkers, which are viewed by some as less effective (even though some targets are not suitable for uncleaveable linker).
Arqule
Arqule and its partners presented results from several studies for the company’s lead agent, ARQ-197. The most important presentation was obviously final results from the randomized phase II in lung cancer, which laid the foundation for an upcoming phase III trial. The other trials included earlier data in additional indications that continued to show ARQ-197’s excellent safety profile even though the drug’s efficacy in these settings should be determined in more advanced studies.
The phase II lung cancer trial evaluated the addition of ARQ-197 to Tarceva vs. Tarceva alone in 2nd/3rd NSCLC patients. Technically, the trial did not meet the primary endpoint, which was showing a statistically significant improvement in progression-free survival (PFS, the time patients live without disease progression). However, as I previously discussed, the study generated a clear efficacy signal using two predefined and acceptable subset analyses.
The first analysis took into considerations imbalances in the genetic profile of patients between the two arms. Although usually, such retrospective analyses should be approached with skepticism, the two markers used for stratifying patients (EGFR and KRAS mutation) are well validated for predicting response to Tarceva. Therefore, the imbalances created an built-in bias in favor of the control arm. The second analysis, which was planned in advance, distinguished between two subtypes of NSCLC and found that in patients with non-squamous tumors the combination led to a statistically significant difference. This stratification is also widely acceptable, as the two subtypes are already known to differ in terms of response to certain agents, particularly Alimta.
The updated results included some interesting points that further establish ARQ-197 as one of the most promising agents for lung cancer. There was a clear trend towards better overall survival in non-squamous patients, although the trial was not powered to show a statistically significant difference. In addition, investigators presented interesting findings on the drug’s ability to delay the formation of new metastases. This property could be a huge advantage in earlier treatment lines, but it will have to be proven prospectively in larger studies. Lastly, in patients from the Tarceva arm who switched to the combination arm after progressing on Tarceva alone, there was a clear signal including 2 partial responses and several cases of prolonged disease stabilization. Interestingly, the two partial responses were in patients with high MET expression. All the findings above are retrospective or anecdotal, and by themselves cannot justify moving into phase III, but they provide supportive evidence that is in line with the known biologic function of MET.
The efficacy signal, coupled with the excellent safety profile led Arqule’s partner, Daiichi Sankyo, to launch a pivotal phase III trial in lung cancer. The study was designed based on the efficacy signal from the phase II, as it will enroll only non-squamous patients. The primary endpoint for the study is overall survival, which is harder to achieve but it is still considered the most relevant endpoint. In order to avoid the imbalances of the phase II, the mutation status of patients must be known at the point of randomization. The trial will be conducted under SPA, which significantly increases chances of approval if the trial meets the primary endpoint.
Metmab as a competitor
The session in which ARQ-197 was presented at ESMO included also results for Roche/Genentech’s Metmab in a very similar patient population but with many differences in terms of trial design and subset analysis. Like ARQ-197, MetMab targets MET but in a totally different approach. It is a monoclonal antibody that binds the external side of the receptor on cells, as opposed to ARQ-197 that inhibits signaling in the internal side of the target.
In the overall population, there was no difference between the two arms, which were balanced with respect to EGFR and KRAS mutations. This is in contrast to the signal ARQ-197 generated adjusting mutation imbalances (though retrospectively). Nevertheless, MetMab generated a clear efficacy signal in a subset of patients whose tumors expressed high levels of MET, even though this signal was just almost statistically significant. This is also in contrast to Arqule’s subset analysis which managed to achieve statistical significance based on a totally different stratification.
Patients with high MET expression had a PFS of 12.4 weeks vs. 6.4 weeks in the Tarceva arm. In terms of overall survival, the addition of MetMab in these patients led to a marginal improvement from 7.4 months to 7.7 months. Looking at the data, it seems that this number does not do justice to MetMab, as the curves look fairly separated. This kind of inconsistency might be explained by the relatively low sample size (65 MET positive patients in total). The most striking observation was that patients with low MET expression seemed to do worse on the MeMab+ Tarceva combination both in terms of PFS and overall survival.
MetMab’s data set further reinforces MET as a viable target, even though there are many open questions that arise from both studies. It is also very hard to compare ARQ-197 and MetMab, due to the substantial differences between the two trials. To begin with, investigators in each study used a totally different approach for distinguishing between MET positive and negative tumors. Roche/Genentech conducted its subset analysis based on MET expression, whereas Arqule used plain histology for identifying the right patients. Unlike ARQ-197, MetMab did not have superior activity in non-squamous patients, whereas ARQ-197 activity seemed to correlate with very high MET expression (although the data in the presentation is limited). In addition, the MetMab study did not include crossover, and therefore could not generate the anecdotal signs of activity like in the ARQ-197 study.
Due to all those differences and the fact that ARQ-197 and MetMab have never been compared directly, it is very hard to compare the two. MetMab’s results are more positive than negative for ARQ-197 since they provide another validation for MET, without showing clear superiority over ARQ-197. The apparent differences in activity could beeventually explained by the distinct modes of action the two compounds have.
The only troubling sign for Arqule is MetMab’s potential deleterious effect on MET negative/low tumors. The ARQ-197 presentation does not include this analysis, unfortunately, but one can assume that had such a signal been obtained, patients with MET negative tumors would have been excluded from the current ARQ-197 phase III trial. There is always the risk that Roche’s approach for quantifying MET expression is more reliable than that used with ARQ-197.
Another interesting trial that could shed more light on MET as a target for lung cancer is that of Aveo’s AV-299 (AVEO), currently in phase II. AV-299 targets the MET pathway using a third approach as it targets the ligand that activates MET as opposed to the receptor itself. Until recently, AV-299 was partnered with Merck, which gave back rights for the products to Aveo last month. The news sent Aveo’s shares higher, which is very rare in cases of termination by a large partner. Arqule’s and Roche’s data could explain the market’s paradoxical reaction.
http://www.hammerstockblog.com/winners-of-esmo-2010/
zu AVEO:
Wir haben eine private Kapitalerhöhung. Sehe ich unterm Strich mit Wohlwollen.
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced the execution of a securities purchase agreement with a select group of institutional and accredited investors providing for a private placement, or PIPE financing. Upon the closing of the PIPE financing, AVEO will receive gross proceeds of approximately $61 million resulting from the sale of 4.5 million shares of common stock at a purchase price of $13.50 per share, which represents an approximate 7% discount to the closing price on October 28, 2010. The closing of the PIPE financing is subject to standard closing conditions. J.P. Morgan Securities LLC and Canaccord Genuity Inc. served as joint lead placement agents for the financing. RBC Capital Markets Corporation served as co-placement agent.
Wir haben eine private Kapitalerhöhung. Sehe ich unterm Strich mit Wohlwollen.
CAMBRIDGE, Mass.--(BUSINESS WIRE)--AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO), a biopharmaceutical company focused on discovering, developing and commercializing cancer therapeutics, today announced the execution of a securities purchase agreement with a select group of institutional and accredited investors providing for a private placement, or PIPE financing. Upon the closing of the PIPE financing, AVEO will receive gross proceeds of approximately $61 million resulting from the sale of 4.5 million shares of common stock at a purchase price of $13.50 per share, which represents an approximate 7% discount to the closing price on October 28, 2010. The closing of the PIPE financing is subject to standard closing conditions. J.P. Morgan Securities LLC and Canaccord Genuity Inc. served as joint lead placement agents for the financing. RBC Capital Markets Corporation served as co-placement agent.
kleine Anmerkung zu NeurogesX:
Der Qutenza-Umsatz ist vom 2. zum 3. Quartal um sagenhafte 597% gestiegen...
Leider bedeutet das in absoluten Zahlen nur, dass statt 33.000,00 Dollar in Q2 jetzt 197.000,00 Dollar erlöst worden sind, womit klargestellt ist, dass diese 597% von mir ironisch gemeint sind...
(schon niedlich, wenn man im Zusammenhang mit Finanberichten eines börsennotierten Unternehmens mal nicht mit Mio. oder Mrd. operiert, sondern mit doch eher gewöhnlichen Geldbeträgen)
Ich bin absolut bereit, dem Unternehmen hier mehr Zeit einzuräumenm, um die Dinge ins Laufen zu bekommen und ich denke, dass man die Umsatzzahlen kaum vor dem zweiten Halbjahr 2011 ernst nehmen kann. Dennoch bin ich über den zähen Start der Vermarktung etwas enttäuscht.
Da die Kostenbasis des Unternehmens auf der anderen Seite schon sehr beachtlich ist, muss 2011, spätestens aber das Jahr 2012 die Wende bringen, sonst kommen hier unerfreuliche Entwicklungen auf die Eigentümer zu.
Der Qutenza-Umsatz ist vom 2. zum 3. Quartal um sagenhafte 597% gestiegen...
Leider bedeutet das in absoluten Zahlen nur, dass statt 33.000,00 Dollar in Q2 jetzt 197.000,00 Dollar erlöst worden sind, womit klargestellt ist, dass diese 597% von mir ironisch gemeint sind...
(schon niedlich, wenn man im Zusammenhang mit Finanberichten eines börsennotierten Unternehmens mal nicht mit Mio. oder Mrd. operiert, sondern mit doch eher gewöhnlichen Geldbeträgen)
Ich bin absolut bereit, dem Unternehmen hier mehr Zeit einzuräumenm, um die Dinge ins Laufen zu bekommen und ich denke, dass man die Umsatzzahlen kaum vor dem zweiten Halbjahr 2011 ernst nehmen kann. Dennoch bin ich über den zähen Start der Vermarktung etwas enttäuscht.
Da die Kostenbasis des Unternehmens auf der anderen Seite schon sehr beachtlich ist, muss 2011, spätestens aber das Jahr 2012 die Wende bringen, sonst kommen hier unerfreuliche Entwicklungen auf die Eigentümer zu.
Regeneron am Montag mit dem Tag der Wahrheit.
Das ist etwas eher, als bisher erwartet. 30% hoch oder 30% runter? Wird spannend.
TARRYTOWN, N.Y., Nov. 19, 2010 /PRNewswire-FirstCall/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq:REGN - News) today announced that it will hold a teleconference and webcast at 8:30 a.m. Eastern Time on Monday, November 22, to discuss results of two Phase 3 studies with VEGF Trap-Eye in Wet Age-related Macular Degeneration, VIEW 1 and VIEW 2, and its VEGF Trap-Eye program. A press release will be issued on Monday prior to the call.
Das ist etwas eher, als bisher erwartet. 30% hoch oder 30% runter? Wird spannend.
TARRYTOWN, N.Y., Nov. 19, 2010 /PRNewswire-FirstCall/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq:REGN - News) today announced that it will hold a teleconference and webcast at 8:30 a.m. Eastern Time on Monday, November 22, to discuss results of two Phase 3 studies with VEGF Trap-Eye in Wet Age-related Macular Degeneration, VIEW 1 and VIEW 2, and its VEGF Trap-Eye program. A press release will be issued on Monday prior to the call.
Antwort auf Beitrag Nr.: 40.558.448 von SLGramann am 20.11.10 09:19:04
Regeneron und Trap Eye:
Bayer and Regeneron Report Positive Top-Line Results of Two Phase 3 Studies with VEGF Trap-Eye in Wet Age-related Macular Degeneration
In both studies, all regimens of VEGF Trap-Eye, including VEGF Trap-Eye dosed every two months, achieved primary endpoint compared to ranibizumab dosed every month
Regulatory applications for marketing approval planned in first-half of 2011
TARRYTOWN, N.Y. and BERLIN, Nov. 22, 2010 /PRNewswire-FirstCall/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) and Bayer HealthCare today announced that in two parallel Phase 3 studies in patients with the neovascular form of age-related macular degeneration (wet AMD), all regimens of VEGF Trap-Eye (aflibercept ophthalmic solution), including VEGF Trap-Eye dosed every two months, successfully met the primary endpoint compared to the current standard of care, ranibizumab dosed every month. The primary endpoint was statistical non-inferiority in the proportion of patients who maintained (or improved) vision over 52 weeks compared to ranibizumab.
Further results will be presented at the Angiogenesis Conference in February 2011. Bayer HealthCare and Regeneron are planning to submit regulatory applications for marketing approval in Europe and the U.S. in the first-half of 2011 based on the positive results of the VIEW 1 and VIEW 2 trials.
In the North American VIEW 1 study, 96 percent of patients receiving VEGF Trap-Eye 0.5mg monthly, 95 percent of patients receiving VEGF Trap-Eye 2mg monthly, and 95 percent of patients receiving VEGF Trap-Eye 2mg every two months achieved maintenance of vision compared to 94 percent of patients receiving ranibizumab 0.5mg dosed every month. In the international VIEW 2 study, 96 percent of patients receiving VEGF Trap-Eye 0.5mg monthly, 96 percent of patients receiving VEGF Trap-Eye 2mg monthly, and 96 percent of patients receiving VEGF Trap-Eye 2mg every two months achieved maintenance of vision compared to 94 percent of patients receiving ranibizumab 0.5mg dosed every month. Visual acuity was measured as a score based on the total number of letters read correctly on the Early Treatment Diabetic Retinopathy Study (ETDRS) eye chart, a standard chart used in research to measure visual acuity, over 52 weeks. Maintenance of vision was defined as losing fewer than three lines (equivalent to 15 letters) on the ETDRS eye chart.
"The currently available anti-VEGF therapies have significantly advanced the treatment of wet AMD, actually improving vision in many patients. However, monthly injections are required to optimize and maintain vision gain over the long-term," said Ursula Schmidt-Erfurth, M.D., Professor and Chair of the Department of Ophthalmology at the University Eye Hospital in Vienna, Austria and the VIEW 2 Principal Investigator. "The results of the VIEW studies indicate that VEGF Trap-Eye could establish a new treatment paradigm for the management of patients with wet AMD --- predictable every-other-month dosing without the need for intervening monitoring or dosing visits."
"In an effort to avoid the inconvenience of monthly office visits and the burden of monthly injections into the eye for their wet AMD patients, retinal specialists have tried to extend the benefits of the existing anti-VEGF therapy with less frequent dosing. A growing body of data suggests that this practice may result in inconsistent visual acuity outcomes," said Jeffrey Heier, M.D., a clinical ophthalmologist and retinal specialist at Ophthalmic Consultants of Boston, Assistant Professor of ophthalmology at Tufts School of Medicine, and Chair of the Steering Committee for the VIEW 1 trial. "A critical goal of these studies was to demonstrate that VEGF Trap-Eye could achieve robust improvements in vision and maintain them over time with a more convenient every-other-month dose. Achievement of this goal could be important for patients, care givers, and physicians."
In the VIEW 1 study, patients receiving VEGF Trap-Eye 2mg monthly achieved a statistically significant greater mean improvement in visual acuity at week 52 versus baseline (secondary endpoint), compared to ranibizumab 0.5mg monthly; patients receiving VEGF Trap-Eye 2mg monthly on average gained 10.9 letters, compared to a mean 8.1 letter gain with ranibizumab 0.5mg dosed every month (p<0.01). All other dose groups of VEGF Trap-Eye in the VIEW 1 study and all dose groups in the VIEW 2 study were not statistically different from ranibizumab in this secondary endpoint.
A generally favorable safety profile was observed for both VEGF Trap-Eye and ranibizumab. The incidence of ocular treatment emergent adverse events was balanced across all four treatment groups in both studies, with the most frequent events associated with the injection procedure, the underlying disease, and/or the aging process. The most frequent ocular adverse events were conjunctival hemorrhage, macular degeneration, eye pain, retinal hemorrhage, and vitreous floaters. The most frequent serious non-ocular adverse events were typical of those reported in this elderly population who receive intravitreal treatment for wet AMD; the most frequently reported events were falls, pneumonia, myocardial infarction, atrial fibrillation, breast cancer, and acute coronary syndrome. There were no notable differences among the study arms.
In the second year of the studies, patients in VIEW 1 and VIEW 2 will continue to be treated with the same dose per injection as in the first year but administered only every three months, or more often for any worsening of AMD, based on protocol-defined criteria (called "quarterly capped PRN" dosing).
http://www.prnewswire.com/news-releases/bayer-and-regeneron-…
Bayer macht Fortschritte bei Entwicklung von Augenheilmittel
Montag, 22. November 2010, 09:03 Uhr
Diesen Artikel drucken[-] Text [+] New York (Reuters) - Bayer und sein US-Partner Regeneron sind bei der Entwicklung eines Augenheilmittels einen weiteren Schritt vorangekommen.
Das Medikament sei in zwei Phase-III-Studien genauso effektiv und sicher wie das Konkurrenzprodukt Lucentis von Roche gewesen und müsse nur halb zu häufig gespritzt werden, teilte Regeneron am Montag mit. Mit dem Mittel VEGF Trap-Eye soll die feuchte altersbedingte Makula-Degeneration (AMD) behandelt werden. Diese Erkrankung ist eine der häufigsten Ursachen von Erblindung.
Der Chef des US-Biotechnologiekonzerns Leonard Schleifer hofft, dass das Mittel zeigt, dass es "das Beste in seiner Klasse" sein kann. Neue Wirkstoffe müssen drei Phasen der klinischen Erprobung am Menschen vor einem Zulassungsantrag erfolgreich durchlaufen.
Bayer will sein Augenmittel VEGF Trap-Eye 2011 auf den Markt bringen
BERLIN/TARRYTOWN (awp international) - Der Chemie- und Pharmakonzern Bayer will nach aktuellen Studiendaten sein gemeinsam mit dem US-Biotechnologieunternehmen Regeneron entwickeltes Augenmittel VEGF Trap-Eye im nächsten Jahr auf den Markt bringen. Das Mittel habe in zwei Phase-III-Studien bei feuchter altersbedingter Makula-Degeneration positive Ergebnisse erzielt, teilten die beiden Unternehmen am Montag in Berlin und Tarrytown mit.
Weitere Daten sollten auf der Konferenz "Angiogenesis 2011" im Februar in Miami vorgestellt werden. Basierend auf den aktuellen Studiendaten sowie früheren Ergebnissen wollen die beiden Unternehmen die Zulassung für das Medikament in Europa und den USA in der ersten Hälfte des kommenden Jahres beantragen.
Die altersbedingte Makula-Degeneration ist eine der häufigsten Ursachen von Erblindung. Die Makula ist ein kleiner Bereich im hinteren Teil des Auges, durch den man feine Einzelheiten klar erkennen kann. Wenn die Makula nicht richtig funktioniert, nimmt man das als Schleier oder Dunkelheit in der Mitte unseres Blickfelds wahr.
Regeneron und Trap Eye:
Bayer and Regeneron Report Positive Top-Line Results of Two Phase 3 Studies with VEGF Trap-Eye in Wet Age-related Macular Degeneration
In both studies, all regimens of VEGF Trap-Eye, including VEGF Trap-Eye dosed every two months, achieved primary endpoint compared to ranibizumab dosed every month
Regulatory applications for marketing approval planned in first-half of 2011
TARRYTOWN, N.Y. and BERLIN, Nov. 22, 2010 /PRNewswire-FirstCall/ -- Regeneron Pharmaceuticals, Inc. (Nasdaq: REGN) and Bayer HealthCare today announced that in two parallel Phase 3 studies in patients with the neovascular form of age-related macular degeneration (wet AMD), all regimens of VEGF Trap-Eye (aflibercept ophthalmic solution), including VEGF Trap-Eye dosed every two months, successfully met the primary endpoint compared to the current standard of care, ranibizumab dosed every month. The primary endpoint was statistical non-inferiority in the proportion of patients who maintained (or improved) vision over 52 weeks compared to ranibizumab.
Further results will be presented at the Angiogenesis Conference in February 2011. Bayer HealthCare and Regeneron are planning to submit regulatory applications for marketing approval in Europe and the U.S. in the first-half of 2011 based on the positive results of the VIEW 1 and VIEW 2 trials.
In the North American VIEW 1 study, 96 percent of patients receiving VEGF Trap-Eye 0.5mg monthly, 95 percent of patients receiving VEGF Trap-Eye 2mg monthly, and 95 percent of patients receiving VEGF Trap-Eye 2mg every two months achieved maintenance of vision compared to 94 percent of patients receiving ranibizumab 0.5mg dosed every month. In the international VIEW 2 study, 96 percent of patients receiving VEGF Trap-Eye 0.5mg monthly, 96 percent of patients receiving VEGF Trap-Eye 2mg monthly, and 96 percent of patients receiving VEGF Trap-Eye 2mg every two months achieved maintenance of vision compared to 94 percent of patients receiving ranibizumab 0.5mg dosed every month. Visual acuity was measured as a score based on the total number of letters read correctly on the Early Treatment Diabetic Retinopathy Study (ETDRS) eye chart, a standard chart used in research to measure visual acuity, over 52 weeks. Maintenance of vision was defined as losing fewer than three lines (equivalent to 15 letters) on the ETDRS eye chart.
"The currently available anti-VEGF therapies have significantly advanced the treatment of wet AMD, actually improving vision in many patients. However, monthly injections are required to optimize and maintain vision gain over the long-term," said Ursula Schmidt-Erfurth, M.D., Professor and Chair of the Department of Ophthalmology at the University Eye Hospital in Vienna, Austria and the VIEW 2 Principal Investigator. "The results of the VIEW studies indicate that VEGF Trap-Eye could establish a new treatment paradigm for the management of patients with wet AMD --- predictable every-other-month dosing without the need for intervening monitoring or dosing visits."
"In an effort to avoid the inconvenience of monthly office visits and the burden of monthly injections into the eye for their wet AMD patients, retinal specialists have tried to extend the benefits of the existing anti-VEGF therapy with less frequent dosing. A growing body of data suggests that this practice may result in inconsistent visual acuity outcomes," said Jeffrey Heier, M.D., a clinical ophthalmologist and retinal specialist at Ophthalmic Consultants of Boston, Assistant Professor of ophthalmology at Tufts School of Medicine, and Chair of the Steering Committee for the VIEW 1 trial. "A critical goal of these studies was to demonstrate that VEGF Trap-Eye could achieve robust improvements in vision and maintain them over time with a more convenient every-other-month dose. Achievement of this goal could be important for patients, care givers, and physicians."
In the VIEW 1 study, patients receiving VEGF Trap-Eye 2mg monthly achieved a statistically significant greater mean improvement in visual acuity at week 52 versus baseline (secondary endpoint), compared to ranibizumab 0.5mg monthly; patients receiving VEGF Trap-Eye 2mg monthly on average gained 10.9 letters, compared to a mean 8.1 letter gain with ranibizumab 0.5mg dosed every month (p<0.01). All other dose groups of VEGF Trap-Eye in the VIEW 1 study and all dose groups in the VIEW 2 study were not statistically different from ranibizumab in this secondary endpoint.
A generally favorable safety profile was observed for both VEGF Trap-Eye and ranibizumab. The incidence of ocular treatment emergent adverse events was balanced across all four treatment groups in both studies, with the most frequent events associated with the injection procedure, the underlying disease, and/or the aging process. The most frequent ocular adverse events were conjunctival hemorrhage, macular degeneration, eye pain, retinal hemorrhage, and vitreous floaters. The most frequent serious non-ocular adverse events were typical of those reported in this elderly population who receive intravitreal treatment for wet AMD; the most frequently reported events were falls, pneumonia, myocardial infarction, atrial fibrillation, breast cancer, and acute coronary syndrome. There were no notable differences among the study arms.
In the second year of the studies, patients in VIEW 1 and VIEW 2 will continue to be treated with the same dose per injection as in the first year but administered only every three months, or more often for any worsening of AMD, based on protocol-defined criteria (called "quarterly capped PRN" dosing).
http://www.prnewswire.com/news-releases/bayer-and-regeneron-…
Bayer macht Fortschritte bei Entwicklung von Augenheilmittel
Montag, 22. November 2010, 09:03 Uhr
Diesen Artikel drucken[-] Text [+] New York (Reuters) - Bayer und sein US-Partner Regeneron sind bei der Entwicklung eines Augenheilmittels einen weiteren Schritt vorangekommen.
Das Medikament sei in zwei Phase-III-Studien genauso effektiv und sicher wie das Konkurrenzprodukt Lucentis von Roche gewesen und müsse nur halb zu häufig gespritzt werden, teilte Regeneron am Montag mit. Mit dem Mittel VEGF Trap-Eye soll die feuchte altersbedingte Makula-Degeneration (AMD) behandelt werden. Diese Erkrankung ist eine der häufigsten Ursachen von Erblindung.
Der Chef des US-Biotechnologiekonzerns Leonard Schleifer hofft, dass das Mittel zeigt, dass es "das Beste in seiner Klasse" sein kann. Neue Wirkstoffe müssen drei Phasen der klinischen Erprobung am Menschen vor einem Zulassungsantrag erfolgreich durchlaufen.
Bayer will sein Augenmittel VEGF Trap-Eye 2011 auf den Markt bringen
BERLIN/TARRYTOWN (awp international) - Der Chemie- und Pharmakonzern Bayer will nach aktuellen Studiendaten sein gemeinsam mit dem US-Biotechnologieunternehmen Regeneron entwickeltes Augenmittel VEGF Trap-Eye im nächsten Jahr auf den Markt bringen. Das Mittel habe in zwei Phase-III-Studien bei feuchter altersbedingter Makula-Degeneration positive Ergebnisse erzielt, teilten die beiden Unternehmen am Montag in Berlin und Tarrytown mit.
Weitere Daten sollten auf der Konferenz "Angiogenesis 2011" im Februar in Miami vorgestellt werden. Basierend auf den aktuellen Studiendaten sowie früheren Ergebnissen wollen die beiden Unternehmen die Zulassung für das Medikament in Europa und den USA in der ersten Hälfte des kommenden Jahres beantragen.
Die altersbedingte Makula-Degeneration ist eine der häufigsten Ursachen von Erblindung. Die Makula ist ein kleiner Bereich im hinteren Teil des Auges, durch den man feine Einzelheiten klar erkennen kann. Wenn die Makula nicht richtig funktioniert, nimmt man das als Schleier oder Dunkelheit in der Mitte unseres Blickfelds wahr.
Antwort auf Beitrag Nr.: 40.564.845 von SLGramann am 22.11.10 11:20:25
Zusammenfassung auf Laienniveau:
1.) VEGF Trap-Eye wirkt genauso gut wie Ranibizumab (Lucentis).
2.) Eine Behandlung alle 2 Montate ist ausreichend, während Lucentis zunächst monatlich verabreicht wird und später nach Bedarf.
3.) Die Dosierung von Trap-Eye ist 4x höher. Ob das zu mehr Nebenwirkungen oder anderen Nachteilen führt, kann ich nicht beurteilen.
Mein Fazit: Bayer / Regeneron scheinen hier ein konkurrenzfähiges Produkt auf die Beine gestellt zu haben, das hinsichtlich der Verabreichung Vorteile zu bieten scheint. Gegen Lucentis anzukommen, wird dennoch nicht leicht werden. Dennoch dürfte die Regeneron-Aktie jetzt zunächst mal Aufwärtspotential haben.
Zusammenfassung auf Laienniveau:
1.) VEGF Trap-Eye wirkt genauso gut wie Ranibizumab (Lucentis).
2.) Eine Behandlung alle 2 Montate ist ausreichend, während Lucentis zunächst monatlich verabreicht wird und später nach Bedarf.
3.) Die Dosierung von Trap-Eye ist 4x höher. Ob das zu mehr Nebenwirkungen oder anderen Nachteilen führt, kann ich nicht beurteilen.
Mein Fazit: Bayer / Regeneron scheinen hier ein konkurrenzfähiges Produkt auf die Beine gestellt zu haben, das hinsichtlich der Verabreichung Vorteile zu bieten scheint. Gegen Lucentis anzukommen, wird dennoch nicht leicht werden. Dennoch dürfte die Regeneron-Aktie jetzt zunächst mal Aufwärtspotential haben.
Antwort auf Beitrag Nr.: 40.564.927 von SLGramann am 22.11.10 11:32:30
Frankfurt/New York (Reuters) - Bayer und sein US-Partner Regeneron sind der Marktzulassung eines neuen Augenheilmittels einen großen Schritt näher gekommen.
Das Präparat VEGF Trap-Eye habe in zwei Phase-III-Studien genauso gut gewirkt und sei ebenso sicher wie das umsatzstarke Konkurrenzprodukt Lucentis von Roche, teilten Bayer und die US-Biotechfirma Regeneron am Montag mit. Allerdings müsste es nur halb so häufig gespritzt werden. Auf Grundlage der beiden Studien wollen die Unternehmen nun im ersten Halbjahr 2011 Zulassungsanträge in den USA und in Europa stellen. Bayer traut der Medizin einen jährlichen Spitzenumsatz von 250 bis 500 Millionen Euro im Jahr zu.
An der Börse kam die Nachricht gut an: Bayer-Titel legten 0,8 Prozent zu. Die Analysten von Kepler Equities trauen der Arznei einen Spitzenumsatz von 350 Millionen Euro im Jahr zu. Diese Schätzung könne erheblich erhöht werden, falls das Mittel auch zur Behandlung einer ähnlichen Krankheit, dem sogenannten diabetischen Makula-Ödems (DME) wirke, erklärten die Analysten.Mit dem Mittel VEGF Trap-Eye soll die feuchte altersbedingte Makula-Degeneration (AMD) behandelt werden, eine krankhafte Gefäßneubildung auf dem Augenhintergrund. Die Erkrankung ist eine der häufigsten Ursachen für die Erblindung von Menschen über 65. Neue Medikamente müssen drei Phasen der klinischen Erprobung am Menschen bestehen. Für die Behandlung des Makula-Ödems befindet sich das Präparat aktuell in der zweiten Phase der klinischen Entwicklung.
Regeneron-Chef Leonard Schleifer hofft nach den AMD-Studien, dass das Mittel "das Beste in seiner Klasse" wird. An den beiden Tests nahmen jeweils mehr als 1200 Patienten teil. Mit dem Konkurrenzprodukt Lucentis erwirtschaftete Roche im ersten Halbjahr 2010 allein in den USA einen Umsatz von 697 Millionen Franken. Außerhalb der USA wird das Präparat vom Baseler Rivalen Novartis vertrieben.
Frankfurt/New York (Reuters) - Bayer und sein US-Partner Regeneron sind der Marktzulassung eines neuen Augenheilmittels einen großen Schritt näher gekommen.
Das Präparat VEGF Trap-Eye habe in zwei Phase-III-Studien genauso gut gewirkt und sei ebenso sicher wie das umsatzstarke Konkurrenzprodukt Lucentis von Roche, teilten Bayer und die US-Biotechfirma Regeneron am Montag mit. Allerdings müsste es nur halb so häufig gespritzt werden. Auf Grundlage der beiden Studien wollen die Unternehmen nun im ersten Halbjahr 2011 Zulassungsanträge in den USA und in Europa stellen. Bayer traut der Medizin einen jährlichen Spitzenumsatz von 250 bis 500 Millionen Euro im Jahr zu.
An der Börse kam die Nachricht gut an: Bayer-Titel legten 0,8 Prozent zu. Die Analysten von Kepler Equities trauen der Arznei einen Spitzenumsatz von 350 Millionen Euro im Jahr zu. Diese Schätzung könne erheblich erhöht werden, falls das Mittel auch zur Behandlung einer ähnlichen Krankheit, dem sogenannten diabetischen Makula-Ödems (DME) wirke, erklärten die Analysten.Mit dem Mittel VEGF Trap-Eye soll die feuchte altersbedingte Makula-Degeneration (AMD) behandelt werden, eine krankhafte Gefäßneubildung auf dem Augenhintergrund. Die Erkrankung ist eine der häufigsten Ursachen für die Erblindung von Menschen über 65. Neue Medikamente müssen drei Phasen der klinischen Erprobung am Menschen bestehen. Für die Behandlung des Makula-Ödems befindet sich das Präparat aktuell in der zweiten Phase der klinischen Entwicklung.
Regeneron-Chef Leonard Schleifer hofft nach den AMD-Studien, dass das Mittel "das Beste in seiner Klasse" wird. An den beiden Tests nahmen jeweils mehr als 1200 Patienten teil. Mit dem Konkurrenzprodukt Lucentis erwirtschaftete Roche im ersten Halbjahr 2010 allein in den USA einen Umsatz von 697 Millionen Franken. Außerhalb der USA wird das Präparat vom Baseler Rivalen Novartis vertrieben.
hallo,
kann mir mal jemand erklären, sind diese ergebnisse tatsächlich SCHLECHT?
http://finance.yahoo.com/news/Celldex-Therapeutics-Reports-b…
hmmm, was ist da nur faul. schreckt die vorstellung von eigener PIII die investoren so ab? (im CC wurde erwähnt, dass man grundsätzlich North America alleine plane, für ex-NA aber mit partner).
oder???
was haltet ihr von dem ansatz, dass man PFS als primären endpunkt für die PIII hernehmen will, weil natürlich schneller verfügbar als OS?
kann mir mal jemand erklären, sind diese ergebnisse tatsächlich SCHLECHT?
http://finance.yahoo.com/news/Celldex-Therapeutics-Reports-b…
hmmm, was ist da nur faul. schreckt die vorstellung von eigener PIII die investoren so ab? (im CC wurde erwähnt, dass man grundsätzlich North America alleine plane, für ex-NA aber mit partner).
oder???
was haltet ihr von dem ansatz, dass man PFS als primären endpunkt für die PIII hernehmen will, weil natürlich schneller verfügbar als OS?
Ohad Hammer feiert in einem neuen Artikel die Auferstehung von Exelixis XL 184:
Exelixis - Back on Track
After a challenging year, Exelixis (EXEL) is finishing 2010 on a positive note, with the help of promising data for its lead agent, XL184. Last week at the EORTC conference, the company published data from a large phase II study in multiple cancer types. The results came at a crucial time for Exelixis, as many were questioning the value of XL184 following BMS’ (BMY) decision to opt-out of its development. As discussed in a previous post, when a partner like BMS dumps a late stage clinical asset after a licensing payment of over $150M, the alarm bells start ringing.
XL184 inhibits several targets, primarily VEGFR2 and Met. As a multi-targeted kinase inhibitor, the drug has some overlap with other drugs, primarily VEGFR inhibitors. There are two drugs that inhibit VEGFR (in addition to other targets) currently in the market as well as a long list of VEGFR inhibitors in late stage clinical testing from GSK (GSK), AstraZeneca (AZN), BMS and Aveo (AVEO). The main question regarding XL184 is whether the drug has a differentiated clinical profile in comparison to other VEGFR inhibitors. Based on recent data presented at EORTC the answer is a resounding “YES”.
hier gehts weiter:
http://www.hammerstockblog.com/exelixis-back-on-track/
Exelixis - Back on Track
After a challenging year, Exelixis (EXEL) is finishing 2010 on a positive note, with the help of promising data for its lead agent, XL184. Last week at the EORTC conference, the company published data from a large phase II study in multiple cancer types. The results came at a crucial time for Exelixis, as many were questioning the value of XL184 following BMS’ (BMY) decision to opt-out of its development. As discussed in a previous post, when a partner like BMS dumps a late stage clinical asset after a licensing payment of over $150M, the alarm bells start ringing.
XL184 inhibits several targets, primarily VEGFR2 and Met. As a multi-targeted kinase inhibitor, the drug has some overlap with other drugs, primarily VEGFR inhibitors. There are two drugs that inhibit VEGFR (in addition to other targets) currently in the market as well as a long list of VEGFR inhibitors in late stage clinical testing from GSK (GSK), AstraZeneca (AZN), BMS and Aveo (AVEO). The main question regarding XL184 is whether the drug has a differentiated clinical profile in comparison to other VEGFR inhibitors. Based on recent data presented at EORTC the answer is a resounding “YES”.
hier gehts weiter:
http://www.hammerstockblog.com/exelixis-back-on-track/
Onyx bestätigt die wirklich starken Daten von Carfilzomib!
Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX) today announced positive complete results from the Phase 2b 003-A1 study of single-agent carfilzomib, a next generation proteasome inhibitor, in patients with relapsed and refractory multiple myeloma. Carfilzomib achieved an overall response rate (ORR) (partial response or greater) of 24.1 percent and a median duration of response (DOR) of 8.3 months in patients who entered the study after receiving a median of five prior lines of therapy (corresponding to a median of 13 anti-myeloma agents) and whose disease was refractory to their last therapeutic regimen. In addition, patients enrolled in the study had progressive disease upon entering the trial. The clinical benefit rate (CBR) (minimal response or greater) in the study population was 34.2 percent. The median overall survival (OS) was 15.5 months. Overall survival for responding patients (≥ minimal response) has not yet been reached; however, based on current data is expected to exceed 19 months.
http://www.onyx-pharm.com/view.cfm/711/Onyx-Pharmaceuticals-…
Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX) today announced positive complete results from the Phase 2b 003-A1 study of single-agent carfilzomib, a next generation proteasome inhibitor, in patients with relapsed and refractory multiple myeloma. Carfilzomib achieved an overall response rate (ORR) (partial response or greater) of 24.1 percent and a median duration of response (DOR) of 8.3 months in patients who entered the study after receiving a median of five prior lines of therapy (corresponding to a median of 13 anti-myeloma agents) and whose disease was refractory to their last therapeutic regimen. In addition, patients enrolled in the study had progressive disease upon entering the trial. The clinical benefit rate (CBR) (minimal response or greater) in the study population was 34.2 percent. The median overall survival (OS) was 15.5 months. Overall survival for responding patients (≥ minimal response) has not yet been reached; however, based on current data is expected to exceed 19 months.
http://www.onyx-pharm.com/view.cfm/711/Onyx-Pharmaceuticals-…
Antwort auf Beitrag Nr.: 40.663.727 von SLGramann am 07.12.10 15:39:34Hier eine Aufbereitung von Fierce. Ich glaube, dass carfilzomib ein echter Knaller werden könnte. Für Onyx vielleicht wichtiger als Nexavar.
Onyx shares surge on fresh round of promising carfilzomib data
December 7, 2010 — 1:10pm ET | By John Carroll
Building on an upbeat side effect profile for carfilzomib, Onyx Pharma has laid out a promising set of extended survival data on the experimental cancer therapy. Yesterday the biotech company noted that in a Phase IIb study the drug did not spur peripheral neuropathy in patients, a common side effect seen with drugs now in use for advanced multiple myeloma. Today researchers added that the drug extended survival to 15.5 months and the news swiftly boosted its shares by 18 percent.
That survival rate "compared favorably to what one would expect, which is less than nine months in this patient population," Onyx Chief Medical Officer Michael Kauffman told Reuters. The study involved 266 cancer patients with treatment-resistant cases of multiple myeloma. In a subset analysis, the biotech noted that 20 percent of patients who had failed Velcade responded to carfilzomib. Onyx plans to file for an FDA approval in the middle of next year.
"In our view, this exceeds expectations, and it's also notable that these results were generated with what looks like a low dose of carfilzomib (probably the lowest effective dose)," JP Morgan analyst Corey Kasimov said in a research note.
"There is a significant need for new treatment options for these patients with refractory multiple myeloma who have exhausted all other options," said Dr. David Siegel. "This heavily pretreated patient population has received all classes of approved and commonly used myeloma therapies, and the durable responses and tolerability demonstrate carfilzomib's potential as a promising treatment."
Onyx shares surge on fresh round of promising carfilzomib data
December 7, 2010 — 1:10pm ET | By John Carroll
Building on an upbeat side effect profile for carfilzomib, Onyx Pharma has laid out a promising set of extended survival data on the experimental cancer therapy. Yesterday the biotech company noted that in a Phase IIb study the drug did not spur peripheral neuropathy in patients, a common side effect seen with drugs now in use for advanced multiple myeloma. Today researchers added that the drug extended survival to 15.5 months and the news swiftly boosted its shares by 18 percent.
That survival rate "compared favorably to what one would expect, which is less than nine months in this patient population," Onyx Chief Medical Officer Michael Kauffman told Reuters. The study involved 266 cancer patients with treatment-resistant cases of multiple myeloma. In a subset analysis, the biotech noted that 20 percent of patients who had failed Velcade responded to carfilzomib. Onyx plans to file for an FDA approval in the middle of next year.
"In our view, this exceeds expectations, and it's also notable that these results were generated with what looks like a low dose of carfilzomib (probably the lowest effective dose)," JP Morgan analyst Corey Kasimov said in a research note.
"There is a significant need for new treatment options for these patients with refractory multiple myeloma who have exhausted all other options," said Dr. David Siegel. "This heavily pretreated patient population has received all classes of approved and commonly used myeloma therapies, and the durable responses and tolerability demonstrate carfilzomib's potential as a promising treatment."
hallo,
hat sich hier jemand mal Basilea näher angesehen?
danke
hat sich hier jemand mal Basilea näher angesehen?
danke
Ein neuer Artikel von Ohad Hammer zu SGEN:
http://www.hammerstockblog.com/the-winner-of-ash-2010-seattl…
Wie immer herrscht diesbezüglich strengste Lesepflicht.
http://www.hammerstockblog.com/the-winner-of-ash-2010-seattl…
Wie immer herrscht diesbezüglich strengste Lesepflicht.
So, endlich sind die Daten zu Incytes 18424 draußen!
December 20, 2010 04:05 PM Eastern Time
Incyte Announces Positive Top-Line Results from COMFORT-I Pivotal Phase III Trial of INCB18424 in Myelofibrosis, a Debilitating, Life-Threatening Blood Cancer
* Trial meets primary and key secondary endpoints
* Safety and tolerability consistent with data from prior studies
* Full results to be submitted for presentation at the 2011 American Society of
Clinical Oncology Annual Meeting
* New Drug Application submission on track for second quarter of 2011
Company to Host Conference Call Today at 5:00 p.m. ET
WILMINGTON, Del.--(BUSINESS WIRE)--Incyte Corporation (Nasdaq:INCY) announced today positive top-line results from COMFORT-I, the pivotal Phase III clinical trial of INCB18424 (also known as INC424) in patients with myelofibrosis (MF) being conducted under a Food and Drug Administration (FDA) Special Protocol Assessment (SPA) Agreement. COMFORT-I (COntrolled MyeloFibrosis Study with ORal JAK Inhibitor Treatment) is a double-blind, placebo-controlled Phase III trial involving 309 patients. The primary endpoint was the response rate defined as the percentage of patients achieving a 35% or greater reduction in spleen volume at 24 weeks as measured by magnetic resonance imaging, or computerized tomography, comparing the rates in patients receiving INCB18424 or placebo. The response rate was 42% in patients randomized to INCB18424 versus less than 1% of patients randomized to placebo; thus a high level of statistical significance (p < 0.0001) was achieved.
High levels of statistical significance were also achieved for the key secondary endpoints based on symptomatic improvement as measured by the modified Myelofibrosis Symptom Assessment Form Diary. Response rates among patients receiving INCB18424 were similar to those previously reported with INCB18424 in the Phase II trial while much lower response rates were reported for patients receiving placebo in COMFORT-I.
The safety profile of INCB18424 was consistent with previous studies, which included reversible thrombocytopenia and anemia.
Incyte intends to submit the data from COMFORT-I for presentation at the 2011 American Society of Clinical Oncology Annual Meeting.
Paul Friedman, M.D., President and CEO of Incyte stated, "These highly significant results from COMFORT-I are similar to what was seen in the Phase II trial recently published in the New England Journal of Medicine. We look forward to continuing our work with the FDA and our investigators as we strive to make this new medicine available to patients with myelofibrosis as rapidly as possible. In this regard, we are proceeding with the preparation of a New Drug Application and believe INCB18424 has the potential to become the first FDA-approved treatment for this debilitating, life-threatening disease.”
“In addition to meeting the efficacy endpoints in this controlled trial, the safety and tolerability profile suggests that INCB18424 could become the first widely available medication to relieve the debilitating symptoms in patients with myelofibrosis,” added Richard Levy, M.D., Executive Vice President and Chief Drug Development and Medical Officer at Incyte.
Based on the SPA agreement, COMFORT-I is the only pivotal trial required for approval in the U.S. Data from a second Phase III trial, COMFORT-II, which is the pivotal trial for European Marketing Authorization, comparing INCB18424 to best available therapy, is being conducted by Novartis in Europe and is also expected to provide supportive data for approval and labeling in the U.S. Results of COMFORT-II are expected early next year. Incyte and Novartis announced a collaboration and license agreement in November 2009 in which Incyte retained exclusive rights to develop and commercialize INCB18424 in the U.S. and Novartis received exclusive rights to develop and commercialize INCB18424 for territories outside the U.S. Both the FDA and the European Medicines Agency have granted INCB18424 orphan drug status for MF.
About COMFORT-I and COMFORT-II
COMFORT-I is a randomized (1:1), double-blind Phase III trial comparing the efficacy and safety of INCB18424 to placebo in 309 patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF) or post-essential thrombocythemia myelofibrosis (PET-MF) and involved over 100 clinical sites in the U.S., Australia and Canada. To be eligible for the study, patients had to have a palpated spleen length of 5 cm or greater and be classified as intermediate 2 or high risk according to the International Working Group (IWG) criteria1. COMFORT-I is scheduled to continue until either INCB18424 receives marketing approval or the last randomized patient remaining in the study has completed week 144 (36 months).
COMFORT-II is a second Phase III trial being conducted by Novartis in Europe. It is a randomized (2:1), controlled, open-label study designed to evaluate the efficacy, safety and tolerability of INCB18424 as compared to the best-available therapy in patients with PMF, PPV-MF or PET-MF. COMFORT-II enrolled 219 patients and involves approximately 55 clinical sites in 9 European countries: Belgium, Austria, France, Italy, Germany, Sweden, the Netherlands, Spain and the U.K. The primary efficacy endpoint in COMFORT–II is the proportion of patients achieving at least 35% reduction in spleen volume from baseline to week 48.
Neither COMFORT-I nor COMFORT-II were powered to demonstrate differences in survival.
------------------
Bin mir jetzt nicht so sicher, ob 42% soooo gut sind, aber wohl gut genug für eine Zulassung denk ich jedenfalls. Kurs ist derzeit nachbörslich ca. 5% im Plus, was wenig euphorisch daherkommt, aber eh nicht viel heißt. Ein Erfolg ist sowieso ziemlich stark eingepreist.
In dem Zusammenhang ist auch die Aufnahme von YM´s in Hammers Depot bedenkenswert. Da geht es nämlich (vor allem) auch um einen JAK-Inhibitor der in der Indikation Myelofibrose entwickelt wird. Aus diesem pdf kann man zum Thema und insbesondere über die Konkurrenzsituation einiges lernen:
http://www.ymbiosciences.com/upload_files/YMcorporate_presen…
Ich selbst habe als ziemlicher Hammer-Nachäffer YM ins Depot genommen.
-------------------------
Mit der Meldung von Incyte ist m.E. das Jahr 2010 durch. Es war alles in allem sehr erfolgreich, was die Studienergebnisse angeht. Ich denke da insbesondere an IMGN, SGEN, REGN, ARQL und INCY.
Hi ipollit, ich hoffe, 2011 legst Du ein neues Biotech-Depot auf!
December 20, 2010 04:05 PM Eastern Time
Incyte Announces Positive Top-Line Results from COMFORT-I Pivotal Phase III Trial of INCB18424 in Myelofibrosis, a Debilitating, Life-Threatening Blood Cancer
* Trial meets primary and key secondary endpoints
* Safety and tolerability consistent with data from prior studies
* Full results to be submitted for presentation at the 2011 American Society of
Clinical Oncology Annual Meeting
* New Drug Application submission on track for second quarter of 2011
Company to Host Conference Call Today at 5:00 p.m. ET
WILMINGTON, Del.--(BUSINESS WIRE)--Incyte Corporation (Nasdaq:INCY) announced today positive top-line results from COMFORT-I, the pivotal Phase III clinical trial of INCB18424 (also known as INC424) in patients with myelofibrosis (MF) being conducted under a Food and Drug Administration (FDA) Special Protocol Assessment (SPA) Agreement. COMFORT-I (COntrolled MyeloFibrosis Study with ORal JAK Inhibitor Treatment) is a double-blind, placebo-controlled Phase III trial involving 309 patients. The primary endpoint was the response rate defined as the percentage of patients achieving a 35% or greater reduction in spleen volume at 24 weeks as measured by magnetic resonance imaging, or computerized tomography, comparing the rates in patients receiving INCB18424 or placebo. The response rate was 42% in patients randomized to INCB18424 versus less than 1% of patients randomized to placebo; thus a high level of statistical significance (p < 0.0001) was achieved.
High levels of statistical significance were also achieved for the key secondary endpoints based on symptomatic improvement as measured by the modified Myelofibrosis Symptom Assessment Form Diary. Response rates among patients receiving INCB18424 were similar to those previously reported with INCB18424 in the Phase II trial while much lower response rates were reported for patients receiving placebo in COMFORT-I.
The safety profile of INCB18424 was consistent with previous studies, which included reversible thrombocytopenia and anemia.
Incyte intends to submit the data from COMFORT-I for presentation at the 2011 American Society of Clinical Oncology Annual Meeting.
Paul Friedman, M.D., President and CEO of Incyte stated, "These highly significant results from COMFORT-I are similar to what was seen in the Phase II trial recently published in the New England Journal of Medicine. We look forward to continuing our work with the FDA and our investigators as we strive to make this new medicine available to patients with myelofibrosis as rapidly as possible. In this regard, we are proceeding with the preparation of a New Drug Application and believe INCB18424 has the potential to become the first FDA-approved treatment for this debilitating, life-threatening disease.”
“In addition to meeting the efficacy endpoints in this controlled trial, the safety and tolerability profile suggests that INCB18424 could become the first widely available medication to relieve the debilitating symptoms in patients with myelofibrosis,” added Richard Levy, M.D., Executive Vice President and Chief Drug Development and Medical Officer at Incyte.
Based on the SPA agreement, COMFORT-I is the only pivotal trial required for approval in the U.S. Data from a second Phase III trial, COMFORT-II, which is the pivotal trial for European Marketing Authorization, comparing INCB18424 to best available therapy, is being conducted by Novartis in Europe and is also expected to provide supportive data for approval and labeling in the U.S. Results of COMFORT-II are expected early next year. Incyte and Novartis announced a collaboration and license agreement in November 2009 in which Incyte retained exclusive rights to develop and commercialize INCB18424 in the U.S. and Novartis received exclusive rights to develop and commercialize INCB18424 for territories outside the U.S. Both the FDA and the European Medicines Agency have granted INCB18424 orphan drug status for MF.
About COMFORT-I and COMFORT-II
COMFORT-I is a randomized (1:1), double-blind Phase III trial comparing the efficacy and safety of INCB18424 to placebo in 309 patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF) or post-essential thrombocythemia myelofibrosis (PET-MF) and involved over 100 clinical sites in the U.S., Australia and Canada. To be eligible for the study, patients had to have a palpated spleen length of 5 cm or greater and be classified as intermediate 2 or high risk according to the International Working Group (IWG) criteria1. COMFORT-I is scheduled to continue until either INCB18424 receives marketing approval or the last randomized patient remaining in the study has completed week 144 (36 months).
COMFORT-II is a second Phase III trial being conducted by Novartis in Europe. It is a randomized (2:1), controlled, open-label study designed to evaluate the efficacy, safety and tolerability of INCB18424 as compared to the best-available therapy in patients with PMF, PPV-MF or PET-MF. COMFORT-II enrolled 219 patients and involves approximately 55 clinical sites in 9 European countries: Belgium, Austria, France, Italy, Germany, Sweden, the Netherlands, Spain and the U.K. The primary efficacy endpoint in COMFORT–II is the proportion of patients achieving at least 35% reduction in spleen volume from baseline to week 48.
Neither COMFORT-I nor COMFORT-II were powered to demonstrate differences in survival.
------------------
Bin mir jetzt nicht so sicher, ob 42% soooo gut sind, aber wohl gut genug für eine Zulassung denk ich jedenfalls. Kurs ist derzeit nachbörslich ca. 5% im Plus, was wenig euphorisch daherkommt, aber eh nicht viel heißt. Ein Erfolg ist sowieso ziemlich stark eingepreist.
In dem Zusammenhang ist auch die Aufnahme von YM´s in Hammers Depot bedenkenswert. Da geht es nämlich (vor allem) auch um einen JAK-Inhibitor der in der Indikation Myelofibrose entwickelt wird. Aus diesem pdf kann man zum Thema und insbesondere über die Konkurrenzsituation einiges lernen:
http://www.ymbiosciences.com/upload_files/YMcorporate_presen…
Ich selbst habe als ziemlicher Hammer-Nachäffer YM ins Depot genommen.
-------------------------
Mit der Meldung von Incyte ist m.E. das Jahr 2010 durch. Es war alles in allem sehr erfolgreich, was die Studienergebnisse angeht. Ich denke da insbesondere an IMGN, SGEN, REGN, ARQL und INCY.
Hi ipollit, ich hoffe, 2011 legst Du ein neues Biotech-Depot auf!
Incyte verarbeitet die Daten des Myelofibrose-Trials und ist derzeit so ziemlich auf Allzeithoch (abgesehen von irgendeinem Quatsch im Blasen-Jahr 2000).
Indes beträgt die Marktkapitalisierung nun aber auch schon 2 Milliarden Dollar.
Stellt sich jemand die Frage, wie weit das noch gehen kann?
Eines Tages wird INCY 18424 KOnkurrenz bekommen.
Mal folgender Kommentar ohne Wertung:
one of the main problems with myelofibrosis patients is anemia. The vast number of blood transfusions they must have causes iron overload, organ failure, etc. Not to mention garden variety problems associated with anemia. Ever have a transfusion? These patients are often there once every week to two weeks. It can take half a day to have 2 to 3 units infused. The spleen can also be problematic. But the main problem is anemia. The incyte drug does NOTHING for anemia...in fact, at ASH it was revealed that patients who were not previously anemic BECAME anemic on the incyte drug and had to stop the trial. The drug that will become the REAL gold standard will address anemia as well as the spleen.
-------
YMI adressiert dieses Feld scheinbar.
Mh...
Indes beträgt die Marktkapitalisierung nun aber auch schon 2 Milliarden Dollar.
Stellt sich jemand die Frage, wie weit das noch gehen kann?
Eines Tages wird INCY 18424 KOnkurrenz bekommen.
Mal folgender Kommentar ohne Wertung:
one of the main problems with myelofibrosis patients is anemia. The vast number of blood transfusions they must have causes iron overload, organ failure, etc. Not to mention garden variety problems associated with anemia. Ever have a transfusion? These patients are often there once every week to two weeks. It can take half a day to have 2 to 3 units infused. The spleen can also be problematic. But the main problem is anemia. The incyte drug does NOTHING for anemia...in fact, at ASH it was revealed that patients who were not previously anemic BECAME anemic on the incyte drug and had to stop the trial. The drug that will become the REAL gold standard will address anemia as well as the spleen.
-------
YMI adressiert dieses Feld scheinbar.
Mh...
Ohad Hammer hat sich zum Thema Incyte vs. YM eingelassen. Der Text enthält einige sehr bemerkenswerte Passagen.
Für mich selbst habe ich die Entscheidung getroffen, auch bei Incyte weiter dabeizubleiben. Das Potential des 18424 ist, wenn man PV und ET dazunimmt, so groß, dass die 2 Mrd. Marktkapitalisierung in der Tat nicht das Ende der Fahnenstange sein müssen. Außerdem könnte der 18424 noch in weiteren Indikationen Erfoge zeigen (bspw. auch bei Psoriasis als topical medication). Auch der Rest der Pipeline ist nicht nichts. Möglicherweise kann Incyte in 3 Jahren bis zu 4 Mrd. Dollar wert sein (nur meine Meinung).
Hier der Link zum Artikel:
http://www.hammerstockblog.com/incyte-concludes-an-exception…
Und hier der Text:
(Hervorhebungen von mir)
Incyte Concludes an Exceptional Year
Earlier this week, Incyte (INCY) announced positive phase III results for its lead agent, INCB424, in myelofibrosis (MF). Although the full data set was not published, it will almost certainly lead to FDA approval, opening up a $200-$300 market in the US alone. Another similar phase III trial which will be reported in the coming months should support approval in Europe as well.
Unprecedented results
Based on the press release and conference call last Monday, results were basically in line with published phase II results which were extremely positive. The primary endpoint (percentage of patients achieving a 35% reduction in spleen volume) was easily met, as 42% of patients in the drug arm responded vs. less than 1% in the placebo arm. INCB424 was also highly superior to placebo in terms of reduction of symptoms, which represents a highly unmet need in these patients.
With the positive data, Incyte is concluding a year packed with achievements. These include removal of the short term debt overhang (discussed here), $69M in milestone payments from partners, positive phase II data with INCB050 and INCB424 in rheumatoid arthritis and PV (polycythemia vera), respectively, and initiation of a global phase III in PV.
Competition intensifies
Incyte’s drug will probably be the first ever FDA approved drug for myelofibrosis, however, investors should expect competition to emerge several years down the road. INCB424’s direct competitors are drugs with similar mode of action from YM Biosciences (YMI), Sanofi (SNY) and Onyx (ONXX), all of which are Jak inhibitors. Celgene’s (CELG) pomalidomide is in phase III testing, but seems to have a different and more subtle effect in myelofibrosis.
Until recently, Incyte’s leadership position was unquestionable, as it had what seemed to be a best in class drug with a 3 year lead over competition. Therefore, by the time a competitor hits the market, even with comparable activity, it will encounter a well-established agent as the standard of care. The only major threat to Incyte could be a compound with a differentiated clinical profile in terms of superior safety or efficacy. This is exactly what YM Biosciences claims to have.
At the recent ASH conference, YM Biosciences presented results for its Jak inhibitor, CYT387, in myelofibrosis. Last year, it was Incyte who stole the show with stellar results in MF. This year it was YM Biosciences’ controversial presentation that got people’s attention.
CYT387 was originally developed by Australia-based Cytopia, which was acquired by YM Biosciences last year. At ASH, investigators presented results from 60 patients, which showed the drug definitely has activity in terms of spleen reduction and symptom alleviation. Although it is hard to reach definitive conclusions with respect to these endpoints in comparison to Incyte’s drug, the data set included allegedly an additional effect on anemia that has not been observed with INCB424.
Anemia is a common complication in MF patients, which often requires patients to receive blood transfusions. To date, Jak inhibitors exhibited little to no effect on anemia, and in some cases even caused anemia in non anemic patients. In contrast, the CYT387 data set included a 50% anemia response, which, if real, could become an important competitive advantage.
So, is CYT387’s anemia effect real?
It depends on who you ask.
The primary investigator for CYT378 clearly highlighted that this is a clear differentiating factor for CYT378, as it was evaluated using similar criteria to those used in Incyte’s trial. Many, including Incyte, claim that it is impossible to compare the anemia effect of the two drugs since YM Biosciences used different definitions for assessing anemia. When asked about this issue during a presentation conducted by YM Biosciences, Dr. Ayalew Tefferi from the Mayo clinic, who also participated in the INCB424 study, replied by calling it “dishonest and inaccurate information”.
Jonathan Aschoff from Brean Murray issued a note claiming that the two studies used different criteria for defining anemia and transfusion independence. According to Aschoff, when the INCB424 data is normalized according to the CYT387 criteria, the two drugs seem comparable in terms of anemia resolution.
Incyte at ASH 2010
Regardless of the CYT387 debate, the meeting was a positive event for Incyte, which presented impressive data for INCB424 in two additional indications: polycythemia vera (PV) and essential thrombocythemia (ET). PV and ET are two blood disorders with cancer features, similarly to myelofibrosis. In PV patients, the drug led to a profound reduction of spleen size in the majority of patients with enlarged spleen, a hallmark complication of the disease. In 39 patients with ET, a disease characterized by over production of platelets, 79% achieved a marked reduction in platelet count and 49% saw normalization in platelet count. Of note, patients were refractory or intolerant to hydroxyurea, a commonly used drug for these conditions.
These results in PV laid the groundwork for the recently initiated phase III trial in Europe and the US. Similarly to INCB424’s development program in MF, the PV program is very attractive given the lack of approved treatments and the dramatic effect INCB424 has. This enables Incyte and its partner Novartis (NVS) to get the drug on the market using a relatively small (300 patients) and short study (~2.5 years). Assuming results are positive, INCB424 is looking at a market opportunity of $200M-$300M in the US alone.
Portfolio updates
Last week, we added YM Biosciences following the data at ASH. The jury is still out on the anemia question despite investigators’ clear enthusiasm. Regardless, the drug is clearly active in myelofibrosis with potential utility in other myeloproliferative diseases such as PV and ET as well as inflammatory disease. The global market for myeloproliferative diseases is estimated at ~$1.5B while the market for inflammatory diseases is substantially larger. With a market cap of under $200M, even if YM Biosciences captures a 5% market share of the Jak inhibitors market, the company represents an attractive investment opportunity. The data at ASH should enable YM Biosciences to strike a lucrative licensing deal in 2011.
This week, we are selling the second position in Immunogen (IMGN) we initiated a year ago. We are retaining our larger core position in the company.
Für mich selbst habe ich die Entscheidung getroffen, auch bei Incyte weiter dabeizubleiben. Das Potential des 18424 ist, wenn man PV und ET dazunimmt, so groß, dass die 2 Mrd. Marktkapitalisierung in der Tat nicht das Ende der Fahnenstange sein müssen. Außerdem könnte der 18424 noch in weiteren Indikationen Erfoge zeigen (bspw. auch bei Psoriasis als topical medication). Auch der Rest der Pipeline ist nicht nichts. Möglicherweise kann Incyte in 3 Jahren bis zu 4 Mrd. Dollar wert sein (nur meine Meinung).
Hier der Link zum Artikel:
http://www.hammerstockblog.com/incyte-concludes-an-exception…
Und hier der Text:
(Hervorhebungen von mir)
Incyte Concludes an Exceptional Year
Earlier this week, Incyte (INCY) announced positive phase III results for its lead agent, INCB424, in myelofibrosis (MF). Although the full data set was not published, it will almost certainly lead to FDA approval, opening up a $200-$300 market in the US alone. Another similar phase III trial which will be reported in the coming months should support approval in Europe as well.
Unprecedented results
Based on the press release and conference call last Monday, results were basically in line with published phase II results which were extremely positive. The primary endpoint (percentage of patients achieving a 35% reduction in spleen volume) was easily met, as 42% of patients in the drug arm responded vs. less than 1% in the placebo arm. INCB424 was also highly superior to placebo in terms of reduction of symptoms, which represents a highly unmet need in these patients.
With the positive data, Incyte is concluding a year packed with achievements. These include removal of the short term debt overhang (discussed here), $69M in milestone payments from partners, positive phase II data with INCB050 and INCB424 in rheumatoid arthritis and PV (polycythemia vera), respectively, and initiation of a global phase III in PV.
Competition intensifies
Incyte’s drug will probably be the first ever FDA approved drug for myelofibrosis, however, investors should expect competition to emerge several years down the road. INCB424’s direct competitors are drugs with similar mode of action from YM Biosciences (YMI), Sanofi (SNY) and Onyx (ONXX), all of which are Jak inhibitors. Celgene’s (CELG) pomalidomide is in phase III testing, but seems to have a different and more subtle effect in myelofibrosis.
Until recently, Incyte’s leadership position was unquestionable, as it had what seemed to be a best in class drug with a 3 year lead over competition. Therefore, by the time a competitor hits the market, even with comparable activity, it will encounter a well-established agent as the standard of care. The only major threat to Incyte could be a compound with a differentiated clinical profile in terms of superior safety or efficacy. This is exactly what YM Biosciences claims to have.
At the recent ASH conference, YM Biosciences presented results for its Jak inhibitor, CYT387, in myelofibrosis. Last year, it was Incyte who stole the show with stellar results in MF. This year it was YM Biosciences’ controversial presentation that got people’s attention.
CYT387 was originally developed by Australia-based Cytopia, which was acquired by YM Biosciences last year. At ASH, investigators presented results from 60 patients, which showed the drug definitely has activity in terms of spleen reduction and symptom alleviation. Although it is hard to reach definitive conclusions with respect to these endpoints in comparison to Incyte’s drug, the data set included allegedly an additional effect on anemia that has not been observed with INCB424.
Anemia is a common complication in MF patients, which often requires patients to receive blood transfusions. To date, Jak inhibitors exhibited little to no effect on anemia, and in some cases even caused anemia in non anemic patients. In contrast, the CYT387 data set included a 50% anemia response, which, if real, could become an important competitive advantage.
So, is CYT387’s anemia effect real?
It depends on who you ask.
The primary investigator for CYT378 clearly highlighted that this is a clear differentiating factor for CYT378, as it was evaluated using similar criteria to those used in Incyte’s trial. Many, including Incyte, claim that it is impossible to compare the anemia effect of the two drugs since YM Biosciences used different definitions for assessing anemia. When asked about this issue during a presentation conducted by YM Biosciences, Dr. Ayalew Tefferi from the Mayo clinic, who also participated in the INCB424 study, replied by calling it “dishonest and inaccurate information”.
Jonathan Aschoff from Brean Murray issued a note claiming that the two studies used different criteria for defining anemia and transfusion independence. According to Aschoff, when the INCB424 data is normalized according to the CYT387 criteria, the two drugs seem comparable in terms of anemia resolution.
Incyte at ASH 2010
Regardless of the CYT387 debate, the meeting was a positive event for Incyte, which presented impressive data for INCB424 in two additional indications: polycythemia vera (PV) and essential thrombocythemia (ET). PV and ET are two blood disorders with cancer features, similarly to myelofibrosis. In PV patients, the drug led to a profound reduction of spleen size in the majority of patients with enlarged spleen, a hallmark complication of the disease. In 39 patients with ET, a disease characterized by over production of platelets, 79% achieved a marked reduction in platelet count and 49% saw normalization in platelet count. Of note, patients were refractory or intolerant to hydroxyurea, a commonly used drug for these conditions.
These results in PV laid the groundwork for the recently initiated phase III trial in Europe and the US. Similarly to INCB424’s development program in MF, the PV program is very attractive given the lack of approved treatments and the dramatic effect INCB424 has. This enables Incyte and its partner Novartis (NVS) to get the drug on the market using a relatively small (300 patients) and short study (~2.5 years). Assuming results are positive, INCB424 is looking at a market opportunity of $200M-$300M in the US alone.
Portfolio updates
Last week, we added YM Biosciences following the data at ASH. The jury is still out on the anemia question despite investigators’ clear enthusiasm. Regardless, the drug is clearly active in myelofibrosis with potential utility in other myeloproliferative diseases such as PV and ET as well as inflammatory disease. The global market for myeloproliferative diseases is estimated at ~$1.5B while the market for inflammatory diseases is substantially larger. With a market cap of under $200M, even if YM Biosciences captures a 5% market share of the Jak inhibitors market, the company represents an attractive investment opportunity. The data at ASH should enable YM Biosciences to strike a lucrative licensing deal in 2011.
This week, we are selling the second position in Immunogen (IMGN) we initiated a year ago. We are retaining our larger core position in the company.
gibts hier n nachfolgethread für 2011?
Antwort auf Beitrag Nr.: 41.038.564 von PathFinder2 am 14.02.11 21:07:13Leider bisher nicht. ipollit ist laut Profil seit Mitte Dezember hier nicht mehr online gewesen.
Interessante Entwicklungen hat es seit dem durchaus gegeben, bspw. hat Regeneron mit VEGF-Trap-Eye gute Daten geliefert.
Interessante Entwicklungen hat es seit dem durchaus gegeben, bspw. hat Regeneron mit VEGF-Trap-Eye gute Daten geliefert.
Antwort auf Beitrag Nr.: 41.039.682 von SLGramann am 15.02.11 07:34:18wie wäre es mit einem 2012 depot oder gar depot spiel??
Antwort auf Beitrag Nr.: 42.526.321 von pokemon am 28.12.11 15:59:04Einen neuen Thread werde ich wohl nicht füllen können, dafür fehlt mir die Zeit. Ohne SLGramann wäre dieser Thread ja auch schon untergegangen. Mein Depot lasse ich weiterlaufen... momentan sieht es so aus, wobei es auch einige "Leichen" enthält:
8,6% Regeneron
8,4% Cubist
8,1% Incyte
7,5% Onyx
6,9% Pharmacyclics
5,6% Micromet
5,3% AVEO
4,5% Exelixis
4,3% Seattle Genetics
4,2% Synta
3,9% ArQule
3,3% ViroPharma
3,3% Isis
2,8% Celldex
2,7% Alnylam
2,7% Intermune
2,6% Array
2,6% Trius
2,5% Rigel
2,3% Ziopharm
2,2% Genmab
1,4% AMAG
1,2% Allos
1,1% Endocyte
0,9% Arena
0,5% Targacept
0,4% NeuroSearch
0,3% NeurogesX
mfg ipollit
8,6% Regeneron
8,4% Cubist
8,1% Incyte
7,5% Onyx
6,9% Pharmacyclics
5,6% Micromet
5,3% AVEO
4,5% Exelixis
4,3% Seattle Genetics
4,2% Synta
3,9% ArQule
3,3% ViroPharma
3,3% Isis
2,8% Celldex
2,7% Alnylam
2,7% Intermune
2,6% Array
2,6% Trius
2,5% Rigel
2,3% Ziopharm
2,2% Genmab
1,4% AMAG
1,2% Allos
1,1% Endocyte
0,9% Arena
0,5% Targacept
0,4% NeuroSearch
0,3% NeurogesX
mfg ipollit
Antwort auf Beitrag Nr.: 42.527.878 von ipollit am 28.12.11 22:26:04da kenne ich leider die wenigsten
mein europ. Biotech Depot besteht aus
80 % Mologen
10% Evotec
10% Bavarian Nordic
Clinuvel überlege ich aktuell zu kaufen
werde mir auch mal die US Biofirmen jetzt anschauen. Danke für die Aufstellung!
mein europ. Biotech Depot besteht aus
80 % Mologen
10% Evotec
10% Bavarian Nordic
Clinuvel überlege ich aktuell zu kaufen
werde mir auch mal die US Biofirmen jetzt anschauen. Danke für die Aufstellung!
Antwort auf Beitrag Nr.: 42.527.878 von ipollit am 28.12.11 22:26:04Hi ipollit, schade, dass Du keine Zeit hast. Deine Thread war immer eine wertvolle Informationsquelle, weil Du einen sehr breiten Ansatz hattest und hier immer Fundamentaldaten die Hauptrolle gespielt haben, statt des unsäglichen Geschwätzes über aktuelle Kurse...
Nun, ich gehe mit folgenden Werten in das Jahr 2012, nachdem ich in den letzten Wochen aufgeräumt habe:
AVEO: 6%
CLDX: 4%
EXEL: 7%
Galapagos: 20% (mal was in Europa gefunden)
MITI: 10%
MorphoSys: 16% (alte Liebe, die rostet und nervt )
PCYC: 34% (für mich der heißeste Wert der nächsten Jahre - ich hoffe, ich irre mich hier nicht, das wäre bitter!)
YMI: 3%
Wünsche Dir ein gutes und erfolgreiches 2012.
Gruß
SLG
@pokemon, Du mit Deinen Mologens! Völlig wahnsinnig, so ein Risiko zu fahren, ohne irgendwelche relevanten Daten und Fakten zu haben. Die haben noch gar nichts geliefert und Du gehst faktisch all in. Wirst wahrscheinlich ganz bös Lehrgeld zahlen.
Nun, ich gehe mit folgenden Werten in das Jahr 2012, nachdem ich in den letzten Wochen aufgeräumt habe:
AVEO: 6%
CLDX: 4%
EXEL: 7%
Galapagos: 20% (mal was in Europa gefunden)
MITI: 10%
MorphoSys: 16% (alte Liebe, die rostet und nervt
)PCYC: 34% (für mich der heißeste Wert der nächsten Jahre - ich hoffe, ich irre mich hier nicht, das wäre bitter!)
YMI: 3%
Wünsche Dir ein gutes und erfolgreiches 2012.
Gruß
SLG
@pokemon, Du mit Deinen Mologens! Völlig wahnsinnig, so ein Risiko zu fahren, ohne irgendwelche relevanten Daten und Fakten zu haben. Die haben noch gar nichts geliefert und Du gehst faktisch all in. Wirst wahrscheinlich ganz bös Lehrgeld zahlen.
@SLG
da hast du grds. recht. aber ich interpretiere die bisherigen bekanntgegebenen klinischen daten und den wissenschaftlichen ansatz anders. ich sehe hier ein riesen potential...die mehrheit der marktteilnehmer ist da wohl anderer ansicht.
p.s. the winner takes it all!
da hast du grds. recht. aber ich interpretiere die bisherigen bekanntgegebenen klinischen daten und den wissenschaftlichen ansatz anders. ich sehe hier ein riesen potential...die mehrheit der marktteilnehmer ist da wohl anderer ansicht.
p.s. the winner takes it all!
hi SLGramann! Die Informationen kamen allerdings fast alle von dir. Welche Quellen nutzt du denn so? Gibt es außer den folgenden weitere interessante Quellen?
tägliche News...
http://www.fiercebiotech.com/
Boards...
http://investorshub.advfn.com/boards/board.aspx?board_id=141…
http://www.siliconinvestor.com/home.aspx?forumid=102&sort=5&… (u.a. den Biotech Charity Contest)
Blogs...
http://www.hammerstockblog.com (alternativ mit anderen Kommentaren http://seekingalpha.com/author/ohad-hammer/articles
http://pharmastrategyblog.com
http://www.thestreet.com/author/1352996/adam-feuerstein/all.…
http://seekingalpha.com/author/ep-vantage/articles
http://seekingalpha.com/author/jason-chew/articles
http://www.prohostbiotech.com
http://biohealthinvestor.com
Reports...
http://www.healthace.com/rssfeed/Oncology_Report_mmddyyM.pdf (mit mm=Monat, dd=Tag und yy=Jahr... z.B. 122011M)
Kurse, Daten...
http://finance.yahoo.com/
http://de.advfn.com/p.php?pid=staticchart&s=N^XXXX&p=0&t=1&d… (realtime mit XXXX=Symbol)
****
Ob Galapagos mal eine Ausnahme unter den europäischen Biotechs darstellt? Bisher hat bei mir alles gefloppt, was aus Europa kam.
PCYC... halte ich auch für einen der besten Werte und möchte ich auch noch ein wenig aufstocken. Allerdings sind 1/3 nicht gerade wenig. Wenn bei denen doch mal eine unerwartete Nebenwirkung auftritt, kann sich das ganze schnell in Luft auflösen. Daher versuche ich, die Positionen nicht zu groß werden zu lassen. So habe ich beispielsweise den Einbruch in TRGT oder ECYT nur minimal gemerkt. Andererseits profitiere ich von den Einzelwerten auch nicht so stark. Naja, viel Glück mit PCYC!
AVEO... momentan beobachte ich ANTH. Dabei habe ich auf ihub gelesen, dass die AVEOs PII von Tivozanib in Russland durchgeführt worden ist. Das hat mich ein wenig verunsichert, da leider alle Studiendaten außerhalb Nordamerika und EU mit großer Vorsicht zu betrachten sind (wie zuletzt bei TRGT). Wie siehst du das?
Danke, wünsche dir ebenfalls ein erfolgreiches und gutes neues Jahr!
mfg ipollit
tägliche News...
http://www.fiercebiotech.com/
Boards...
http://investorshub.advfn.com/boards/board.aspx?board_id=141…
http://www.siliconinvestor.com/home.aspx?forumid=102&sort=5&… (u.a. den Biotech Charity Contest)
Blogs...
http://www.hammerstockblog.com (alternativ mit anderen Kommentaren http://seekingalpha.com/author/ohad-hammer/articles
http://pharmastrategyblog.com
http://www.thestreet.com/author/1352996/adam-feuerstein/all.…
http://seekingalpha.com/author/ep-vantage/articles
http://seekingalpha.com/author/jason-chew/articles
http://www.prohostbiotech.com
http://biohealthinvestor.com
Reports...
http://www.healthace.com/rssfeed/Oncology_Report_mmddyyM.pdf (mit mm=Monat, dd=Tag und yy=Jahr... z.B. 122011M)
Kurse, Daten...
http://finance.yahoo.com/
http://de.advfn.com/p.php?pid=staticchart&s=N^XXXX&p=0&t=1&d… (realtime mit XXXX=Symbol)
****
Ob Galapagos mal eine Ausnahme unter den europäischen Biotechs darstellt? Bisher hat bei mir alles gefloppt, was aus Europa kam.
PCYC... halte ich auch für einen der besten Werte und möchte ich auch noch ein wenig aufstocken. Allerdings sind 1/3 nicht gerade wenig. Wenn bei denen doch mal eine unerwartete Nebenwirkung auftritt, kann sich das ganze schnell in Luft auflösen. Daher versuche ich, die Positionen nicht zu groß werden zu lassen. So habe ich beispielsweise den Einbruch in TRGT oder ECYT nur minimal gemerkt. Andererseits profitiere ich von den Einzelwerten auch nicht so stark. Naja, viel Glück mit PCYC!
AVEO... momentan beobachte ich ANTH. Dabei habe ich auf ihub gelesen, dass die AVEOs PII von Tivozanib in Russland durchgeführt worden ist. Das hat mich ein wenig verunsichert, da leider alle Studiendaten außerhalb Nordamerika und EU mit großer Vorsicht zu betrachten sind (wie zuletzt bei TRGT). Wie siehst du das?
Danke, wünsche dir ebenfalls ein erfolgreiches und gutes neues Jahr!
mfg ipollit
Antwort auf Beitrag Nr.: 42.529.597 von ipollit am 29.12.11 13:23:37Hi ipollit, alle Infoquellen die ich nutze, hast Du bereits genannt - und noch einige mehr.
Zu Galapagos habe ich vor kurzem einen Thread angestoßen, der sofort kompetente Teilnehmer gefunden hat.
Vielleicht magst Du mal schauen:
Thread: Galapagos NV
AVEO: Das wusste ich nicht, dass das in Russland gelaufen ist. Grundsätzlich sehe ich so etwas auch kritisch. Ich stelle aber fest, dass recht viele Trials einen Schwerpunkt in Osteuropa haben. Über die Gründe kann man spekulieren.
Im Fall AVEO mache ich es mir etwas bequem und verlasse mich auch etwas auf die Expertise von Astellas. Ich gehe fest davon aus, dass die sich die Daten genau angeschaut haben.
PCYC: Ja, die Gewichtung ist sehr hoch und ich würde das keinem zur Nachahmung empfehlen. PCYC hat eigentlich nur den BTK-Inhibitor als echtes Asset und die Marktkapitalisierung ist bei einer Milliarde. Wenn das Projekt schief geht, steht man auf einen Schlag nackt da.
Auf der anderen Seite sind die Daten bis jetzt offensichtlich erstklassig. Das Nebenwirkungsprofil scheint gut auszusehen. Das Marktpotential ist sehr, sehr groß. Der Deal mit J&J ist ganz außergewöhnlich gut. Und - das könnte langfristig eine ganz, ganz fette Sahnehaube werden - der Signalweg könnte auch bei Autoimmunerkrankungen taugen, die im Partnerdeal nicht drin sind.
Kurz und gut: Persönlich habe ich noch nie ein so großes Potential gesehen, wie hier. In der Vergangenheit hat es so was natürlich schon gegeben, aber da war ich noch nicht dabei. Ich denke mir, so oder ähnlich hat es sicher mal bei Celgene angefangen.
Klar, da ist jetzt ne Menge Gier im Spiel. Ich hoffe hier auf so etwas wie den Jackpot. Die Art Größenwahn wird im Leben ja aber meist bestraft.
Ich glaub, dank PCYC werde ich das Beten lernen.
ECYT: Katastrophe. Schwer zu verstehen. Will das alles mal in Ruhe nachvollziehen, hatte dazu aber einfach noch keine Lust. War schon eine herbe Enttäuschung!
Gruß
SLG
Zu Galapagos habe ich vor kurzem einen Thread angestoßen, der sofort kompetente Teilnehmer gefunden hat.
Vielleicht magst Du mal schauen:
Thread: Galapagos NV
AVEO: Das wusste ich nicht, dass das in Russland gelaufen ist. Grundsätzlich sehe ich so etwas auch kritisch. Ich stelle aber fest, dass recht viele Trials einen Schwerpunkt in Osteuropa haben. Über die Gründe kann man spekulieren.
Im Fall AVEO mache ich es mir etwas bequem und verlasse mich auch etwas auf die Expertise von Astellas. Ich gehe fest davon aus, dass die sich die Daten genau angeschaut haben.
PCYC: Ja, die Gewichtung ist sehr hoch und ich würde das keinem zur Nachahmung empfehlen. PCYC hat eigentlich nur den BTK-Inhibitor als echtes Asset und die Marktkapitalisierung ist bei einer Milliarde. Wenn das Projekt schief geht, steht man auf einen Schlag nackt da.
Auf der anderen Seite sind die Daten bis jetzt offensichtlich erstklassig. Das Nebenwirkungsprofil scheint gut auszusehen. Das Marktpotential ist sehr, sehr groß. Der Deal mit J&J ist ganz außergewöhnlich gut. Und - das könnte langfristig eine ganz, ganz fette Sahnehaube werden - der Signalweg könnte auch bei Autoimmunerkrankungen taugen, die im Partnerdeal nicht drin sind.
Kurz und gut: Persönlich habe ich noch nie ein so großes Potential gesehen, wie hier. In der Vergangenheit hat es so was natürlich schon gegeben, aber da war ich noch nicht dabei. Ich denke mir, so oder ähnlich hat es sicher mal bei Celgene angefangen.
Klar, da ist jetzt ne Menge Gier im Spiel. Ich hoffe hier auf so etwas wie den Jackpot. Die Art Größenwahn wird im Leben ja aber meist bestraft.
Ich glaub, dank PCYC werde ich das Beten lernen.
ECYT: Katastrophe. Schwer zu verstehen. Will das alles mal in Ruhe nachvollziehen, hatte dazu aber einfach noch keine Lust. War schon eine herbe Enttäuschung!
Gruß
SLG
Antwort auf Beitrag Nr.: 42.531.616 von SLGramann am 29.12.11 21:22:47bei warren buffet, george soros und co war es der wessentliche startbaustein...einmal jackpot, dann läßt es sich später gut einkaufen ohne große schmerzen... und dann sind die so konservativ auf einmal
im wirklichen leben gibt es eben nur wenige jack potz gewinner.
im poker ist es besonders gefährlich, manchmal!
wünsch dir für 2012 viel glück und den jack pott

und dann sind die so konservativ auf einmal im wirklichen leben gibt es eben nur wenige jack potz gewinner.
im poker ist es besonders gefährlich, manchmal!
wünsch dir für 2012 viel glück und den jack pott


AVEO... direkt am ersten Tag die ersten (aus meiner Sicht) positiven Studienergebnisse! Ob der Markt hier aber nicht noch bessere Ergebnisse erwartet hat? Für ONXX sollte es nicht zu negativ sein, da Nexavar inzwischen Leberkrebs als wichtigste Indikation hat.
AVEO and Astellas Announce Tivozanib Successfully Demonstrated Progression-Free Survival Superiority over Sorafenib in Patients with Advanced Renal Cell Cancer in Phase 3 TIVO-1 Trial
CAMBRIDGE, Mass. & TOKYO--(BUSINESS WIRE)-- AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO - News) and Astellas Pharma Inc. (TSE: 4503) today announced that tivozanib demonstrated superiority over sorafenib in the primary endpoint of progression-free survival (PFS) in TIVO-1, a global, randomized Phase 3 clinical trial evaluating the efficacy and safety of investigational drug tivozanib compared to sorafenib in 517 patients with advanced renal cell carcinoma (RCC). TIVO-1 is the first registration study in first-line RCC that is comparing an investigational agent against an approved VEGF therapy.
All patients in TIVO-1 had clear cell RCC, had undergone a prior nephrectomy, and had not previously been treated with either a VEGF or mTOR therapy. Based on the top-line analysis of events in TIVO-1, determined by a blinded, independent review committee, key top-line findings include:
tivozanib demonstrated a statistically significant improvement in PFS with a median PFS of 11.9 months compared to a median PFS of 9.1 months for sorafenib in the overall study population
tivozanib demonstrated a statistically significant improvement in PFS with a median PFS of 12.7 months compared to a median PFS of 9.1 months for sorafenib in the pre-specified subpopulation of patients who were treatment naïve (no prior systemic anti-cancer therapy); this subpopulation was approximately 70% of the total study population
tivozanib demonstrated a well-tolerated safety profile consistent with the Phase 2 experience; the most commonly reported side effect was hypertension, a well established on-target and manageable effect of VEGFR inhibitors
Based on these data, AVEO and Astellas currently plan to submit for marketing approval of tivozanib in the United States and Europe in 2012, subject to final collection and analyses of all available data from the trial.

mfg ipollit
AVEO and Astellas Announce Tivozanib Successfully Demonstrated Progression-Free Survival Superiority over Sorafenib in Patients with Advanced Renal Cell Cancer in Phase 3 TIVO-1 Trial
CAMBRIDGE, Mass. & TOKYO--(BUSINESS WIRE)-- AVEO Pharmaceuticals, Inc. (NASDAQ: AVEO - News) and Astellas Pharma Inc. (TSE: 4503) today announced that tivozanib demonstrated superiority over sorafenib in the primary endpoint of progression-free survival (PFS) in TIVO-1, a global, randomized Phase 3 clinical trial evaluating the efficacy and safety of investigational drug tivozanib compared to sorafenib in 517 patients with advanced renal cell carcinoma (RCC). TIVO-1 is the first registration study in first-line RCC that is comparing an investigational agent against an approved VEGF therapy.
All patients in TIVO-1 had clear cell RCC, had undergone a prior nephrectomy, and had not previously been treated with either a VEGF or mTOR therapy. Based on the top-line analysis of events in TIVO-1, determined by a blinded, independent review committee, key top-line findings include:
tivozanib demonstrated a statistically significant improvement in PFS with a median PFS of 11.9 months compared to a median PFS of 9.1 months for sorafenib in the overall study population
tivozanib demonstrated a statistically significant improvement in PFS with a median PFS of 12.7 months compared to a median PFS of 9.1 months for sorafenib in the pre-specified subpopulation of patients who were treatment naïve (no prior systemic anti-cancer therapy); this subpopulation was approximately 70% of the total study population
tivozanib demonstrated a well-tolerated safety profile consistent with the Phase 2 experience; the most commonly reported side effect was hypertension, a well established on-target and manageable effect of VEGFR inhibitors
Based on these data, AVEO and Astellas currently plan to submit for marketing approval of tivozanib in the United States and Europe in 2012, subject to final collection and analyses of all available data from the trial.
mfg ipollit
Antwort auf Beitrag Nr.: 42.542.981 von ipollit am 03.01.12 12:45:23Finde die Ergebnisse auch wirklich gut, auch wenn sie nicht ganz an die P II rankommen (was man aber fast erwarten musste).
Die Nexavar-Daten sind aber hier viel besser, als AVEO in früheren Vergleichen kommuniziert hat.
Dass es trotzdem einen statistisch signifikanten Vorsprung für Tivozanib gibt, ist sehr, sehr wichtig.
Tja, hat der Markt mehr erwartet? Eigentlich wäre das frech, denn soweit man das aus der Pressemeldung ersehen kann, ist das strategische Ziel voll erreicht:
Tivozanib ist besser als Nexavar und aller Voraussicht nach auf dem Weg zum Markt. In einem Jahr dürfte AVEO "commercial stage" sein.
Die Nexavar-Daten sind aber hier viel besser, als AVEO in früheren Vergleichen kommuniziert hat.
Dass es trotzdem einen statistisch signifikanten Vorsprung für Tivozanib gibt, ist sehr, sehr wichtig.
Tja, hat der Markt mehr erwartet? Eigentlich wäre das frech, denn soweit man das aus der Pressemeldung ersehen kann, ist das strategische Ziel voll erreicht:
Tivozanib ist besser als Nexavar und aller Voraussicht nach auf dem Weg zum Markt. In einem Jahr dürfte AVEO "commercial stage" sein.
Antwort auf Beitrag Nr.: 42.542.981 von ipollit am 03.01.12 12:45:23Aveo Pharmaceuticals Inc. (AVEO), maker of an experimental medicine for kidney cancer, declined the most in four months after data from a late-stage trial missed investors’ “ultra-high expectations,” an analyst said.
Aveo dropped 6.2 percent to $16.13 at 9:37 a.m. New York time, after earlier sinking to $16.10 for the biggest intraday decline since Sept. 9. The Cambridge, Massachusetts-based company’s shares rose 18 percent in 2011.
Aveo’s tivozanib helped patients with advanced renal cell carcinoma live for 11.9 months without their disease progressing, compared with 9.1 months for those on Nexavar, a medicine sold by Onyx Pharmaceuticals Inc. and Bayer AG (BAYN), Aveo said today in a statement. Investors may have expected progression-free survival of at least 13 months on tivozanib, Jason Kantor, an analyst with RBC Capital Markets in San Francisco, wrote today in a research note.
“The data looks good from both a regulatory and commercial perspective,” Kantor wrote. “However, it may have slightly missed the high efficacy hurdle set by the Street.”
Aveo’s medicine was “well-tolerated,” showing a safety profile similar to earlier results. The 517-patient study was from the third and final phase of trials generally required for regulatory approval. Aveo and partner Astellas Pharma Inc. (4503), of Tokyo, plan to apply for approval in the U.S. and Europe this year, they said in the statement.
------------------
Wenn wir jetzt eine ausgeprägte Kursschwäche bekommen sollten, wäre das eher eine Gelegenheit zum Aufstocken imo.
Aveo dropped 6.2 percent to $16.13 at 9:37 a.m. New York time, after earlier sinking to $16.10 for the biggest intraday decline since Sept. 9. The Cambridge, Massachusetts-based company’s shares rose 18 percent in 2011.
Aveo’s tivozanib helped patients with advanced renal cell carcinoma live for 11.9 months without their disease progressing, compared with 9.1 months for those on Nexavar, a medicine sold by Onyx Pharmaceuticals Inc. and Bayer AG (BAYN), Aveo said today in a statement. Investors may have expected progression-free survival of at least 13 months on tivozanib, Jason Kantor, an analyst with RBC Capital Markets in San Francisco, wrote today in a research note.
“The data looks good from both a regulatory and commercial perspective,” Kantor wrote. “However, it may have slightly missed the high efficacy hurdle set by the Street.”
Aveo’s medicine was “well-tolerated,” showing a safety profile similar to earlier results. The 517-patient study was from the third and final phase of trials generally required for regulatory approval. Aveo and partner Astellas Pharma Inc. (4503), of Tokyo, plan to apply for approval in the U.S. and Europe this year, they said in the statement.
------------------
Wenn wir jetzt eine ausgeprägte Kursschwäche bekommen sollten, wäre das eher eine Gelegenheit zum Aufstocken imo.
Antwort auf Beitrag Nr.: 42.543.957 von SLGramann am 03.01.12 15:50:04dachte bei dem behandlungsansatz liegt der standartbehandlungserfolg bei 9 bis 12 monaten.
das ergebnis ist dann im studienvergleich hier beträchtlich, aber allgemein gesehen me2?
repektable börsenbewertung von 750 mio!
das ergebnis ist dann im studienvergleich hier beträchtlich, aber allgemein gesehen me2?
repektable börsenbewertung von 750 mio!
Antwort auf Beitrag Nr.: 42.543.957 von SLGramann am 03.01.12 15:50:04heute bereits - 15%
akt. bei 14 1/2
das 12 monatstief liegt bei 13 $, das hoch bei 21,5 $!!
ist das ein kaufzeitpunkt?
akt. bei 14 1/2
das 12 monatstief liegt bei 13 $, das hoch bei 21,5 $!!
ist das ein kaufzeitpunkt?
Antwort auf Beitrag Nr.: 42.545.011 von pokemon am 03.01.12 18:29:08Meiner Meinung nach ist AVEO ein Kauf.
Aber man muss Geduld mitbringen und Garantien gibts eh keine.
Hier eine Zusammenfassung der Situation von Minyanville:
In the drug development business, sometimes being good isn’t good enough. Aveo Pharmaceuticals (AVEO) said Tuesday that its experimental kidney cancer treatment worked better than an already approved drug. But some investors had much higher hopes for the study results.
Shares of Aveo fell more than 14% to $14.76 in midday trading Tuesday. The stock has dropped 23% in the past six months.
The company and development partner Astellas Pharma said their drug tivozanib helped patients with advanced kidney cancer in a late-stage study survive almost 12 months on average without the disease progressing. That was better than the 9-month average for patients on Nexavar, an approved drug sold by Onyx Pharmaceuticals (ONXX) and Bayer. Onyx shares fell 3% to $42.67. The stock is up 21% in the past six months.
Some investors were hoping tivozanib would beat Nexavar more handily, says RBC Capital Markets analyst Jason Kantor.
Though tivozanib “may have slightly missed the high efficacy hurdle set by the Street,” Kantor says he still believes the drug has a good shot at being approved and being a commercial success. He recommends buying the stock.
Aveo plans to submit applications this year to sell the drug in the US and Europe. But Nexavar isn’t the only drug tivozanib will have to go up against -- it also will compete with Pfizer’s (PFE) Sutent, which is the best-selling medicine for advanced kidney cancer. (The Pfizer drug had $870 million in sales through the first nine months of last year.)
Earlier company research from Aveo suggested its drug was more effective and caused fewer side effects than Sutent, but Pfizer looms large for the company in another respect. The big pharmaceutical maker recently won backing of a US Food and Drug Administration panel of experts who recommended that Pfizer’s drug Inlyta, or axitinib, be approved for sale.
Pfizer’s drug is being considered for approval as a secondary treatment for kidney cancer but Canaccord Genuity analyst George Farmer says he believes the drug may also be used by doctors as a first-line treatment, creating even more competition.
“We see a challenging marketing battle ahead with axitinib,” Farmer says. He says he expects the Pfizer drug will be approved in the US by a late April deadline set by the FDA.
Still, he recommends buying Aveo’s stock.
The release of information from Aveo Tuesday was limited. Full results will be disclosed at a major cancer conference in June.
Aber man muss Geduld mitbringen und Garantien gibts eh keine.
Hier eine Zusammenfassung der Situation von Minyanville:
In the drug development business, sometimes being good isn’t good enough. Aveo Pharmaceuticals (AVEO) said Tuesday that its experimental kidney cancer treatment worked better than an already approved drug. But some investors had much higher hopes for the study results.
Shares of Aveo fell more than 14% to $14.76 in midday trading Tuesday. The stock has dropped 23% in the past six months.
The company and development partner Astellas Pharma said their drug tivozanib helped patients with advanced kidney cancer in a late-stage study survive almost 12 months on average without the disease progressing. That was better than the 9-month average for patients on Nexavar, an approved drug sold by Onyx Pharmaceuticals (ONXX) and Bayer. Onyx shares fell 3% to $42.67. The stock is up 21% in the past six months.
Some investors were hoping tivozanib would beat Nexavar more handily, says RBC Capital Markets analyst Jason Kantor.
Though tivozanib “may have slightly missed the high efficacy hurdle set by the Street,” Kantor says he still believes the drug has a good shot at being approved and being a commercial success. He recommends buying the stock.
Aveo plans to submit applications this year to sell the drug in the US and Europe. But Nexavar isn’t the only drug tivozanib will have to go up against -- it also will compete with Pfizer’s (PFE) Sutent, which is the best-selling medicine for advanced kidney cancer. (The Pfizer drug had $870 million in sales through the first nine months of last year.)
Earlier company research from Aveo suggested its drug was more effective and caused fewer side effects than Sutent, but Pfizer looms large for the company in another respect. The big pharmaceutical maker recently won backing of a US Food and Drug Administration panel of experts who recommended that Pfizer’s drug Inlyta, or axitinib, be approved for sale.
Pfizer’s drug is being considered for approval as a secondary treatment for kidney cancer but Canaccord Genuity analyst George Farmer says he believes the drug may also be used by doctors as a first-line treatment, creating even more competition.
“We see a challenging marketing battle ahead with axitinib,” Farmer says. He says he expects the Pfizer drug will be approved in the US by a late April deadline set by the FDA.
Still, he recommends buying Aveo’s stock.
The release of information from Aveo Tuesday was limited. Full results will be disclosed at a major cancer conference in June.
danke für den update.
Antwort auf Beitrag Nr.: 42.545.844 von SLGramann am 03.01.12 20:28:19denke die zeiten haben sich geändert. was früher mal ausreichend gewesen wäre wird heute abgestraft!! sehr schade!!
die neue preispolitik der gesetzgeber verlangen mehr für die preishoheit bei neuzulassungen!
ich kaufe mir doch kein aveo. viel glück beim weiteren investment
fokussiere mich lieber auf produkte mit wirklich neuartigen angehensweisen!
die neue preispolitik der gesetzgeber verlangen mehr für die preishoheit bei neuzulassungen!
ich kaufe mir doch kein aveo. viel glück beim weiteren investment
fokussiere mich lieber auf produkte mit wirklich neuartigen angehensweisen!
was meinst du zu dndn, beeindruckende kurse diese woche!
Ohad Hammer mit einer ziemlich kritischen Sicht auf die AVEO-Daten:
AVEO - More open questions than answers
Earlier this week, AVEO (AVEO) announced topline results from a phase III trial for tivozanib in renal cancer. Although technically the trial met its primary endpoint and could probably lead to approval, results were far from satisfactory.
AVEO intends to position tivozanib as the standard of care treatment for 1st line renal cancer, replacing Pfizer’s (PFE) Sutent. As both drugs hit common targets, AVEO hoped that tivozanib’s selectivity profile would enable it to be superior in terms of side effects and at least comparable to Sutent in terms of efficacy. The underlying issue with AVEO’s clinical strategy was the idea of making tivozanib Sutent’s successor without comparing it directly with the Pfizer drug.
Sutent, which generated $1.06B in sales last year, is the most commonly used first line drug for renal cancer. It was approved based on impressive improvement in progression-free survival (PFS) from 5 months in the control arm to 11 months in the Sutent arm. Although Sutent is considered safer than chemotherapy agents, it is still associated with a great degree of side effects. These include liver toxicity, fatigue and diarrhea.
Shooting for indirect superiority
AVEO started with a large single-arm phase II in 1st line patients. The 272-patient study showed a PFS of 11.8 months in the general population. The activity in the patient population that was studied in Sutent’s phase III trial (clear-cell histology and prior nephrectomy) was even more impressive - 14.8 months. (The Sutent study recruited only clear cell patients, 90% of them had had prior nephrectomy).
The safety profile for tivozanib looked substantially better than that of Sutent, especially side effects that are not related to the drug’s primary mechanism of action.
Based on the results, AVEO launched a phase III trial (TIVO-1) that evaluates tivozanib in 1st line renal cancer. Patients were allowed to have a history of prior systemic therapy but prior Sutent or Nexavar was excluded. Instead of using Sutent, Onyx’s (ONXX) Nexavar was chosen as a comparator. Theoretically, Nexavar is approved for 1st line RCC but it is rarely used in that setting due to its limited efficacy (estimated PFS of just under 6 months).
Having such a weak opponent substantially increased AVEO’s chances of showing superiority over the control arm, which the company believed was enough to gain FDA approval. The company hoped that tivozanib’s results would be strong enough in order to persuade physicians to use the drug instead of Sutent based on high PFS and better tolerability.
Inconclusive data
The initial results published earlier this week showed tivozanib led to a superior PFS compared with Nexavar (11.9 vs. 9.1 months). In treatment-naive patients, who did not receive Sutent or cytokines (not commonly used today), tivozanib’s PFS was 12.7 months vs. 9.1 months for Nexavar.
Technically, the trial was successful but the data are far from satisfactory and cast a real shadow on tivozanib’s prospects in 1st line renal cancer. Numerically, a PFS of 12.7 months is at the lower end of expectations and looks slightly better than Sutent’s 11 months. In fact, Sutent’s 11 months were achieved in a patient population that included 9% patients with no prior nephrectomy. It is plausible that Sutent’s PFS only in patients with a history of prior nephrectomy was slightly higher than 11 months, making the alleged difference smaller.
Even more worrying is the better than expected PFS Nexavar achieved. This implies the patient population in AVEO’s study had a relatively good prognosis compared to other phase III trials. Sutent is considered much more effective than Nexavar in 1st line patients although the two drugs have never been compared directly. Therefore, not only is it hard for AVEO to claim superiority over Sutent, tivozanib might end up being less effective than Sutent. On a more positive note, AVEO reported that tivozanib’s safety profile was consistent with the phase II data, which could be an important differentiator going forward.
New competition on the horizon
Once approved, AVEO’s tivozanib might face competition from additional kinase inhibitors. These include Pfizer’s Inlyta and GSK’s (GSK) Votrient, both of which are expected to have phase III data in 1st line patients in the coming months. Pfizer is evaluating Inlyta against Nexavar for obvious reasons whereas GSK is the only one testing its drug head to head vs. Sutent.
Analysis of phase III results for Inlyta in 2nd line patients, which also employed Nexavar as a control, is another source of concern as to tivozanib’s future. Inlyta had a remarkable effect in a subset of patients who had not been treated with Sutent but with cytokines as 1st line treatment (12.1 vs. 6.5 months). In this case, Nexavar performed as expected, similarly to its original phase III trial. Tivozanib achieved a milder effect in this subset of patients in its recent phase III trial (~11.5 vs. 9.1 months). Normalizing these numbers results in a 86% improvement for Inlyta over Nexavar compared to a 26% improvement for tivozanib over Nexavar in a similar patient population. The fact that the Nexavar arm in AVEO’s study did better than that Nexavar arms in the Inlyta or original Nexavar phase III trials implies that AVEO used patients with better prognosis although no data has been actually published.

Votrient, which is already approved for 1st line renal cancer, is currently being evaluated vs. Sutent in a very large phase III study (927 patients). In a previous phase III trial vs. placebo, Votrient led to a PFS of 11.1 months in 1st line patients, which is almost identical to the 11 months Sutent achieved. Numerically, AVEO’s tivozanib generated superior results (12.7 months) but again, it is still unclear whether AVEO’s trial included a more favorable mix of patients.
Summary
AVEO chose a less effective drug as a comparator in order to increase chances of regulatory approval, however, the somewhat disappointing results cast a shadow on the drug’s value. Based on available data, tivozanib appears to have an excellent safety profile and a certain degree of efficacy but it is hard to claim the drug is comparable, let alone superior, to Sutent.
The drug’s commercial success will now depend on how physicians interpret the PFS data, the weight of side effects in their decision and results for Inlyta and Votrient later this year. So far AVEO’s results are not the landslide the market was expecting.
Tivozanib’s potential spans beyond renal cancer, but the next catalyst is more than a year away. On a more positive note, AVEO has several other clinical programs as well as an impressive discovery engine, both could generate value during 2012. With a market cap of over $600M and 50% of tivozanib marketing rights, AVEO seems fully priced.
Portfolio updates
We are selling our position in Endocyte (ECYT) following the problematic updated results from the phase II trial. On top of the bad overall survival data, which casts a shadow on the entire study, the likelihood of filing in the EU is now approaching zero and the Doxil shortage might lengthen and complicate the ongoing phase III trial.
We are initiating a position in Gilead (GILD).
http://www.hammerstockblog.com/aveo-more-open-questions-than…
---------------
DNDN: Sorry, keine Meinung.
AVEO - More open questions than answers
Earlier this week, AVEO (AVEO) announced topline results from a phase III trial for tivozanib in renal cancer. Although technically the trial met its primary endpoint and could probably lead to approval, results were far from satisfactory.
AVEO intends to position tivozanib as the standard of care treatment for 1st line renal cancer, replacing Pfizer’s (PFE) Sutent. As both drugs hit common targets, AVEO hoped that tivozanib’s selectivity profile would enable it to be superior in terms of side effects and at least comparable to Sutent in terms of efficacy. The underlying issue with AVEO’s clinical strategy was the idea of making tivozanib Sutent’s successor without comparing it directly with the Pfizer drug.
Sutent, which generated $1.06B in sales last year, is the most commonly used first line drug for renal cancer. It was approved based on impressive improvement in progression-free survival (PFS) from 5 months in the control arm to 11 months in the Sutent arm. Although Sutent is considered safer than chemotherapy agents, it is still associated with a great degree of side effects. These include liver toxicity, fatigue and diarrhea.
Shooting for indirect superiority
AVEO started with a large single-arm phase II in 1st line patients. The 272-patient study showed a PFS of 11.8 months in the general population. The activity in the patient population that was studied in Sutent’s phase III trial (clear-cell histology and prior nephrectomy) was even more impressive - 14.8 months. (The Sutent study recruited only clear cell patients, 90% of them had had prior nephrectomy).
The safety profile for tivozanib looked substantially better than that of Sutent, especially side effects that are not related to the drug’s primary mechanism of action.
Based on the results, AVEO launched a phase III trial (TIVO-1) that evaluates tivozanib in 1st line renal cancer. Patients were allowed to have a history of prior systemic therapy but prior Sutent or Nexavar was excluded. Instead of using Sutent, Onyx’s (ONXX) Nexavar was chosen as a comparator. Theoretically, Nexavar is approved for 1st line RCC but it is rarely used in that setting due to its limited efficacy (estimated PFS of just under 6 months).
Having such a weak opponent substantially increased AVEO’s chances of showing superiority over the control arm, which the company believed was enough to gain FDA approval. The company hoped that tivozanib’s results would be strong enough in order to persuade physicians to use the drug instead of Sutent based on high PFS and better tolerability.
Inconclusive data
The initial results published earlier this week showed tivozanib led to a superior PFS compared with Nexavar (11.9 vs. 9.1 months). In treatment-naive patients, who did not receive Sutent or cytokines (not commonly used today), tivozanib’s PFS was 12.7 months vs. 9.1 months for Nexavar.
Technically, the trial was successful but the data are far from satisfactory and cast a real shadow on tivozanib’s prospects in 1st line renal cancer. Numerically, a PFS of 12.7 months is at the lower end of expectations and looks slightly better than Sutent’s 11 months. In fact, Sutent’s 11 months were achieved in a patient population that included 9% patients with no prior nephrectomy. It is plausible that Sutent’s PFS only in patients with a history of prior nephrectomy was slightly higher than 11 months, making the alleged difference smaller.
Even more worrying is the better than expected PFS Nexavar achieved. This implies the patient population in AVEO’s study had a relatively good prognosis compared to other phase III trials. Sutent is considered much more effective than Nexavar in 1st line patients although the two drugs have never been compared directly. Therefore, not only is it hard for AVEO to claim superiority over Sutent, tivozanib might end up being less effective than Sutent. On a more positive note, AVEO reported that tivozanib’s safety profile was consistent with the phase II data, which could be an important differentiator going forward.
New competition on the horizon
Once approved, AVEO’s tivozanib might face competition from additional kinase inhibitors. These include Pfizer’s Inlyta and GSK’s (GSK) Votrient, both of which are expected to have phase III data in 1st line patients in the coming months. Pfizer is evaluating Inlyta against Nexavar for obvious reasons whereas GSK is the only one testing its drug head to head vs. Sutent.
Analysis of phase III results for Inlyta in 2nd line patients, which also employed Nexavar as a control, is another source of concern as to tivozanib’s future. Inlyta had a remarkable effect in a subset of patients who had not been treated with Sutent but with cytokines as 1st line treatment (12.1 vs. 6.5 months). In this case, Nexavar performed as expected, similarly to its original phase III trial. Tivozanib achieved a milder effect in this subset of patients in its recent phase III trial (~11.5 vs. 9.1 months). Normalizing these numbers results in a 86% improvement for Inlyta over Nexavar compared to a 26% improvement for tivozanib over Nexavar in a similar patient population. The fact that the Nexavar arm in AVEO’s study did better than that Nexavar arms in the Inlyta or original Nexavar phase III trials implies that AVEO used patients with better prognosis although no data has been actually published.
Votrient, which is already approved for 1st line renal cancer, is currently being evaluated vs. Sutent in a very large phase III study (927 patients). In a previous phase III trial vs. placebo, Votrient led to a PFS of 11.1 months in 1st line patients, which is almost identical to the 11 months Sutent achieved. Numerically, AVEO’s tivozanib generated superior results (12.7 months) but again, it is still unclear whether AVEO’s trial included a more favorable mix of patients.
Summary
AVEO chose a less effective drug as a comparator in order to increase chances of regulatory approval, however, the somewhat disappointing results cast a shadow on the drug’s value. Based on available data, tivozanib appears to have an excellent safety profile and a certain degree of efficacy but it is hard to claim the drug is comparable, let alone superior, to Sutent.
The drug’s commercial success will now depend on how physicians interpret the PFS data, the weight of side effects in their decision and results for Inlyta and Votrient later this year. So far AVEO’s results are not the landslide the market was expecting.
Tivozanib’s potential spans beyond renal cancer, but the next catalyst is more than a year away. On a more positive note, AVEO has several other clinical programs as well as an impressive discovery engine, both could generate value during 2012. With a market cap of over $600M and 50% of tivozanib marketing rights, AVEO seems fully priced.
Portfolio updates
We are selling our position in Endocyte (ECYT) following the problematic updated results from the phase II trial. On top of the bad overall survival data, which casts a shadow on the entire study, the likelihood of filing in the EU is now approaching zero and the Doxil shortage might lengthen and complicate the ongoing phase III trial.
We are initiating a position in Gilead (GILD).
http://www.hammerstockblog.com/aveo-more-open-questions-than…
---------------
DNDN: Sorry, keine Meinung.
Antwort auf Beitrag Nr.: 42.563.297 von SLGramann am 07.01.12 12:22:19kommentar von OH überrascht mich nicht!
dndn hatte ich mir nach dem einbruch gekauft und momentumweise zugekauft!
denke mologen wird davon profitieren, besonders nach den anstehenden klinischen daten zu MGN1601!
dndn hatte ich mir nach dem einbruch gekauft und momentumweise zugekauft!
denke mologen wird davon profitieren, besonders nach den anstehenden klinischen daten zu MGN1601!
Beitrag zu dieser Diskussion schreiben
Zu dieser Diskussion können keine Beiträge mehr verfasst werden, da der letzte Beitrag vor mehr als zwei Jahren verfasst wurde und die Diskussion daraufhin archiviert wurde.
Bitte wenden Sie sich an feedback@wallstreet-online.de und erfragen Sie die Reaktivierung der Diskussion oder starten Sie eine neue Diskussion.
Meistdiskutiert
| Wertpapier | Beiträge | |
|---|---|---|
| 202 | ||
| 127 | ||
| 81 | ||
| 68 | ||
| 53 | ||
| 40 | ||
| 37 | ||
| 35 | ||
| 35 | ||
| 33 |
| Wertpapier | Beiträge | |
|---|---|---|
| 30 | ||
| 29 | ||
| 28 | ||
| 27 | ||
| 26 | ||
| 26 | ||
| 25 | ||
| 23 | ||
| 20 | ||
| 17 |



















