



Morphosys – fachliche Überlegungen zur Entwicklungspipeline - 500 Beiträge pro Seite (Seite 2)
eröffnet am 19.07.13 21:27:33 von
neuester Beitrag 06.05.20 22:40:51 von
neuester Beitrag 06.05.20 22:40:51 von
Beiträge: 772
ID: 1.183.993
ID: 1.183.993
Aufrufe heute: 1
Gesamt: 149.380
Gesamt: 149.380
Aktive User: 0
ISIN: DE0006632003 · WKN: 663200 · Symbol: MOR
67,68
EUR
0,00 %
0,00 EUR
Letzter Kurs 08:44:15 Lang & Schwarz
Neuigkeiten
23.04.24 · wallstreetONLINE Redaktion |
| Morphosys Aktien jetzt im kostenlosen Demokonto handeln!Anzeige |
08:00 Uhr · wO Chartvergleich |
23.04.24 · wO Newsflash |
 Gewinn, Umsatz über Erwartung: Novartis hebt Prognosen an: Starke Medikamentenverkäufe treiben Wachstum
Gewinn, Umsatz über Erwartung: Novartis hebt Prognosen an: Starke Medikamentenverkäufe treiben WachstumWerte aus der Branche Biotechnologie
| Wertpapier | Kurs | Perf. % |
|---|---|---|
| 53,50 | +98,15 | |
| 1,3500 | +29,82 | |
| 4,5700 | +24,18 | |
| 1,8300 | +23,65 | |
| 0,7158 | +22,48 |
| Wertpapier | Kurs | Perf. % |
|---|---|---|
| 5,4000 | -15,23 | |
| 0,7008 | -15,56 | |
| 0,7603 | -15,56 | |
| 2,8600 | -19,21 | |
| 3,1800 | -54,24 |
Antwort auf Beitrag Nr.: 52.564.379 von Ville7 am 08.06.16 01:33:35Ergänzend zu meinem Vergleich zwischen Dara und MOR202 Monotherapie: dieser hängt auch noch an anderer Stelle: bei Dara waren die 8mg und die 16mg Gruppen gleichverteilt. Bei MOR202 sind 25% der Patienten in der 8mg Gruppe und 75% der Patienten in der 16mg Gruppe. Auch von dieser Seite aus ist ein Vergleich höchst unsauber, da er potentiell der MOR202 Studie durch höhere durchschnittliche Dosierung einen Vorteil gewährt.
Antwort auf Beitrag Nr.: 52.564.379 von Ville7 am 08.06.16 01:33:35
Nein, das geht natürlich nicht. Ich konnte noch nie verstehen, warum man die MOR202+DEX-Kombi als Monotherapie bezeichnet hat. Aus meiner Sicht grenzwertig und einer Irreführung nahe.
Da die Antikörper aber allesamt in Kombination mit LEN oder POM (+ jeweils DEX) eingesetzt, messe ich den Mono-Daten bzw. Mono+DEX-Daten keine größere Bedeutung bei. Ich schau mir gleich die klinisch relevanten Kombi-Daten an.
Zur mDoR gibt es sowohl für MOR202 als auch für Biotest´s BT-062 leider noch keine validen Daten. Man kann nur, wie auch Du es gemacht hast, auf Basis von Kongresspräsentationen selber Grobabschätzungen durchführen.
Zitat von Ville7: Joschka, re MOR202:
Fragen: Ist es valide, wenn Morphosys seinen MOR202+Dex Trial zu Daratumumab ohne Dex vergleicht? Kann man beides als Monotherapie zählen, so wie Morphosys das macht? Erhöht Dex die Wirksamkeit des anti-CD38 Antikörpers nicht?
Nein, das geht natürlich nicht. Ich konnte noch nie verstehen, warum man die MOR202+DEX-Kombi als Monotherapie bezeichnet hat. Aus meiner Sicht grenzwertig und einer Irreführung nahe.
Da die Antikörper aber allesamt in Kombination mit LEN oder POM (+ jeweils DEX) eingesetzt, messe ich den Mono-Daten bzw. Mono+DEX-Daten keine größere Bedeutung bei. Ich schau mir gleich die klinisch relevanten Kombi-Daten an.
Zur mDoR gibt es sowohl für MOR202 als auch für Biotest´s BT-062 leider noch keine validen Daten. Man kann nur, wie auch Du es gemacht hast, auf Basis von Kongresspräsentationen selber Grobabschätzungen durchführen.
Antwort auf Beitrag Nr.: 52.564.388 von Ville7 am 08.06.16 01:50:14
In der gegebenen Dosis eindeutig JA!
Im Übrigen ist die Bezeichnung als low dose irreführend (ich glaube, in der letzten Meldung oder im Poster war davon die Rede), in der "Mono"studie verwendet Morphosys die übliche DEX-Kombidosierung.
Aber, wie gesagt, die Mono-Daten sind eigentlich nicht von Belang. Es geht um die Wirkung der klinisch relevanten Kombi-Verabreichung ... um positive wie negative Synergieeffekte.
Vor allem sind weitere Studiendaten erforderlich, die bisherigen Kollektive sind viel zu klein.
Zitat von Ville7: Dexamethasone
...
Wirksamkeiterhöhend? Ja oder Nein?
In der gegebenen Dosis eindeutig JA!
Im Übrigen ist die Bezeichnung als low dose irreführend (ich glaube, in der letzten Meldung oder im Poster war davon die Rede), in der "Mono"studie verwendet Morphosys die übliche DEX-Kombidosierung.
Aber, wie gesagt, die Mono-Daten sind eigentlich nicht von Belang. Es geht um die Wirkung der klinisch relevanten Kombi-Verabreichung ... um positive wie negative Synergieeffekte.
Vor allem sind weitere Studiendaten erforderlich, die bisherigen Kollektive sind viel zu klein.
@Ville
Ich habe noch schnell die große Unternehmenspräsentation zur ASCO-Konferenz durchgesehen. Dort ist alles richtig dargestellt.
Ich habe noch schnell die große Unternehmenspräsentation zur ASCO-Konferenz durchgesehen. Dort ist alles richtig dargestellt.
Ville und Joschka,
Sehr schöne Diskussion. Danke, dass ihr Euch hier die Zeit nehmt und die Dinge kritisch hinterfragt. Für mich als absoluten Laien ist das eine große Hilfe, mir ein Bild zu machen.
Sehr schöne Diskussion. Danke, dass ihr Euch hier die Zeit nehmt und die Dinge kritisch hinterfragt. Für mich als absoluten Laien ist das eine große Hilfe, mir ein Bild zu machen.
Trading Spotlight
Antwort auf Beitrag Nr.: 52.564.421 von Joschka Schröder am 08.06.16 03:52:27
Hi Joschka,
auch in dieser Präsentation finde ich die Überschrift "MOR202 alone and in combination with pomalidomide or lenalidomide in relapsed or refractory multiple myeloma" irreführend. Die jeweilige Kombi mit DEX wird unterschlagen.
Zudem verstehe ich nicht, wieso auf Folie 20
"Assess the safety profile and establish the maximum tolerated dose (MTD) and/or recommended
dose of MOR202 in patients with R-R MM:
* As
monotherapy
* In combination with
Dex
* In combination with pomalidomide (POM)/
Dex
* In combination with lenalidomide (LEN)/
Dex"
Hat Morphosys auch eine Gruppe MOR202 ohne DEX gehabt? Ich finde dazu in den folgenden Folien keine Daten. Folie 21 gibt ein 3-3 Design aus. Also keine Monotherapie getestet.
https://www.morphosys.de/sites/default/files/phone-conferenc…
Die Kommunikation scheint teilweise recht schlampig zu sein.
Bei den mündlich gesprochenen Sätzen, die ich dazu ab und an in den IR Präsentationen gehört habe, sogar grenzwertig irreführend. Könnte mir vorstellen, dass eine derartige Irreführung in USA sogar Grundlage für einen Class-Action lawsuit sein könnte.
Zitat von Joschka Schröder: @Ville
Ich habe noch schnell die große Unternehmenspräsentation zur ASCO-Konferenz durchgesehen. Dort ist alles richtig dargestellt.
Hi Joschka,
auch in dieser Präsentation finde ich die Überschrift "MOR202 alone and in combination with pomalidomide or lenalidomide in relapsed or refractory multiple myeloma" irreführend. Die jeweilige Kombi mit DEX wird unterschlagen.
Zudem verstehe ich nicht, wieso auf Folie 20
"Assess the safety profile and establish the maximum tolerated dose (MTD) and/or recommended
dose of MOR202 in patients with R-R MM:
* As
monotherapy
* In combination with
Dex
* In combination with pomalidomide (POM)/
Dex
* In combination with lenalidomide (LEN)/
Dex"
Hat Morphosys auch eine Gruppe MOR202 ohne DEX gehabt? Ich finde dazu in den folgenden Folien keine Daten. Folie 21 gibt ein 3-3 Design aus. Also keine Monotherapie getestet.
https://www.morphosys.de/sites/default/files/phone-conferenc…
Die Kommunikation scheint teilweise recht schlampig zu sein.
Bei den mündlich gesprochenen Sätzen, die ich dazu ab und an in den IR Präsentationen gehört habe, sogar grenzwertig irreführend. Könnte mir vorstellen, dass eine derartige Irreführung in USA sogar Grundlage für einen Class-Action lawsuit sein könnte.
Antwort auf Beitrag Nr.: 52.564.553 von Ville7 am 08.06.16 07:08:59Korrektur: "3+3 design" hat nichts mit der Anzahl getesteter Arme zu tun.

Joschka, ich glaube ich habe des Rätsels Lösung gefunden. Und Morphosys' Aussagen sind wohl doch nicht (so) irreführend.
Dara hat in seinen "Monotherapie"-Studien auch mit DEX gearbeitet. Zumindest bei der ersten Studie im 3+3 Design.
https://clinicaltrials.gov/ct2/show/NCT00574288?term=daratum…
"Prophylactic steroids were administered to reduce the incidence of IREs (up to a maximum dose equivalent of 27 mg of dexamethasone per week)"
(Morphosys arbeitet in der POM oder LEN Kombi mit 40mg bei Patienten <75 Jahre und 20mg bei >75 Jahre)
Und es gab laut Clinicaltrials auch einen Part A (0,1mg/kg) und Part B (4mg/kg) von MOR202 mit reiner Monotherapie ohne DEX. Einmal alle zwei Wochen (Part A) verabreicht und einmal wöchentlich (Part B). Die Daten dazu wurden entweder nur nie veröffentlicht oder werden nicht mehr genannt. Erst ab Part C beginnen die veröffentlichten Daten.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4480519/
In der größeren Phase II Studie (SIRIUS) hat man bei Genmab dann nicht mehr mit DEX gearbeitet, sondern mit Methylprednisolone, zudem mit Diphenhydramine einem Antihistaminika und Acetaminophen gegen Schmerzen und Fieber. Methylprednisolone (ich nenne es im weiteren PRED) scheint von der Wirkung her vergleichbar mit DEX und auch aktiv in MM zu sein wie ich heute Nacht in dem Link gepostet hatte. Die durchschnittliche Dosis von Methylprednisolone im SIRIUS trial finde ich bisher nirgend genannt. Und auch nicht, ob DEX oder PRED mehr cytotoxisch sind.
Man verwendet den Zusatz DEX oder PRED wohl nicht um eine Wirkung auf den Tumor zu bekommen, sondern um die Infusionsreaktionen zu managen /zu unterdrücken. Und wie wir wissen hat Dara damit ein viel größeres Problem als MOR202.
Somit scheinen die "Monotherapie"-Ergebnisse doch halbwegs vergleichbar zu sein und MOR202 schneidet diesbezüglich bisher nicht schlechter ab.
Dara hat in seinen "Monotherapie"-Studien auch mit DEX gearbeitet. Zumindest bei der ersten Studie im 3+3 Design.
https://clinicaltrials.gov/ct2/show/NCT00574288?term=daratum…
"Prophylactic steroids were administered to reduce the incidence of IREs (up to a maximum dose equivalent of 27 mg of dexamethasone per week)"
(Morphosys arbeitet in der POM oder LEN Kombi mit 40mg bei Patienten <75 Jahre und 20mg bei >75 Jahre)
Und es gab laut Clinicaltrials auch einen Part A (0,1mg/kg) und Part B (4mg/kg) von MOR202 mit reiner Monotherapie ohne DEX. Einmal alle zwei Wochen (Part A) verabreicht und einmal wöchentlich (Part B). Die Daten dazu wurden entweder nur nie veröffentlicht oder werden nicht mehr genannt. Erst ab Part C beginnen die veröffentlichten Daten.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4480519/
In der größeren Phase II Studie (SIRIUS) hat man bei Genmab dann nicht mehr mit DEX gearbeitet, sondern mit Methylprednisolone, zudem mit Diphenhydramine einem Antihistaminika und Acetaminophen gegen Schmerzen und Fieber. Methylprednisolone (ich nenne es im weiteren PRED) scheint von der Wirkung her vergleichbar mit DEX und auch aktiv in MM zu sein wie ich heute Nacht in dem Link gepostet hatte. Die durchschnittliche Dosis von Methylprednisolone im SIRIUS trial finde ich bisher nirgend genannt. Und auch nicht, ob DEX oder PRED mehr cytotoxisch sind.
Man verwendet den Zusatz DEX oder PRED wohl nicht um eine Wirkung auf den Tumor zu bekommen, sondern um die Infusionsreaktionen zu managen /zu unterdrücken. Und wie wir wissen hat Dara damit ein viel größeres Problem als MOR202.
Somit scheinen die "Monotherapie"-Ergebnisse doch halbwegs vergleichbar zu sein und MOR202 schneidet diesbezüglich bisher nicht schlechter ab.
Hier noch der Link zur MOR202 Studie:
https://clinicaltrials.gov/ct2/show/NCT01421186?term=MOR202&…
https://clinicaltrials.gov/ct2/show/NCT01421186?term=MOR202&…
MOR202 hat zwei shots on goal gegenüber Dara:
1. gleiche oder nur schwach geringere Wirksamkeit und gleiche Sicherheit + geringere Infusionsreaktionen, dadurch schnellere Verabreichung -> Arzt wird ganz klar MOR202 bevorzugen.
2. gleiche oder nur schwach geringere Wirksamkeit bzgl. Tumorshrinkage, aber höhere mDoR -> möglicherweise wäre MOR202 dann in den bedeuterenden measures PFS und / oder OS gegenüber Dara überlegen
Einstellen ist gar kein Thema mehr. Aber ohne Partner sollte man m.E. in keine P3 gehen.
1. gleiche oder nur schwach geringere Wirksamkeit und gleiche Sicherheit + geringere Infusionsreaktionen, dadurch schnellere Verabreichung -> Arzt wird ganz klar MOR202 bevorzugen.
2. gleiche oder nur schwach geringere Wirksamkeit bzgl. Tumorshrinkage, aber höhere mDoR -> möglicherweise wäre MOR202 dann in den bedeuterenden measures PFS und / oder OS gegenüber Dara überlegen
Einstellen ist gar kein Thema mehr. Aber ohne Partner sollte man m.E. in keine P3 gehen.
Antwort auf Beitrag Nr.: 52.564.409 von Joschka Schröder am 08.06.16 03:14:29
Ich bin sehr gespannt, irgendwann die kompletten Kombidaten mit den Dara Daten vergleichen zu können. Schaut bisher ja auch nicht sooo schlecht aus. Außerdem gibt es lt. Firma einen klaren präklinischen rationale für eine Synergie mit IMiDs.
Ich switche daher meine Einstellung zu MOR202 von negativ auf neutral/leicht positiv.
Zitat von Joschka Schröder: Aber, wie gesagt, die Mono-Daten sind eigentlich nicht von Belang. Es geht um die Wirkung der klinisch relevanten Kombi-Verabreichung ... um positive wie negative Synergieeffekte.
Vor allem sind weitere Studiendaten erforderlich, die bisherigen Kollektive sind viel zu klein.
Ich bin sehr gespannt, irgendwann die kompletten Kombidaten mit den Dara Daten vergleichen zu können. Schaut bisher ja auch nicht sooo schlecht aus. Außerdem gibt es lt. Firma einen klaren präklinischen rationale für eine Synergie mit IMiDs.
Ich switche daher meine Einstellung zu MOR202 von negativ auf neutral/leicht positiv.
Antwort auf Beitrag Nr.: 52.555.256 von Joschka Schröder am 06.06.16 22:38:31
re MOR208: Ich habe nicht den geringsten Zweifel, dass die MOR208 Kombi in DLBCL die Kombi mit Ritiximab höchstsignifikant schlägt - und zwar in allen angegebenen primären und sekundären Endpunkten.
https://clinicaltrials.gov/ct2/show/NCT02763319?term=MOR208&…
Ich habe nur keinen Überblick - auch angesichts der Konkurrenzsituation von so vielen Agents - was dann für ein mögliches Marktpotential daraus für MOR208 resultieren könnte.
Zitat von Joschka Schröder: ...
MOR208
Hauptindikation ist ja die DLBCL. Hier waren die Ansprechraten des Gesamtkollektivs (inkl. der nicht evaluierbaren Patienten) ja bereits aus dem Abstract bekannt. Sie entsprechen in etwa den Ergebnissen, die mit Konkurrenzpräparaten wie Adcetris (anti-CD30), IMGN529 (anti-CD37) und SAR3419 (anti-CD19) erzielt werden können. Ein Highlight ist aber die Dauer der klinischen Antwort (DoR): 20 Monate sind absolut top. Im Abstract waren es noch 13,7 Monate und beim letzten ASCO-Meeting 7,7 Monate. Zum Vergleich: DoR Adcetris = 1,6 Monate!
Erfreulich auch die 40 %-Quote bzgl. des progressionsfreien Überlebens von 1 Jahr. Auch hier als Anhaltspunkt: Das mittlere PFS in der Adcetris-Studie betrug zuletzt 1,4 Monate im Gesamtkollektiv und 4 Monate bei den CD30-positiven Patienten.
...
re MOR208: Ich habe nicht den geringsten Zweifel, dass die MOR208 Kombi in DLBCL die Kombi mit Ritiximab höchstsignifikant schlägt - und zwar in allen angegebenen primären und sekundären Endpunkten.
https://clinicaltrials.gov/ct2/show/NCT02763319?term=MOR208&…
Ich habe nur keinen Überblick - auch angesichts der Konkurrenzsituation von so vielen Agents - was dann für ein mögliches Marktpotential daraus für MOR208 resultieren könnte.
Joschka, noch eine Frage zu MOR208.
Kannst du dir erklären, wieso Morphosys 20Monate mDoR angibt? Ich sehe auf dem Schaubild nur ca. 14 Monate, wenn ich den Median bei DLBCL nehme (rote Balken) - siehe lila umkreist:
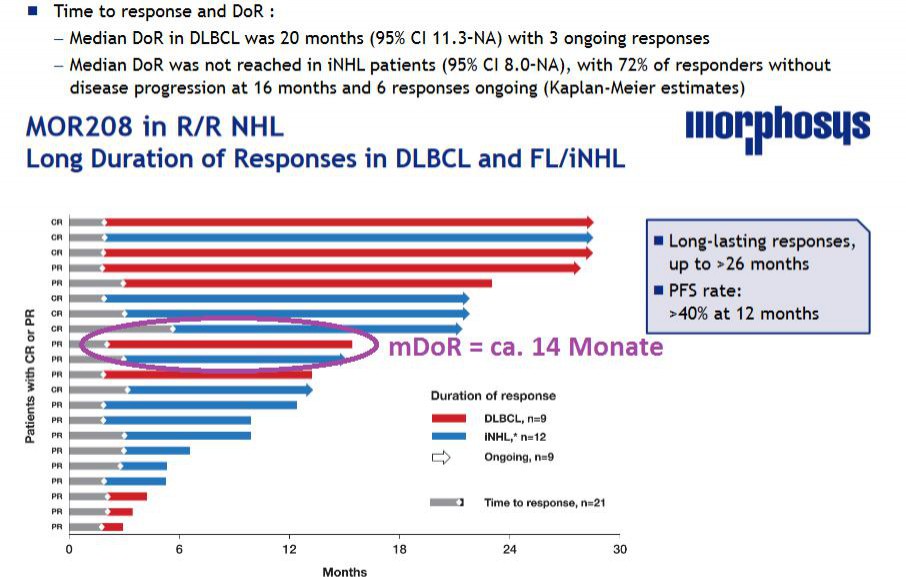
Kannst du dir erklären, wieso Morphosys 20Monate mDoR angibt? Ich sehe auf dem Schaubild nur ca. 14 Monate, wenn ich den Median bei DLBCL nehme (rote Balken) - siehe lila umkreist:

Antwort auf Beitrag Nr.: 52.565.363 von Ville7 am 08.06.16 09:03:57Ville, Du bist ja schlimmer als ich 
zu Deinem Beitrag 513:
Des Rätsels Lösung könnte in dem zusätzlichen Patienten liegen, der in der Graphik nicht aufgeführt ist (s. Fußnote der Abb. 4 im MOR202-Poster). Wenn es sich um einen DLBCL-Patienten handelt, verschiebt sich die mDoR in der Abb. entsprechend.
Ist aber ein guter Hinweis, da müßte man konkret noch einmal bei Morphosys nachfragen, ob es sich tatsächlich so verhält.
zu Deinem Beitrag 508:
Dexamethason ist deutlich potenter als Methlyprednisolon. Ich müßte noch einmal in der Äquivalenztabelle von Kaiser nachsehen, aber 4 mg Methylprednisolon sollten in etwa so potent sein wie 0,75 mg Dexamethason. Dies müssen wir bei einem Vergleich der Dara- und MOR202-Studien berücksichtigen. Ebenso natürlich die Zusammenstellung des Patientenkollektivs (Anzahl der Vorbehandlungen etc.) Hast Du einen Link zu den Kortikosteroiddaten der Siriusstudie? Ansonsten müßte ich selbst in den entsprechenden Datenbanken suchen.

zu Deinem Beitrag 513:
Des Rätsels Lösung könnte in dem zusätzlichen Patienten liegen, der in der Graphik nicht aufgeführt ist (s. Fußnote der Abb. 4 im MOR202-Poster). Wenn es sich um einen DLBCL-Patienten handelt, verschiebt sich die mDoR in der Abb. entsprechend.
Ist aber ein guter Hinweis, da müßte man konkret noch einmal bei Morphosys nachfragen, ob es sich tatsächlich so verhält.
zu Deinem Beitrag 508:
Dexamethason ist deutlich potenter als Methlyprednisolon. Ich müßte noch einmal in der Äquivalenztabelle von Kaiser nachsehen, aber 4 mg Methylprednisolon sollten in etwa so potent sein wie 0,75 mg Dexamethason. Dies müssen wir bei einem Vergleich der Dara- und MOR202-Studien berücksichtigen. Ebenso natürlich die Zusammenstellung des Patientenkollektivs (Anzahl der Vorbehandlungen etc.) Hast Du einen Link zu den Kortikosteroiddaten der Siriusstudie? Ansonsten müßte ich selbst in den entsprechenden Datenbanken suchen.
Antwort auf Beitrag Nr.: 52.567.943 von Joschka Schröder am 08.06.16 13:09:18
Die Studienergebnisse sind im April 2016 in The Lancet veröffentlicht worden. Werde versuchen, sie später im Hinblick auf die angesprochenen Punkt durchzusehen.
Zitat von Joschka Schröder: Hast Du einen Link zu den Kortikosteroiddaten der Siriusstudie? Ansonsten müßte ich selbst in den entsprechenden Datenbanken suchen.
Die Studienergebnisse sind im April 2016 in The Lancet veröffentlicht worden. Werde versuchen, sie später im Hinblick auf die angesprochenen Punkt durchzusehen.
Antwort auf Beitrag Nr.: 52.567.943 von Joschka Schröder am 08.06.16 13:09:18
zu 1. Es ist nicht möglich, dass es der fehlende Patient ist. Denn dieser hat erst nach 17 Monaten eine Response gehabt, die DoR müsste somit sehr kurz sein - unterhalb des 14 Monate Medians. Wenn er hinzugenommen worden wäre hätte das den aktuellen Median für DoR sogar auf 12-13 Monate gesenkt.
Wenn ein Patient in der Rechnung fehlt und den Median erhöhen soll, dann müsste er sich über dem jetzigen Median einsortieren und zwar so, dass bei dann 10 Patienten die Patienten an Pos. 5 und 6 im Schnitt die angebenen 20 Monate DoR haben.
Das ist aber mit nur einem fehlenden Patienten gar nicht möglich - egal was er für eine DoR hat.
Selbst wenn sie den Patienten mit der allerlängsten DoR nicht dargestellt hätten würde sich maximal folgendes DoR errechnen: Mittelwert aus aktueller Pos.5 (derzeitiger Median, neue Pos. 6 = 14 Monate) und aktueller Pos. 4 (dann Pos. 5 mit DoR hier ca. 20Monate) = ca. 17 Monate.
Ich habe daher eher das Gefühl, dass es sich hier um einen ganz peinlichen Fehler handelt und der mDoR wie früher kommuniziert bei 13,7 Monaten (abgelesen entspricht das meinen 14 Monaten). Und mit dem zusätzlichen late Responder sogar aktuell niedriger ist.
Anfrage bei MOR läuft.
zu 2. Ich kann dich leider mit Fach-Infos am Wenigsten versorgen - komme nur an freie Texte und die geben meist nicht die Tiefe wieder. Habe keine Zugänge zu kostenpflichtigen Arzt- oder Wissenschaftsportalen. Ich denke da hast du den perfekten Zugang.
Zitat von Joschka Schröder: Ville, Du bist ja schlimmer als ich
zu Deinem Beitrag 513:
Des Rätsels Lösung könnte in dem zusätzlichen Patienten liegen, der in der Graphik nicht aufgeführt ist (s. Fußnote der Abb. 4 im MOR202-Poster). Wenn es sich um einen DLBCL-Patienten handelt, verschiebt sich die mDoR in der Abb. entsprechend.
Ist aber ein guter Hinweis, da müßte man konkret noch einmal bei Morphosys nachfragen, ob es sich tatsächlich so verhält.
zu Deinem Beitrag 508:
Dexamethason ist deutlich potenter als Methlyprednisolon. Ich müßte noch einmal in der Äquivalenztabelle von Kaiser nachsehen, aber 4 mg Methylprednisolon sollten in etwa so potent sein wie 0,75 mg Dexamethason. Dies müssen wir bei einem Vergleich der Dara- und MOR202-Studien berücksichtigen. Ebenso natürlich die Zusammenstellung des Patientenkollektivs (Anzahl der Vorbehandlungen etc.) Hast Du einen Link zu den Kortikosteroiddaten der Siriusstudie? Ansonsten müßte ich selbst in den entsprechenden Datenbanken suchen.
zu 1. Es ist nicht möglich, dass es der fehlende Patient ist. Denn dieser hat erst nach 17 Monaten eine Response gehabt, die DoR müsste somit sehr kurz sein - unterhalb des 14 Monate Medians. Wenn er hinzugenommen worden wäre hätte das den aktuellen Median für DoR sogar auf 12-13 Monate gesenkt.
Wenn ein Patient in der Rechnung fehlt und den Median erhöhen soll, dann müsste er sich über dem jetzigen Median einsortieren und zwar so, dass bei dann 10 Patienten die Patienten an Pos. 5 und 6 im Schnitt die angebenen 20 Monate DoR haben.
Das ist aber mit nur einem fehlenden Patienten gar nicht möglich - egal was er für eine DoR hat.
Selbst wenn sie den Patienten mit der allerlängsten DoR nicht dargestellt hätten würde sich maximal folgendes DoR errechnen: Mittelwert aus aktueller Pos.5 (derzeitiger Median, neue Pos. 6 = 14 Monate) und aktueller Pos. 4 (dann Pos. 5 mit DoR hier ca. 20Monate) = ca. 17 Monate.
Ich habe daher eher das Gefühl, dass es sich hier um einen ganz peinlichen Fehler handelt und der mDoR wie früher kommuniziert bei 13,7 Monaten (abgelesen entspricht das meinen 14 Monaten). Und mit dem zusätzlichen late Responder sogar aktuell niedriger ist.
Anfrage bei MOR läuft.
zu 2. Ich kann dich leider mit Fach-Infos am Wenigsten versorgen - komme nur an freie Texte und die geben meist nicht die Tiefe wieder. Habe keine Zugänge zu kostenpflichtigen Arzt- oder Wissenschaftsportalen. Ich denke da hast du den perfekten Zugang.

Dexamethason hat längeres half life und ist deswegen u.a. "potenter". Aber biologische Wirkung ist im Prinzip vergleichbar mit prednison. Für meisten Indikationen handelt sich um persönlichen Geschmack des artztes was verschreiben wird. Evtl vorhandene Unterschiede müsste mann direkt experimental festellen. Da nutzen Korrelationen wenig. Hauptdefizit bei MOR202 bleibt kleines kolektiv, meine Meinung nach. Das durfte sich bis ende der jahres ändern.Vielleicht erleben wir mit bisschen gluck ( und Gusel. daten)noch endjahres Rally
Antwort auf Beitrag Nr.: 52.557.113 von Ville7 am 07.06.16 09:07:40Aber ohne Partner in eine zweite P3 (neben MOr208) zu gehen sehe ich aus Sicht eines Investors als sehr hohes Risiko an und habe die Befürchtung, dass man nun massiv verwässern muss. Ich hoffe daher, dass sich für eine P3 noch ein neuer Partner finden lässt.
Auf der HV wurde gesagt das man P3 Studien für MOR208 und auch MOR202 mit Partner machen will.
Auf der HV wurde gesagt das man P3 Studien für MOR208 und auch MOR202 mit Partner machen will.
Antwort auf Beitrag Nr.: 52.573.190 von Maslacak am 08.06.16 21:44:39
Da muss ich Dir leider widersprechen. Es ist richtig, dass die Halbwertszeit des Dexamethasons (Plasma-HWZ 3,5 Std.) etwas länger ist als diejenige des Prednisolons/Prednisons (Plasma-HWZ 2-3 Std.), dies erklärt aber nicht die um den Faktor 7 stärkere genomische Wirkung des Dexamethason!
Dexamethason stimuliert die spezifischen intrazellulären Glukokortikoidrezeptoren deutlich stärker (und auch nachhaltiger -> biologische Halbwertszeit 36-72 Std. versus 18-36 Std.) als es Prednison/Prednisolon vermag.
Hinsichtlich der nicht-genomischen Wirkungen (Interaktion auf Zellmembranebene), die in diesem Zusammenhang aber weit weniger von Bedeutung sind, sehen die Verhältnisse wieder anders aus.: Hier ist die Wirkung des Dexamethason nur um den Faktor 0,2 stärker als die des Prednisolon.
Lange Rede, kurzer Sinn: Die Frage nach dem jeweils verwendeten Präparat ist von großer Bedeutung.
Dexamethason setzt man in der Kombitherapie wegen seiner therapeutischen Potenz ein. Noch vor 20 Jahren bestand die Standardtherapie des Multiplen Myeloms in einer Kombination aus Dexamethason + Alkylanzien. Rezidive wurden mit einer Kombination aus Dexamethason, Adriamycin und Vincristin (VAD-Schma) behandelt, wobei in der Primärtherapie auch oft Dexamethason alleine eingesetzt worden ist.
Zitat von Maslacak: Dexamethason hat längeres half life und ist deswegen u.a. "potenter". Aber biologische Wirkung ist im Prinzip vergleichbar mit prednison. Für meisten Indikationen handelt sich um persönlichen Geschmack des artztes was verschreiben wird.
Da muss ich Dir leider widersprechen. Es ist richtig, dass die Halbwertszeit des Dexamethasons (Plasma-HWZ 3,5 Std.) etwas länger ist als diejenige des Prednisolons/Prednisons (Plasma-HWZ 2-3 Std.), dies erklärt aber nicht die um den Faktor 7 stärkere genomische Wirkung des Dexamethason!
Dexamethason stimuliert die spezifischen intrazellulären Glukokortikoidrezeptoren deutlich stärker (und auch nachhaltiger -> biologische Halbwertszeit 36-72 Std. versus 18-36 Std.) als es Prednison/Prednisolon vermag.
Hinsichtlich der nicht-genomischen Wirkungen (Interaktion auf Zellmembranebene), die in diesem Zusammenhang aber weit weniger von Bedeutung sind, sehen die Verhältnisse wieder anders aus.: Hier ist die Wirkung des Dexamethason nur um den Faktor 0,2 stärker als die des Prednisolon.
Lange Rede, kurzer Sinn: Die Frage nach dem jeweils verwendeten Präparat ist von großer Bedeutung.
Dexamethason setzt man in der Kombitherapie wegen seiner therapeutischen Potenz ein. Noch vor 20 Jahren bestand die Standardtherapie des Multiplen Myeloms in einer Kombination aus Dexamethason + Alkylanzien. Rezidive wurden mit einer Kombination aus Dexamethason, Adriamycin und Vincristin (VAD-Schma) behandelt, wobei in der Primärtherapie auch oft Dexamethason alleine eingesetzt worden ist.
Antwort auf Beitrag Nr.: 52.572.992 von Ville7 am 08.06.16 21:23:48
Das ist richtig! Großartig, dass Du Dir die Abbildung so genau angesehen hast!
Zitat von Ville7: Selbst wenn sie den Patienten mit der allerlängsten DoR nicht dargestellt hätten würde sich maximal folgendes DoR errechnen: Mittelwert aus aktueller Pos.5 (derzeitiger Median, neue Pos. 6 = 14 Monate) und aktueller Pos. 4 (dann Pos. 5 mit DoR hier ca. 20Monate) = ca. 17 Monate.
Das ist richtig! Großartig, dass Du Dir die Abbildung so genau angesehen hast!
Antwort auf Beitrag Nr.: 52.573.973 von Joschka Schröder am 09.06.16 02:21:57
Die IR antwortet, das die Kaplan Meier Methode verwendet wurde. ville sollte ja wohl die gleich Antwort schon haben. Bin kein Experte, wie sich danach der Median ermittelt, auch mal schnelle googeln bringt da nicht weiter. Da muss ich schon mal länger lesen
Zitat von Joschka Schröder:Zitat von Ville7: Selbst wenn sie den Patienten mit der allerlängsten DoR nicht dargestellt hätten würde sich maximal folgendes DoR errechnen: Mittelwert aus aktueller Pos.5 (derzeitiger Median, neue Pos. 6 = 14 Monate) und aktueller Pos. 4 (dann Pos. 5 mit DoR hier ca. 20Monate) = ca. 17 Monate.
Das ist richtig! Großartig, dass Du Dir die Abbildung so genau angesehen hast!
Die IR antwortet, das die Kaplan Meier Methode verwendet wurde. ville sollte ja wohl die gleich Antwort schon haben. Bin kein Experte, wie sich danach der Median ermittelt, auch mal schnelle googeln bringt da nicht weiter. Da muss ich schon mal länger lesen
danke für die interessanten Beiträge, ich bin bei der Aktie jetzt wieder deutlich ruhiger...
na, da kennt wohl schon jemand die Guselkumabergebnisse
Antwort auf Beitrag Nr.: 52.604.642 von riverstar_de am 13.06.16 15:40:29ich tippe auf eine erneute massive Shortattacke
Antwort auf Beitrag Nr.: 52.605.632 von milchbubi am 13.06.16 17:44:33
schade, Du hattest Recht:Marshall Wace LLP 0,71 %
https://www.bundesanzeiger.de/ebanzwww/wexsservlet?session.s…
Zitat von milchbubi: ich tippe auf eine erneute massive Shortattacke
schade, Du hattest Recht:Marshall Wace LLP 0,71 %
https://www.bundesanzeiger.de/ebanzwww/wexsservlet?session.s…
Antwort auf Beitrag Nr.: 52.613.426 von riverstar_de am 14.06.16 15:29:39ich habs auch grad gesehen....
unglaublich, deren Riecher/Trefferquote.
unglaublich, deren Riecher/Trefferquote.

Antwort auf Beitrag Nr.: 52.613.804 von milchbubi am 14.06.16 16:14:51was meinstdu.kannst du mir das bitte genauer erklären.
Beachtet mal den Threadtitel und verlegt doch bitte die Diskussion in den normalen Thread, im Interesse aller
http://www.wallstreet-online.de/diskussion/1205075-2601-2610…
http://www.wallstreet-online.de/diskussion/1205075-2601-2610…
GSK startet nun eine dritte klinische Phase II-Studie mit MOR103/GSK3196165 -> https://clinicaltrials.gov/ct2/show/NCT02799472?term=GSK3196…
Übersichtliches Konzept, 40 Patienten, im Mai 2017 sollen die Ergebnisdaten vorliegen.
PS: Danke, RichyBerlin für Beitrag 528!
Übersichtliches Konzept, 40 Patienten, im Mai 2017 sollen die Ergebnisdaten vorliegen.
PS: Danke, RichyBerlin für Beitrag 528!
Betrifft indirekt MOR208
18.7.2016
Roche provides update on phase III study of Gazyva/Gazyvaro in people with previously untreated diffuse large B-cell lymphoma
• GOYA study did not meet its primary endpoint of improvement in progression-free survival with Gazyva/Gazyvaro plus CHOP chemotherapy versus MabThera/Rituxan plus CHOP chemotherapy
Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the phase III GOYA study evaluating Gazyva®/Gazyvaro® (obinutuzumab) plus CHOP chemotherapy (G-CHOP) in people with previously untreated diffuse large B-cell lymphoma (DLBCL) did not meet its primary endpoint of significantly reducing the risk of disease worsening or death (progression-free survival; PFS) compared to MabThera/Rituxan (rituximab) plus CHOP chemotherapy (R-CHOP). Adverse events with Gazyva/Gazyvaro and MabThera/Rituxan were consistent with those seen in previous clinical trials when each was combined with various chemotherapies. Data from the GOYA study will be presented at an upcoming medical meeting.
“Two previous studies showed Gazyva/Gazyvaro helped people with previously untreated follicular lymphoma or chronic lymphocytic leukaemia live longer without their disease worsening compared to MabThera/Rituxan, when each was combined with chemotherapy. We were hopeful we could show a similar result for people with diffuse large B-cell lymphoma and once again improve on the standard of care,” said Sandra Horning, MD, Chief Medical Officer and Head of Global Product Development. “We will continue to analyse the GOYA data to better understand the results, and to study other investigational treatments in this disease with the goal of further helping these patients.”
About the GOYA study
GOYA (NCT01287741) is a global phase III open-label, multi-centre, randomised two-arm study examining the efficacy and safety of the combination of Gazyva/Gazyvaro plus CHOP chemotherapy (G-CHOP) compared to MabThera/Rituxan plus CHOP chemotherapy (R-CHOP). GOYA included 1,418 previously untreated patients with CD20-positive DLBCL. The primary endpoint of the study is investigator-assessed PFS, with secondary endpoints including PFS assessed by independent review committee (IRC), response rate (overall response, ORR; and complete response, CR), overall survival (OS), disease free survival (DFS) and safety profile. The GOYA study is being conducted in cooperation with the Fondazione Italiana Linfomi (FIL, Italy).
About Gazyva/Gazyvaro (obinutuzumab)
Gazyva/Gazyvaro is an engineered monoclonal antibody designed to attach to CD20, a protein expressed on certain B cells, but not on stem cells or plasma cells. Gazyva/Gazyvaro is designed to attack and destroy targeted B-cells both directly and together with the body's immune system.
Gazyva/Gazyvaro is currently approved in more than 70 countries in combination with chlorambucil, for people with previously untreated chronic lymphocytic leukaemia. The approvals were based on the CLL11 study, showing significant improvements with Gazyva/Gazyvaro plus chlorambucil across multiple clinical endpoints, including PFS, overall response rate (ORR), complete response rate (CR), and minimal residual disease (MRD) when compared head-to-head with MabThera/Rituxan plus chlorambucil.
In February 2016, Gazyva was approved by the US Food and Drug Administration in combination with bendamustine followed by Gazyva alone for people with follicular lymphoma who did not respond to a Rituxan-containing regimen, or whose follicular lymphoma returned after such treatment. In June 2016, Gazyvaro was approved by the European Commission in combination with bendamustine followed by Gazyvaro maintenance in people with follicular lymphoma who did not respond or who progressed during or up to six months after treatment with MabThera or a MabThera-containing regimen. Both approvals were based on the phase III GADOLIN study, showing a significant improvement in progression-free survival with Gazyva/Gazyvaro-based therapy compared to bendamustine alone. Gazyva is marketed as Gazyvaro in the EU and Switzerland.
In May 2016, the phase III GALLIUM study in people with previously untreated follicular lymphoma met its primary endpoint early. GALLIUM compared the efficacy and safety of Gazyva/Gazyvaro plus chemotherapy (CHOP, CVP or bendamustine) followed by Gazyva/Gazyvaro alone, head-to-head with MabThera/Rituxan plus chemotherapy followed by MabThera/Rituxan alone. Results from a pre-planned interim analysis showed that Gazyva/Gazyvaro-based treatment resulted in superior progression-free survival compared to MabThera/Rituxan-based treatment. Adverse events with either Gazyva/Gazyvaro or MabThera/Rituxan were consistent with those seen in previous clinical trials when each was combined with various chemotherapies. Data from the GALLIUM study will be presented at an upcoming medical meeting and submitted to health authorities for approval consideration.
Additional combination studies investigating Gazyva/Gazyvaro with other approved or investigational medicines, including cancer immunotherapies and small molecule inhibitors, are underway across a range of blood cancers.
About DLBCL
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL(1). DLBCL is an aggressive (fast-growing) type of NHL, which is generally responsive to treatment in the frontline(2). However, as many as 40% of patients will relapse, at which time salvage therapy options are limited and survival is short(2). Approximately 123,000 people worldwide are estimated to be diagnosed with DLBCL each year(3).
18.7.2016
Roche provides update on phase III study of Gazyva/Gazyvaro in people with previously untreated diffuse large B-cell lymphoma
• GOYA study did not meet its primary endpoint of improvement in progression-free survival with Gazyva/Gazyvaro plus CHOP chemotherapy versus MabThera/Rituxan plus CHOP chemotherapy
Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the phase III GOYA study evaluating Gazyva®/Gazyvaro® (obinutuzumab) plus CHOP chemotherapy (G-CHOP) in people with previously untreated diffuse large B-cell lymphoma (DLBCL) did not meet its primary endpoint of significantly reducing the risk of disease worsening or death (progression-free survival; PFS) compared to MabThera/Rituxan (rituximab) plus CHOP chemotherapy (R-CHOP). Adverse events with Gazyva/Gazyvaro and MabThera/Rituxan were consistent with those seen in previous clinical trials when each was combined with various chemotherapies. Data from the GOYA study will be presented at an upcoming medical meeting.
“Two previous studies showed Gazyva/Gazyvaro helped people with previously untreated follicular lymphoma or chronic lymphocytic leukaemia live longer without their disease worsening compared to MabThera/Rituxan, when each was combined with chemotherapy. We were hopeful we could show a similar result for people with diffuse large B-cell lymphoma and once again improve on the standard of care,” said Sandra Horning, MD, Chief Medical Officer and Head of Global Product Development. “We will continue to analyse the GOYA data to better understand the results, and to study other investigational treatments in this disease with the goal of further helping these patients.”
About the GOYA study
GOYA (NCT01287741) is a global phase III open-label, multi-centre, randomised two-arm study examining the efficacy and safety of the combination of Gazyva/Gazyvaro plus CHOP chemotherapy (G-CHOP) compared to MabThera/Rituxan plus CHOP chemotherapy (R-CHOP). GOYA included 1,418 previously untreated patients with CD20-positive DLBCL. The primary endpoint of the study is investigator-assessed PFS, with secondary endpoints including PFS assessed by independent review committee (IRC), response rate (overall response, ORR; and complete response, CR), overall survival (OS), disease free survival (DFS) and safety profile. The GOYA study is being conducted in cooperation with the Fondazione Italiana Linfomi (FIL, Italy).
About Gazyva/Gazyvaro (obinutuzumab)
Gazyva/Gazyvaro is an engineered monoclonal antibody designed to attach to CD20, a protein expressed on certain B cells, but not on stem cells or plasma cells. Gazyva/Gazyvaro is designed to attack and destroy targeted B-cells both directly and together with the body's immune system.
Gazyva/Gazyvaro is currently approved in more than 70 countries in combination with chlorambucil, for people with previously untreated chronic lymphocytic leukaemia. The approvals were based on the CLL11 study, showing significant improvements with Gazyva/Gazyvaro plus chlorambucil across multiple clinical endpoints, including PFS, overall response rate (ORR), complete response rate (CR), and minimal residual disease (MRD) when compared head-to-head with MabThera/Rituxan plus chlorambucil.
In February 2016, Gazyva was approved by the US Food and Drug Administration in combination with bendamustine followed by Gazyva alone for people with follicular lymphoma who did not respond to a Rituxan-containing regimen, or whose follicular lymphoma returned after such treatment. In June 2016, Gazyvaro was approved by the European Commission in combination with bendamustine followed by Gazyvaro maintenance in people with follicular lymphoma who did not respond or who progressed during or up to six months after treatment with MabThera or a MabThera-containing regimen. Both approvals were based on the phase III GADOLIN study, showing a significant improvement in progression-free survival with Gazyva/Gazyvaro-based therapy compared to bendamustine alone. Gazyva is marketed as Gazyvaro in the EU and Switzerland.
In May 2016, the phase III GALLIUM study in people with previously untreated follicular lymphoma met its primary endpoint early. GALLIUM compared the efficacy and safety of Gazyva/Gazyvaro plus chemotherapy (CHOP, CVP or bendamustine) followed by Gazyva/Gazyvaro alone, head-to-head with MabThera/Rituxan plus chemotherapy followed by MabThera/Rituxan alone. Results from a pre-planned interim analysis showed that Gazyva/Gazyvaro-based treatment resulted in superior progression-free survival compared to MabThera/Rituxan-based treatment. Adverse events with either Gazyva/Gazyvaro or MabThera/Rituxan were consistent with those seen in previous clinical trials when each was combined with various chemotherapies. Data from the GALLIUM study will be presented at an upcoming medical meeting and submitted to health authorities for approval consideration.
Additional combination studies investigating Gazyva/Gazyvaro with other approved or investigational medicines, including cancer immunotherapies and small molecule inhibitors, are underway across a range of blood cancers.
About DLBCL
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL(1). DLBCL is an aggressive (fast-growing) type of NHL, which is generally responsive to treatment in the frontline(2). However, as many as 40% of patients will relapse, at which time salvage therapy options are limited and survival is short(2). Approximately 123,000 people worldwide are estimated to be diagnosed with DLBCL each year(3).
Antwort auf Beitrag Nr.: 52.853.326 von Joschka Schröder am 18.07.16 07:44:41inwiefern? mor208 spricht doch das zielmolekül cd 19 an u. der ak von roche cd 20?
Antwort auf Beitrag Nr.: 52.854.616 von milchbubi am 18.07.16 10:29:01Beides sind optimierte Antikörper (einmal gegen CD19, das andere Mal gegen CD20), die in derselben Indikation eingesetzt werden sollen. Die enttäuschenden Ergebnisse der GOYA-Studie verbessern die Marktchancen von MOR208. Insoweit gehören die Ergebnisse in diesen Thread.
MOR208 hat in den bisherigen DLBCL-Studien zu ungewöhnlich langdauernden, d.h. nachhaltigen klinischen Antworten geführt.
MOR208 hat in den bisherigen DLBCL-Studien zu ungewöhnlich langdauernden, d.h. nachhaltigen klinischen Antworten geführt.
Antwort auf Beitrag Nr.: 52.854.754 von Joschka Schröder am 18.07.16 10:46:17Danke für die Erklärung!
GlobeNewswire (Deutschland)·Mehr Nachrichten von GlobeNewswire (Deutschland)
MorphoSys AG: MorphoSys schließt Sicherheitsteil der L-MIND-Kombinationsstudie mit MOR208 bei Patienten mit DLBCL erfolgreich ab
MorphoSys AG / MorphoSys schließt Sicherheitsteil der L-MIND-Kombinationsstudie mit MOR208 bei Patienten mit DLBCL erfolgreich ab . Verarbeitet und übermittelt durch NASDAQ OMX Corporate Solutions. Für den Inhalt der Mitteilung ist der Emittent verantwortlich. Source: Globenewswire
Die MorphoSys AG (Frankfurt: MOR; Prime Standard Segment, TecDAX; OTC: MPSYY) gab heute bekannt, dass der Sicherheitsteil der klinischen Phase 2-Studie von MOR208 in Kombination mit Lenalidomid bei Patienten mit rezidivierendem oder refraktärem diffusen großzelligen B-Zell-Lymphom (DLBCL) erfolgreich abgeschlossen wurde. DLBCL ist die häufigste Form des Non-Hodgkin Lymphoms (NHL). Sechs Patienten wurden während des Sicherheitsteils der L-MIND-Studie mit der empfohlenen Dosis von MOR208 (12 mg/kg) in Kombination mit dem Krebsmedikament Lenalidomid behandelt. Es wurden keine unerwarteten Hinweise in Bezug auf die Sicherheit festgestellt, und die Studie wird wie geplant fortgesetzt.
"Wir setzen die Kombinationsstudie mit MOR208 und Lenalidomid als mögliche neue Behandlung für Patienten mit DLBCL weiter fort. Diese Studie ist Teil unserer umfassenden Entwicklungsstrategie, MOR208 als therapeutischen Antikörper in Kombination mit einer Vielzahl anderer Krebsmedikamente klinisch einzusetzen. Wir sind sehr zufrieden mit den ersten Ergebnissen des Sicherheitsteils der L-MIND-Studie, die keine unerwarteten Sicherheitsrisiken zeigten", kommentiert Dr. Arndt Schottelius, Entwicklungsvorstand der MorphoSys AG. "Die klinische Studie schreitet wie geplant voran, und wir gehen davon aus, 2017 erste Daten zur Wirksamkeit vorstellen zu können."
L-MIND ist eine einarmige, unverblindete und in mehreren Studienzentren durchgeführte klinische Studie. Darin wird der gegen das Zielmolekül CD19 gerichtete Antikörper MOR208 in Kombination mit dem Krebsmedikament Lenalidomid bei rund 80 Patienten erprobt, die beim Eintritt in die Studie mit rezidivierendem bzw. refraktärem DLBCL diagnostiziert worden sind. Zudem müssen die Patienten mindestens eine bzw. maximal zwei Vorbehandlungen erhalten haben, darunter eine gegen das Zielmolekül CD20 gerichtete Therapie, beispielsweise Rituximab. Des Weiteren dürfen die eingeschlossenen Patienten nicht für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation infrage kommen. Der primäre Endpunkt der Studie ist die Gesamtansprechrate (ORR). Die sekundären Endpunkte umfassen die Dauer des Ansprechens (DoR), das progressionsfreie Überleben (PFS) und das Gesamtüberleben (OS), sowie eine Bewertung der Sicherheit der Arzneimittelkombination und die pharmakokinetischen Parameter von MOR208.
Über CD19 und MOR208
CD19 wird stark und homogen auf Tumorzellen von verschiedenen B-Zell-Lymphomen, darunter DLBCL und CLL (chronisch lymphatische Leukämie), exprimiert. CD19 verstärkt die Signalgebung von B-Zell-Rezeptoren (BCR), was einen wichtigen Faktor für das Überleben von B-Zellen darstellt und CD19 somit zu einem potenziellen Zielmolekül bei bösartigen B-Zell-Erkrankungen macht.
MOR208 (vormals Xmab®5574) ist ein Antikörper mit modifiziertem Fc-Teil, der gegen das Zielmolekül CD19 gerichtet ist. Die Modifizierung des Fc-Teils führt zu einer deutlichen Verstärkung der antikörperabhängigen, zellvermittelten zytotoxischen Immunantwort (ADCC) und Phagozytose (ADCP). Zudem führt MOR208 zum direkten Zelltod durch die Bindung an CD19.
MorphoSys untersucht in seinen klinischen Studien den gegen das BCR-relevante Zielmolekül CD19 gerichteten Fc-modifizierten Antikörper MOR208. Damit soll eine neue immuntherapeutische Behandlungsoption für bösartige B-Zell-Erkrankungen entwickelt werden.
Die auf den Jahrestagungen der American Society of Clinical Oncology (ASCO 2016) sowie der European Hematology Association (EHA 2016) vorgestellten aktualisierten Daten zeigten die Ergebnisse der Studien zur Wirksamkeit und Sicherheit von MOR208 bei 92 stark vorbehandelten Patienten mit Non-Hodgkin Lymphom (NHL). Die Gesamtansprechrate (ORR) betrug 36% bei Patienten mit diffusem großzelligem B-Zell-Lymphom (DLBCL) und erreichte 33% bei Patienten mit indolentem NHL (iNHL) (jeweils basierend auf auswertbaren Patienten). Zum Zeitpunkt der Analyse lag die mediane Ansprechdauer (DoR) (gemäß Kaplan-Meier-Methode) in DLBCL bei 20 Monaten, wobei drei Patienten weiterhin auf die Behandlung ansprechen. Die mediane Ansprechdauer wurde bei Patienten mit iNHL nicht erreicht; hier sprachen 72% der Patienten an ohne ein Fortschreiten der Krankheit nach 16 Monaten. Die progressionsfreie Überlebensrate (PFS) nach zwölf Monaten betrug in DLBCL 40% und zeigte dabei ähnliche PFS-Raten bei Rituximab-sensitiven und Rituximab-refraktären Patienten. Im Zusammenhang mit der Behandlung von MOR208 wurden schwerwiegende unerwünschte Ereignisse bei 6% der DLBCL und 4% der iNHL Patienten gemeldet. Im Zusammenhang mit der Behandlung wurden keine Todesfälle berichtet.
MorphoSys in Kürze:
MorphoSys hat mit der HuCAL-Technologie die erfolgreichste Antikörper-Bibliothek der Pharma-Industrie entwickelt. Durch den erfolgreichen Einsatz dieser und weiterer firmeneigener Technologien wurde MorphoSys zu einem Marktführer im Bereich therapeutischer Antikörper, einer der am schnellsten wachsenden Medikamenten-Klassen der Humanmedizin.
Gemeinsam mit seinen Pharma-Partnern hat MorphoSys eine therapeutische Pipeline (https://www.morphosys.de/pipeline) mit mehr als 100 Antikörper-basierten Medikamenten-Kandidaten unter anderem zur Behandlung von Krebs, rheumatoider Arthritis und Alzheimer aufgebaut. MorphoSys ist auf die Entwicklung neuer Antikörper-Technologien und Wirkstoffe spezialisiert, um die Medikamente von morgen herzustellen. MorphoSys ist an der Frankfurter Börse unter dem Symbol "MOR" notiert. Aktuelle Informationen zu MorphoSys finden Sie unter www.morphosys.de (http://www.morphosys.de).
Diese Veröffentlichung enthält bestimmte in die Zukunft gerichtete Aussagen, die den MorphoSys-Konzern betreffen. Diese spiegeln die Meinung von MorphoSys zum Datum dieser Mitteilung wider und beinhalten bestimmte Risiken und Unsicherheiten. Sollten sich die den Annahmen der Gesellschaft zugrunde liegenden Verhältnisse ändern, so können die tatsächlichen Ergebnisse und Maßnahmen von den erwarteten Ergebnissen und Maßnahmen abweichen. MorphoSys beabsichtigt nicht, diese in die Zukunft gerichteten Aussagen zu aktualisieren, soweit sie den Wortlaut dieser Pressemitteilung betreffen.
Für weitere Informationen kontaktieren Sie bitte:
MorphoSys AG
Dr. Claudia Gutjahr-Löser
Head of Corporate Communications & IR
MorphoSys AG: MorphoSys schließt Sicherheitsteil der L-MIND-Kombinationsstudie mit MOR208 bei Patienten mit DLBCL erfolgreich ab
MorphoSys AG / MorphoSys schließt Sicherheitsteil der L-MIND-Kombinationsstudie mit MOR208 bei Patienten mit DLBCL erfolgreich ab . Verarbeitet und übermittelt durch NASDAQ OMX Corporate Solutions. Für den Inhalt der Mitteilung ist der Emittent verantwortlich. Source: Globenewswire
Die MorphoSys AG (Frankfurt: MOR; Prime Standard Segment, TecDAX; OTC: MPSYY) gab heute bekannt, dass der Sicherheitsteil der klinischen Phase 2-Studie von MOR208 in Kombination mit Lenalidomid bei Patienten mit rezidivierendem oder refraktärem diffusen großzelligen B-Zell-Lymphom (DLBCL) erfolgreich abgeschlossen wurde. DLBCL ist die häufigste Form des Non-Hodgkin Lymphoms (NHL). Sechs Patienten wurden während des Sicherheitsteils der L-MIND-Studie mit der empfohlenen Dosis von MOR208 (12 mg/kg) in Kombination mit dem Krebsmedikament Lenalidomid behandelt. Es wurden keine unerwarteten Hinweise in Bezug auf die Sicherheit festgestellt, und die Studie wird wie geplant fortgesetzt.
"Wir setzen die Kombinationsstudie mit MOR208 und Lenalidomid als mögliche neue Behandlung für Patienten mit DLBCL weiter fort. Diese Studie ist Teil unserer umfassenden Entwicklungsstrategie, MOR208 als therapeutischen Antikörper in Kombination mit einer Vielzahl anderer Krebsmedikamente klinisch einzusetzen. Wir sind sehr zufrieden mit den ersten Ergebnissen des Sicherheitsteils der L-MIND-Studie, die keine unerwarteten Sicherheitsrisiken zeigten", kommentiert Dr. Arndt Schottelius, Entwicklungsvorstand der MorphoSys AG. "Die klinische Studie schreitet wie geplant voran, und wir gehen davon aus, 2017 erste Daten zur Wirksamkeit vorstellen zu können."
L-MIND ist eine einarmige, unverblindete und in mehreren Studienzentren durchgeführte klinische Studie. Darin wird der gegen das Zielmolekül CD19 gerichtete Antikörper MOR208 in Kombination mit dem Krebsmedikament Lenalidomid bei rund 80 Patienten erprobt, die beim Eintritt in die Studie mit rezidivierendem bzw. refraktärem DLBCL diagnostiziert worden sind. Zudem müssen die Patienten mindestens eine bzw. maximal zwei Vorbehandlungen erhalten haben, darunter eine gegen das Zielmolekül CD20 gerichtete Therapie, beispielsweise Rituximab. Des Weiteren dürfen die eingeschlossenen Patienten nicht für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation infrage kommen. Der primäre Endpunkt der Studie ist die Gesamtansprechrate (ORR). Die sekundären Endpunkte umfassen die Dauer des Ansprechens (DoR), das progressionsfreie Überleben (PFS) und das Gesamtüberleben (OS), sowie eine Bewertung der Sicherheit der Arzneimittelkombination und die pharmakokinetischen Parameter von MOR208.
Über CD19 und MOR208
CD19 wird stark und homogen auf Tumorzellen von verschiedenen B-Zell-Lymphomen, darunter DLBCL und CLL (chronisch lymphatische Leukämie), exprimiert. CD19 verstärkt die Signalgebung von B-Zell-Rezeptoren (BCR), was einen wichtigen Faktor für das Überleben von B-Zellen darstellt und CD19 somit zu einem potenziellen Zielmolekül bei bösartigen B-Zell-Erkrankungen macht.
MOR208 (vormals Xmab®5574) ist ein Antikörper mit modifiziertem Fc-Teil, der gegen das Zielmolekül CD19 gerichtet ist. Die Modifizierung des Fc-Teils führt zu einer deutlichen Verstärkung der antikörperabhängigen, zellvermittelten zytotoxischen Immunantwort (ADCC) und Phagozytose (ADCP). Zudem führt MOR208 zum direkten Zelltod durch die Bindung an CD19.
MorphoSys untersucht in seinen klinischen Studien den gegen das BCR-relevante Zielmolekül CD19 gerichteten Fc-modifizierten Antikörper MOR208. Damit soll eine neue immuntherapeutische Behandlungsoption für bösartige B-Zell-Erkrankungen entwickelt werden.
Die auf den Jahrestagungen der American Society of Clinical Oncology (ASCO 2016) sowie der European Hematology Association (EHA 2016) vorgestellten aktualisierten Daten zeigten die Ergebnisse der Studien zur Wirksamkeit und Sicherheit von MOR208 bei 92 stark vorbehandelten Patienten mit Non-Hodgkin Lymphom (NHL). Die Gesamtansprechrate (ORR) betrug 36% bei Patienten mit diffusem großzelligem B-Zell-Lymphom (DLBCL) und erreichte 33% bei Patienten mit indolentem NHL (iNHL) (jeweils basierend auf auswertbaren Patienten). Zum Zeitpunkt der Analyse lag die mediane Ansprechdauer (DoR) (gemäß Kaplan-Meier-Methode) in DLBCL bei 20 Monaten, wobei drei Patienten weiterhin auf die Behandlung ansprechen. Die mediane Ansprechdauer wurde bei Patienten mit iNHL nicht erreicht; hier sprachen 72% der Patienten an ohne ein Fortschreiten der Krankheit nach 16 Monaten. Die progressionsfreie Überlebensrate (PFS) nach zwölf Monaten betrug in DLBCL 40% und zeigte dabei ähnliche PFS-Raten bei Rituximab-sensitiven und Rituximab-refraktären Patienten. Im Zusammenhang mit der Behandlung von MOR208 wurden schwerwiegende unerwünschte Ereignisse bei 6% der DLBCL und 4% der iNHL Patienten gemeldet. Im Zusammenhang mit der Behandlung wurden keine Todesfälle berichtet.
MorphoSys in Kürze:
MorphoSys hat mit der HuCAL-Technologie die erfolgreichste Antikörper-Bibliothek der Pharma-Industrie entwickelt. Durch den erfolgreichen Einsatz dieser und weiterer firmeneigener Technologien wurde MorphoSys zu einem Marktführer im Bereich therapeutischer Antikörper, einer der am schnellsten wachsenden Medikamenten-Klassen der Humanmedizin.
Gemeinsam mit seinen Pharma-Partnern hat MorphoSys eine therapeutische Pipeline (https://www.morphosys.de/pipeline) mit mehr als 100 Antikörper-basierten Medikamenten-Kandidaten unter anderem zur Behandlung von Krebs, rheumatoider Arthritis und Alzheimer aufgebaut. MorphoSys ist auf die Entwicklung neuer Antikörper-Technologien und Wirkstoffe spezialisiert, um die Medikamente von morgen herzustellen. MorphoSys ist an der Frankfurter Börse unter dem Symbol "MOR" notiert. Aktuelle Informationen zu MorphoSys finden Sie unter www.morphosys.de (http://www.morphosys.de).
Diese Veröffentlichung enthält bestimmte in die Zukunft gerichtete Aussagen, die den MorphoSys-Konzern betreffen. Diese spiegeln die Meinung von MorphoSys zum Datum dieser Mitteilung wider und beinhalten bestimmte Risiken und Unsicherheiten. Sollten sich die den Annahmen der Gesellschaft zugrunde liegenden Verhältnisse ändern, so können die tatsächlichen Ergebnisse und Maßnahmen von den erwarteten Ergebnissen und Maßnahmen abweichen. MorphoSys beabsichtigt nicht, diese in die Zukunft gerichteten Aussagen zu aktualisieren, soweit sie den Wortlaut dieser Pressemitteilung betreffen.
Für weitere Informationen kontaktieren Sie bitte:
MorphoSys AG
Dr. Claudia Gutjahr-Löser
Head of Corporate Communications & IR
Antwort auf Beitrag Nr.: 52.854.754 von Joschka Schröder am 18.07.16 10:46:17
Die aktuelle MOR208 DLBCL soll doch 2017 in eine P3 Studie überführt werden?
Wie sollte die deiner Meinung nach konzipiert werden?
Zitat von Joschka Schröder: Beides sind optimierte Antikörper (einmal gegen CD19, das andere Mal gegen CD20), die in derselben Indikation eingesetzt werden sollen. Die enttäuschenden Ergebnisse der GOYA-Studie verbessern die Marktchancen von MOR208. Insoweit gehören die Ergebnisse in diesen Thread.
MOR208 hat in den bisherigen DLBCL-Studien zu ungewöhnlich langdauernden, d.h. nachhaltigen klinischen Antworten geführt.
Die aktuelle MOR208 DLBCL soll doch 2017 in eine P3 Studie überführt werden?
Wie sollte die deiner Meinung nach konzipiert werden?
Der ursprünglich geplanten Kombinationsbehandlung aus MOR208 und Idelalisib dürfte nun nichts mehr im Weg stehen -> http://www.akdae.de/Arzneimittelsicherheit/DSM/Archiv/2016-2…
off topic:
Weiß jemand, was aus dem geschätzten user "SLGramann" geworden ist?
Weiß jemand, was aus dem geschätzten user "SLGramann" geworden ist?
Antwort auf Beitrag Nr.: 53.123.550 von Joschka Schröder am 24.08.16 08:18:31
m 13.07.16 schon im anderen Thread gepostet :-)
Zitat von Joschka Schröder: Der ursprünglich geplanten Kombinationsbehandlung aus MOR208 und Idelalisib dürfte nun nichts mehr im Weg stehen -> http://www.akdae.de/Arzneimittelsicherheit/DSM/Archiv/2016-2…
m 13.07.16 schon im anderen Thread gepostet :-)
Antwort auf Beitrag Nr.: 53.132.655 von riverstar_de am 25.08.16 08:52:56
Zitat von riverstar_de:Zitat von Joschka Schröder: Der ursprünglich geplanten Kombinationsbehandlung aus MOR208 und Idelalisib dürfte nun nichts mehr im Weg stehen -> http://www.akdae.de/Arzneimittelsicherheit/DSM/Archiv/2016-2…
hatte ich am 13.07.16 schon im anderen Thread gepostet:
http://www.wallstreet-online.de/diskussion/1205075-2761-2770…" target="_blank" rel="nofollow ugc noopener">http://www.wallstreet-online.de/diskussion/1205075-2761-2770…
Antwort auf Beitrag Nr.: 53.132.655 von riverstar_de am 25.08.16 08:52:56Vielen Dank, manchmal weiß ich selbst nicht mehr, was ich bereits gepostet habe ... ich hoffe, das sind keine ersten Anzeichen einer Altersdemenz.
Antwort auf Beitrag Nr.: 53.132.733 von Joschka Schröder am 25.08.16 08:58:57
keine Sorge - ich hatte es gepostet, also alles in Ordnung mit Deinem Hirn.
Zitat von Joschka Schröder: Vielen Dank, manchmal weiß ich selbst nicht mehr, was ich bereits gepostet habe ... ich hoffe, das sind keine ersten Anzeichen einer Altersdemenz.
keine Sorge - ich hatte es gepostet, also alles in Ordnung mit Deinem Hirn.
Ironie der Geschichte, dass gerade AbDSerotec in die Entwicklung neuer anti-sclerostin-Antikörper involviert ist, nachdem man bei BPS804 offenbar nicht die optimale Bindungsstelle getroffen hat ...
https://www.uni-wuerzburg.de/sonstiges/meldungen/single/arti…
https://www.uni-wuerzburg.de/sonstiges/meldungen/single/arti…
Antwort auf Beitrag Nr.: 45.081.753 von Joschka Schröder am 19.07.13 21:27:33Heute wurde also erstmals offiziell bestätigt, was bereits im Eingangsthread vor drei Jahren zu lesen war.
Über das Target wurde ja auch schon etwas geschrieben ->
Ab heute ist nun endlich geklärt, in welcher Indikation MOR106 eingesetzt werden soll. Bei der atopischen Dermatitis handelt es sich zweifellos um eine Indikation mit großem medizinischen Bedarf und entsprechendem wirtschaftlichen Potential. Ein Hautentwicklungskonkurrent für MOR106 wird der Anti-IL4-Rezeptor-ɑ-Antikörper Dupilumab von Sanofi/Gegeneron sein, der die Signalwege von IL 4 und IL 13 hemmt und sich bereits in klinischen Studien bewährt hat.
Zitat von Joschka Schröder: Von meiner Seite aus werde ich versuchen, gelegentlich einige Entwicklungspräparate zu beleuchten, die noch nicht im Fokus der Öffentlichkeit stehen. So hat Morphosys beispielsweise Antikörper u.a. gegen folgende Targets entwickelt:
...
IL-17C (gemeinsam mit Galapagos)
Über das Target wurde ja auch schon etwas geschrieben ->
Zitat von Joschka Schröder: Von einer einfachen Variante des IL-17 würde ich eher nicht sprechen. Die Aminosäuresequenzen von IL-17C und IL-17A, das z.B. durch secukinumab/Cosentyx blockiert wird, stimmen z.B. nur zu 27 % überein und IL-17C bindet nicht bzw. nur mit geringer Affinität an den IL-17A-Rezeptor. Zur Funktion: IL-17C stimuliert u.a. die Expression von GCSF und beta-defensin-2 in Keratozyten. Außerdem stimuliert IL-17C die Bildung von Il-17A. TNF and IL1B wiederum induzieren die IL-17C-Freisetzung aus Epithelzellen. Letztlich bin ich aber überfragt, wenn es um die Beamtwortung der entscheidenden Frage geht, welchen konkreten therapeutischen Vorteil ein anti-IL-17C-Antikörper z.B. im Vergleich zu einem anti-IL-17A-Antikörper haben kann. Tut mir leid, dass ich derzeit nicht mehr zu diesem Thema…
Ab heute ist nun endlich geklärt, in welcher Indikation MOR106 eingesetzt werden soll. Bei der atopischen Dermatitis handelt es sich zweifellos um eine Indikation mit großem medizinischen Bedarf und entsprechendem wirtschaftlichen Potential. Ein Hautentwicklungskonkurrent für MOR106 wird der Anti-IL4-Rezeptor-ɑ-Antikörper Dupilumab von Sanofi/Gegeneron sein, der die Signalwege von IL 4 und IL 13 hemmt und sich bereits in klinischen Studien bewährt hat.
Antwort auf Beitrag Nr.: 53.198.817 von Joschka Schröder am 03.09.16 11:19:50
Sorry, aber hier muss ich dir widersprechen. Morphosys stellt in der Regel mehrere Kandidaten gegen das vom Partner gestellte Target her. Für die Auswahl des optimalen Kandidaten ist dann der Partner zuständig. Also auch für die Bestimmung der optimalen Bindungsstellen und Bindungsstärke usw. - hier also Novartis.
Da der in der Veröffentlichung der Uni Würzburg angesprochene AK auch nur einer von 10 zur Auswahl gestellten ist, hätte dir das aber eigentlich auffallen müssen.
Zitat von Joschka Schröder: Ironie der Geschichte, dass gerade AbDSerotec in die Entwicklung neuer anti-sclerostin-Antikörper involviert ist, nachdem man bei BPS804 offenbar nicht die optimale Bindungsstelle getroffen hat ...
https://www.uni-wuerzburg.de/sonstiges/meldungen/single/arti…
Sorry, aber hier muss ich dir widersprechen. Morphosys stellt in der Regel mehrere Kandidaten gegen das vom Partner gestellte Target her. Für die Auswahl des optimalen Kandidaten ist dann der Partner zuständig. Also auch für die Bestimmung der optimalen Bindungsstellen und Bindungsstärke usw. - hier also Novartis.
Da der in der Veröffentlichung der Uni Würzburg angesprochene AK auch nur einer von 10 zur Auswahl gestellten ist, hätte dir das aber eigentlich auffallen müssen.
Antwort auf Beitrag Nr.: 53.378.304 von Milestones am 29.09.16 21:08:15Deinen Kommentar kann ich ehrlich gesagt nicht ganz nachvollziehen. Der jeweilige Auftraggeber kann immer nur eine Auswahl aus denjenigen Antikörpern treffen, die ihm vom Vertragspartner zur Verfügung gestellt worden worden sind. Novartis hat ja nicht irgendeine bestimmte Aminosäurequenz bei Morphosys in Auftrag gegeben. Ich glaube, Du hast da möglicherweise falsche Vorstellungen von der Antikörperentwicklung. Davon unabhängig handelt es in jedem Fall um eine Ironie der Geschichte, dass AbDSerotec wieder in die Entwicklung von anti-sclerostin-Mabs involviert ist.
Antwort auf Beitrag Nr.: 53.379.216 von Joschka Schröder am 29.09.16 23:29:06
Das glaube ich eher weniger... Wie dem auch sei, wenn MIR etwas nicht 100%-ig gefiele, was mein Dienstleister liefert, dann ließe ICH ihn weiterarbeiten... Aber Novartis ist da sicherlich kulanter. Sie haben ja auch nur die Kriterien festgelegt... Da kann man schon mal ein Auge zudrücken.
Zitat von Joschka Schröder: Deinen Kommentar kann ich ehrlich gesagt nicht ganz nachvollziehen. Der jeweilige Auftraggeber kann immer nur eine Auswahl aus denjenigen Antikörpern treffen, die ihm vom Vertragspartner zur Verfügung gestellt worden worden sind. Novartis hat ja nicht irgendeine bestimmte Aminosäurequenz bei Morphosys in Auftrag gegeben. Ich glaube, Du hast da möglicherweise falsche Vorstellungen von der Antikörperentwicklung. Davon unabhängig handelt es in jedem Fall um eine Ironie der Geschichte, dass AbDSerotec wieder in die Entwicklung von anti-sclerostin-Mabs involviert ist.
Das glaube ich eher weniger... Wie dem auch sei, wenn MIR etwas nicht 100%-ig gefiele, was mein Dienstleister liefert, dann ließe ICH ihn weiterarbeiten... Aber Novartis ist da sicherlich kulanter. Sie haben ja auch nur die Kriterien festgelegt... Da kann man schon mal ein Auge zudrücken.
Zu den Ergebnissen der Voyage1-Studie:
Positiv ist schon mal, dass die Ergebnisse der X-Plore-Vorgängerstudie übertroffen worden sind, wozu sicherlich auch das optimierte Dosisregime beigetragen hat.
Voyage1 (Phase 3):
PASI 90 nach 16 Wochen: 73,3 % Guselkumab*, 49,7 % Humira**, 2,9 % Placebo
X-Plore (Phase 2b):
PASI 90 nach 16 Wochen: 57 % bzw. 62 % Guselkumab***, 44 % Humira**, 2 % Placebo
* Guselkumab: 100 mg Woche 0, 4 und danach alle 8 Wochen
** Humira 80 mg initial, danach 40 mg wöchentlich
*** Guselkumab: 200 mg Woche 0, 4 und danach 100 mg alle 12 Wochen bzw. 100 mg alle 8 Wochen
Nach aktuellem Sachstand wird Johnson & Johnson das auf längere Sicht nicht mehr konkurrenzfähige Stelara durch Guselkumab ersetzen.
Einen detaillierten Vergleich mit zahlreichen anderen Entwicklungs- und Konkurrenzpräparaten werde ich später nachreichen.
Positiv ist schon mal, dass die Ergebnisse der X-Plore-Vorgängerstudie übertroffen worden sind, wozu sicherlich auch das optimierte Dosisregime beigetragen hat.
Voyage1 (Phase 3):
PASI 90 nach 16 Wochen: 73,3 % Guselkumab*, 49,7 % Humira**, 2,9 % Placebo
X-Plore (Phase 2b):
PASI 90 nach 16 Wochen: 57 % bzw. 62 % Guselkumab***, 44 % Humira**, 2 % Placebo
* Guselkumab: 100 mg Woche 0, 4 und danach alle 8 Wochen
** Humira 80 mg initial, danach 40 mg wöchentlich
*** Guselkumab: 200 mg Woche 0, 4 und danach 100 mg alle 12 Wochen bzw. 100 mg alle 8 Wochen
Nach aktuellem Sachstand wird Johnson & Johnson das auf längere Sicht nicht mehr konkurrenzfähige Stelara durch Guselkumab ersetzen.
Einen detaillierten Vergleich mit zahlreichen anderen Entwicklungs- und Konkurrenzpräparaten werde ich später nachreichen.
Besonders erfreulich fällt der indirekte Vergleich mit den Consentyx-Phase-3-Daten aus (Consentyx = Secukinumab = anti-IL17A-Mab von Novartis).
Auch unter Berücksichtigung des Umstands, dass die Consentyx-Daten nach 12 Wochen erhoben worden sind (statt nach 16 Wochen wie bei Guselkumab), scheint Guselkumab im PASI 90-Vergleich ein ganzes Stück besser anzuschneiden.
Die Auswertung der Secukinumab-Studien bereits nach 12 Wochen relativiert sich infolge der unterschiedlichen Dosisschemata, Secukinumab z.B. Injektionen in Woche 0, 1, 2, 3, 4 und danach alle 4 Wochen, Guselkumab hingegen nur in Woche 0, 4 und danach alle 8 Wochen. Bei einer späteren Vermarktung ist die seltene Guselkumab-Darreichung natürlich von Vorteil.
PASI 90 nach 16 Wochen:
73,3 % Guselkumab, 49,7 % Humira, 2,9 % Placebo
PASI 90 nach 12 Wochen:
59,2 % (300 mg Secukinumab), 59,2 % (150 mg Secukinumab) und 1,2 % (Placebo) in der ERASURE-Studie
45 % (300 mg Secukinumab), 40 % (150 mg Secukinumab) und 0 % (Placebo) in der JUNCTURE-Studie
54,2 % (300 mg Secukinumab), 41,9 % (150 mg Secukinumab) und 20,7 % (Eternacept!) in der FIXTURE-Studie
Der Vergleich mit Humira, Secukinumab und Eternacept fällt für Guselkumab also sehr erfreulich aus.
An dieser Stelle stoppe ich die vergleichende Darstellung erst einmal, weil die Analystengemeinde auch etwas selbstständig erarbeiten sollte. Morgen stelle ich die restlichen Vergleichsdaten ins Netz, wobei natürlich auch andere Antikörpersegmente bzw. Targets berücksichtigt werden sollen.
Die guten Daten der VOYAGE 1-Studie sollten dem Morphosys-Kurs zunächst einmal ordentlich Auftrieb verleihen.
Auch unter Berücksichtigung des Umstands, dass die Consentyx-Daten nach 12 Wochen erhoben worden sind (statt nach 16 Wochen wie bei Guselkumab), scheint Guselkumab im PASI 90-Vergleich ein ganzes Stück besser anzuschneiden.
Die Auswertung der Secukinumab-Studien bereits nach 12 Wochen relativiert sich infolge der unterschiedlichen Dosisschemata, Secukinumab z.B. Injektionen in Woche 0, 1, 2, 3, 4 und danach alle 4 Wochen, Guselkumab hingegen nur in Woche 0, 4 und danach alle 8 Wochen. Bei einer späteren Vermarktung ist die seltene Guselkumab-Darreichung natürlich von Vorteil.
PASI 90 nach 16 Wochen:
73,3 % Guselkumab, 49,7 % Humira, 2,9 % Placebo
PASI 90 nach 12 Wochen:
59,2 % (300 mg Secukinumab), 59,2 % (150 mg Secukinumab) und 1,2 % (Placebo) in der ERASURE-Studie
45 % (300 mg Secukinumab), 40 % (150 mg Secukinumab) und 0 % (Placebo) in der JUNCTURE-Studie
54,2 % (300 mg Secukinumab), 41,9 % (150 mg Secukinumab) und 20,7 % (Eternacept!) in der FIXTURE-Studie
Der Vergleich mit Humira, Secukinumab und Eternacept fällt für Guselkumab also sehr erfreulich aus.
An dieser Stelle stoppe ich die vergleichende Darstellung erst einmal, weil die Analystengemeinde auch etwas selbstständig erarbeiten sollte. Morgen stelle ich die restlichen Vergleichsdaten ins Netz, wobei natürlich auch andere Antikörpersegmente bzw. Targets berücksichtigt werden sollen.
Die guten Daten der VOYAGE 1-Studie sollten dem Morphosys-Kurs zunächst einmal ordentlich Auftrieb verleihen.
Antwort auf Beitrag Nr.: 53.396.844 von Joschka Schröder am 03.10.16 11:52:48Vielen Dank Joschka für deine Mühe und die Ausführungen!
Antwort auf Beitrag Nr.: 53.396.184 von Joschka Schröder am 03.10.16 09:43:41
Stelara/Ustekimumab war in head-to-head-Studien den Antikörpern Secukinumab (anti-IL-17A) und Brodalumab (anti-IL-17A-R) deutlich unterlegen, Johnson&Johnson wird also voll auf Guselkumab setzen, mit Stelara wird man langfristig keinen Blumentopf mehr gewinnen.
Zitat von Joschka Schröder: Nach aktuellem Sachstand wird Johnson & Johnson das auf längere Sicht nicht mehr konkurrenzfähige Stelara durch Guselkumab ersetzen.
Stelara/Ustekimumab war in head-to-head-Studien den Antikörpern Secukinumab (anti-IL-17A) und Brodalumab (anti-IL-17A-R) deutlich unterlegen, Johnson&Johnson wird also voll auf Guselkumab setzen, mit Stelara wird man langfristig keinen Blumentopf mehr gewinnen.
Antwort auf Beitrag Nr.: 53.396.844 von Joschka Schröder am 03.10.16 11:52:48Es muss natürlich Cosentyx heißen, nicht Consentyx (wobei ich "Consentyx" deutlich origineller finde)
Antwort auf Beitrag Nr.: 53.396.844 von Joschka Schröder am 03.10.16 11:52:48
Wieso ist denn bei Secukinumab bei den drei Studien eine Streuung von 14% (59,2 zu 45) bei 300 mg und 19,2% (59,2 zu 40) bei 150 mg? Finde ich ziemlich viel Unterschied.
Und sind 73,3 % zu 59,2 % bei vier Wochen längerer Behandlung wirklich so viel besser?
Zitat von Joschka Schröder: PASI 90 nach 16 Wochen:
73,3 % Guselkumab, 49,7 % Humira, 2,9 % Placebo
PASI 90 nach 12 Wochen:
59,2 % (300 mg Secukinumab), 59,2 % (150 mg Secukinumab) und 1,2 % (Placebo) in der ERASURE-Studie
45 % (300 mg Secukinumab), 40 % (150 mg Secukinumab) und 0 % (Placebo) in der JUNCTURE-Studie
54,2 % (300 mg Secukinumab), 41,9 % (150 mg Secukinumab) und 20,7 % (Eternacept!) in der FIXTURE-Studie.
Wieso ist denn bei Secukinumab bei den drei Studien eine Streuung von 14% (59,2 zu 45) bei 300 mg und 19,2% (59,2 zu 40) bei 150 mg? Finde ich ziemlich viel Unterschied.
Und sind 73,3 % zu 59,2 % bei vier Wochen längerer Behandlung wirklich so viel besser?
Antwort auf Beitrag Nr.: 53.397.354 von hinz12 am 03.10.16 13:38:02Vielen Dank, dass Du die Zahlen so aufmerksam gelesen hast: Ich hatte mich vertippt! Es muss heißen
55 % (300 mg Secukinumab), 40 % (150 mg Secukinumab) und 0 % (Placebo) in der JUNCTURE-Studie
Die Ergebnisse der drei Phase-3-Studien sind weitgehend konsistent!
55 % (300 mg Secukinumab), 40 % (150 mg Secukinumab) und 0 % (Placebo) in der JUNCTURE-Studie
Die Ergebnisse der drei Phase-3-Studien sind weitgehend konsistent!
Nur noch kurz, weil ich leider wenig Zeit habe:
Guselkumabs Konkurrenzpräparat Tildrakizumab, das ebenfalls an IL-23p19 bindet, dürfte aus dem Rennen ausscheiden. Sun Pharma hat aktuell zum EADV-Meeting über Phase-3-Daten berichtet, die nicht konkurrenzfähig scheinen, auch wenn man von Unternehmensseite die Ergebnisse als Erfolg preist. Sun Pharma hat das Präparat im vorletzten Jahr von Merck/MSD einlizensiert und ist kürzlich für den europäischen Markt eine Allianz mit Amirall eingegangen. Merck hatte offenbar frühzeitig Gründe, den Antikörper nicht selbst weiterzuentwickeln. Weitere Details dann morgen.
Guselkumabs Konkurrenzpräparat Tildrakizumab, das ebenfalls an IL-23p19 bindet, dürfte aus dem Rennen ausscheiden. Sun Pharma hat aktuell zum EADV-Meeting über Phase-3-Daten berichtet, die nicht konkurrenzfähig scheinen, auch wenn man von Unternehmensseite die Ergebnisse als Erfolg preist. Sun Pharma hat das Präparat im vorletzten Jahr von Merck/MSD einlizensiert und ist kürzlich für den europäischen Markt eine Allianz mit Amirall eingegangen. Merck hatte offenbar frühzeitig Gründe, den Antikörper nicht selbst weiterzuentwickeln. Weitere Details dann morgen.
Antwort auf Beitrag Nr.: 53.397.450 von Joschka Schröder am 03.10.16 13:52:27
Bitte :-)
Ist denn die Zeile
59,2 % (300 mg Secukinumab), 59,2 % (150 mg Secukinumab) und 1,2 % (Placebo) in der ERASURE-Studie
richtig? Das kann doch eigentlich auch nicht sein, dass die Wirksamkeit von 300 mg und 150 mg gleich?
Zitat von Joschka Schröder: Vielen Dank, dass Du die Zahlen so aufmerksam gelesen hast: Ich hatte mich vertippt! Es muss heißen
55 % (300 mg Secukinumab), 40 % (150 mg Secukinumab) und 0 % (Placebo) in der JUNCTURE-Studie
Die Ergebnisse der drei Phase-3-Studien sind weitgehend konsistent!
Bitte :-)
Ist denn die Zeile
59,2 % (300 mg Secukinumab), 59,2 % (150 mg Secukinumab) und 1,2 % (Placebo) in der ERASURE-Studie
richtig? Das kann doch eigentlich auch nicht sein, dass die Wirksamkeit von 300 mg und 150 mg gleich?
Antwort auf Beitrag Nr.: 53.398.662 von hinz12 am 03.10.16 17:27:05Doch, das stimmt so. Ein Flüchtigkeitsfehler reicht.
Antwort auf Beitrag Nr.: 53.398.815 von Joschka Schröder am 03.10.16 17:47:33Okay
Antwort auf Beitrag Nr.: 53.398.815 von Joschka Schröder am 03.10.16 17:47:33Aber dann bleibt die Frage nach der Streuung von 19,2%. Wie kann das sein?
Antwortest du noch auf meine PM?
Antwortest du noch auf meine PM?
Antwort auf Beitrag Nr.: 53.399.025 von hinz12 am 03.10.16 18:19:14Du meinst den PASI 90-Außreißer in der 150 mg-Gruppe der ERASURE-Studie? Ich weiß es nicht. Das war mir heute auch schon aufgefallen, deshalb meine schnelle Antwort vorhin.
Die ERASURE-Daten hatte ich ursprünglich aus einer eigentlich zuverlässigen Psoriasis-Datenbank in meine eigene eigene Studien-Datenbank übernommen. Wenn mehr Zeit ist, schau ich aber gerne nochmal in der Originalarbeit nach, die müßte auch irgendwo hier herumliegen.
Deine Frage per BM werde ich auch später beantworten. Bitte erinnere mich einfach daran, falls ich es vergesse.
Die ERASURE-Daten hatte ich ursprünglich aus einer eigentlich zuverlässigen Psoriasis-Datenbank in meine eigene eigene Studien-Datenbank übernommen. Wenn mehr Zeit ist, schau ich aber gerne nochmal in der Originalarbeit nach, die müßte auch irgendwo hier herumliegen.
Deine Frage per BM werde ich auch später beantworten. Bitte erinnere mich einfach daran, falls ich es vergesse.
Antwort auf Beitrag Nr.: 53.400.573 von Joschka Schröder am 03.10.16 22:45:34Genau den Ausreißer bei 150 mg in der Erasure-Studie meine ich. Sieht komisch aus.
Hi Joschka,
vielen Dank fuer deine analytischen Beitraege. Wie immer sehr erhellend...
Du haeltst Risankizumab fuer ueberlegen zu guselkumab. Meines Wissens gab es bisher erst phase 2 ergebnisse von Risankizumab, bitte korrigieren falls nicht korrekt.
Jnj hat m.e. drei Vorteile gegenueber diesem kuenftigen Mitbewerber:
1. Man ist ein gewaltiger platzhirsch in ra und psoriasis. Der Marketingmuskel wird das vielleicht schlechtere produkt mglw besser verkaufen
2. Bietet jnj eine klare wechselstrategie an mit NAVIGATE um die stelara "ausscheider" gleich auf guselkumab zu "navigieren" -sofern NAVIGATE erfolgreich ist
3. Man hat einen zeitlichen vorsprung zu Risankizumab
Ich bin zudem sehr gespannt darauf deine aktuelle Einschaetzung zu MOR 202 zu lesen. Fuer mich ist der AK ja ein sehr wahrscheinlicher Einstellungskandidat wegen mangelnder Wirksamkeit. Die geringeren infusion reactions und die schonung von nk zellen sind fuer mich nebelbomben um den kapitalmarkt bei laune zu halten.
vielen Dank fuer deine analytischen Beitraege. Wie immer sehr erhellend...
Du haeltst Risankizumab fuer ueberlegen zu guselkumab. Meines Wissens gab es bisher erst phase 2 ergebnisse von Risankizumab, bitte korrigieren falls nicht korrekt.
Jnj hat m.e. drei Vorteile gegenueber diesem kuenftigen Mitbewerber:
1. Man ist ein gewaltiger platzhirsch in ra und psoriasis. Der Marketingmuskel wird das vielleicht schlechtere produkt mglw besser verkaufen
2. Bietet jnj eine klare wechselstrategie an mit NAVIGATE um die stelara "ausscheider" gleich auf guselkumab zu "navigieren" -sofern NAVIGATE erfolgreich ist
3. Man hat einen zeitlichen vorsprung zu Risankizumab
Ich bin zudem sehr gespannt darauf deine aktuelle Einschaetzung zu MOR 202 zu lesen. Fuer mich ist der AK ja ein sehr wahrscheinlicher Einstellungskandidat wegen mangelnder Wirksamkeit. Die geringeren infusion reactions und die schonung von nk zellen sind fuer mich nebelbomben um den kapitalmarkt bei laune zu halten.
Bei all der Euphorie in Bezug auf die guten Werte von Guselkumab, Risankizumab und dem seit kurzem am Markt befindlichen cosentyx, stellt sich die Realität der Psoriasis-Behandlung momentan folgendermaßen dar:
Zuerst wird mal mit topischen Therapeutika wie Daivonex und Psorcutan therapiert, bei weiter vorhandenen Beschwerden kommt Methotrexat hinzu.
In weniger entwickelten Ländern und bei älteren Hautärzten spielt die PUVA-(Strahlen)-Therapie eine Rolle.
In Deutschland wird oft erst nach Jahren der Schwenk auf die teuren TNF-Antagonisten oder Interleukin-Therapeutika aus Kostengründen vollzogen.
Schon jetzt haben wir auch im Biosimilar-Bereich ernstzunehmende preisliche Alternativen, deren Originatormoleküle am Markt gut eingeführt sind, und deren Sicherheitsprofil den verschreibenden Ärzten bekannt ist (Infliximab/Remicade, Etanercept/Enbrel, möglicherweise bald Adalimumab/Humira)
Im Einzelfall werden diese schon mit einem Discount zum Original von über 70% angeboten (Norwegen).
Aus meiner Sicht werden sich reifere biosimilars in Richtung von commodity-Biotechnologika entwickeln...d.h. "Massenware" zu einem günstigen Preis.
Ab ca. 2024 kommt das erste Interleukin12/23-Biosimilar Ustekinumab hinzu, welches über ein ausgeprägtes Sicherheitsprofil, bis sogar zu Anwendung in der Pädiatrie, verfügt.
Diese preisgünstigen Therapeutika werden aus meiner Sicht erst einmal die nach der Methotrexat-Anwendung folgende Therapieoption aus ökonomischen Gründen sein.
Erst nach deren "Therapieversagen" oder unzureichender Wirksamkeit wird man auf die neueren MABs mit höherem PASI-Wert zurückgreifen.
Wieviel Rest-Umsatz das dann für vielleicht 3 neue Wirkstoffe mit ungeklärterem Nebenwirkungsprofil ist, wird sich zeigen.
Positiv für das Marktpotential vieler Interleukin-Antagonisten dürfte wiederum die Indikationsausweitung auf chronische Polyarthritis und entzündliche Magen-Darmkrankheiten sein (Morbus Crohn, Colitis ulcerosa).
Zuerst wird mal mit topischen Therapeutika wie Daivonex und Psorcutan therapiert, bei weiter vorhandenen Beschwerden kommt Methotrexat hinzu.
In weniger entwickelten Ländern und bei älteren Hautärzten spielt die PUVA-(Strahlen)-Therapie eine Rolle.
In Deutschland wird oft erst nach Jahren der Schwenk auf die teuren TNF-Antagonisten oder Interleukin-Therapeutika aus Kostengründen vollzogen.
Schon jetzt haben wir auch im Biosimilar-Bereich ernstzunehmende preisliche Alternativen, deren Originatormoleküle am Markt gut eingeführt sind, und deren Sicherheitsprofil den verschreibenden Ärzten bekannt ist (Infliximab/Remicade, Etanercept/Enbrel, möglicherweise bald Adalimumab/Humira)
Im Einzelfall werden diese schon mit einem Discount zum Original von über 70% angeboten (Norwegen).
Aus meiner Sicht werden sich reifere biosimilars in Richtung von commodity-Biotechnologika entwickeln...d.h. "Massenware" zu einem günstigen Preis.
Ab ca. 2024 kommt das erste Interleukin12/23-Biosimilar Ustekinumab hinzu, welches über ein ausgeprägtes Sicherheitsprofil, bis sogar zu Anwendung in der Pädiatrie, verfügt.
Diese preisgünstigen Therapeutika werden aus meiner Sicht erst einmal die nach der Methotrexat-Anwendung folgende Therapieoption aus ökonomischen Gründen sein.
Erst nach deren "Therapieversagen" oder unzureichender Wirksamkeit wird man auf die neueren MABs mit höherem PASI-Wert zurückgreifen.
Wieviel Rest-Umsatz das dann für vielleicht 3 neue Wirkstoffe mit ungeklärterem Nebenwirkungsprofil ist, wird sich zeigen.
Positiv für das Marktpotential vieler Interleukin-Antagonisten dürfte wiederum die Indikationsausweitung auf chronische Polyarthritis und entzündliche Magen-Darmkrankheiten sein (Morbus Crohn, Colitis ulcerosa).
Antwort auf Beitrag Nr.: 53.408.469 von Ville7 am 04.10.16 20:39:00
Vielen Dank, das Kompliment gebe ich gerne zurück. Da mir aktuell etwas die Arbeit über den Kopf wächst, nur schnell etwas ins Unreine.
Im Grundsatz sehen wir die Dinge genauso, nur im Detail differiert der Blickwinkel:
zu Punkt 1: Das stimmt im Vergleich zu fast allen anderen Wettbewerbern, bzgl. Risankizumab nicht unbedingt, weil AbbVie im Autoimmunsektor der größte Platzhirsch ist.
zu Punkt 2: Zustimmung
zu Punkt 3: Zustimmung
Zur Frage der Überlegenheit. Bzgl. Risankizumab liegen in der Tat nur wenige Studiendaten vor, die es allerdings in sich haben: Mit nur 2 Injektionen packt Risankizumab nach 12 Wochen 81 % PASI 90 und 50 % PASI 100.
Guselkumab schafft mit 3 Injektionen (in die VOYAGE-1-Studie ist extra eine zusätzliche Zwischendosis integriert worden) nach 16 Wochen 73 % PASI 90. Die zusätzlichen vier Wochen Beobachtungszeit führen zu Verzerrungen!
Ich habe mir extra noch einmal den Anhang der Originalpublikation zur Guselkumab-Phase-II-Studie angesehen. Dort sieht man, dass durch die vier zusätzlichen Beobachtungswochen (Wochen 12 bis 16) der PASI 90-Wert um 12 % größer ausfällt, als dies bei einer 12-Wochen-Auswertung der Fall gewesen wäre. Man kann dies natürlich nicht 1:1 auf die VOYAGE 1-Studie übertragen, die Tendenz ist jedoch kalr: Die Ergebnisse der VOYAGE 1-Studie sind positiv überzeichnet.
Vor diesem Hintergrund kann man - natürlich unter dem Vorbehalt einer späteren Bestätigung in P3 - schon sagen, dass Risankizumab bei der Drei- oder Viermonatsbewertung ein ganzes Stück besser abschneidet als Guselkumab. Hohe PASI-Werte sind insoweit wichtig, als dadurch der teure Einsatz von Biotherapeutika optimiert werden kann (hoher Prozentwert verspricht höhere Erfolgswahrscheinlichkeit).
Richtig ist aber auch, dass es für den Patienten letztlich entscheidend ist, nachhaltig therapiert zu werden. Von einem Verschwinden der Symptomatik für einen Zeitraum von einigen Monaten mit anschließendem Wiederaufflackern der Symptome kann sich niemand etwas kaufen. Entscheidend ist die Nachhaltigkeit der Heilung ... und unter diesem Aspekt ist die Datenlage bei beiden Präparaten, insbesondere aber bei Risankizumab (bei Guselkumab gibt es wenigstens 24 Wochen-Daten) bislang einfach zu dürftig, um sich ein Urteil bilden zu können.
Per Saldo besitzt Risankizumab auf Basis der aktuellen Datenlage eine Favoritenstellung, für eine endgültige Beurteilung bedarf es aber deutlich mehr Daten aus längeren Beobachtungszeiträumen. Wäre diese Zusammenfassung für Dich ok?
Zu MOR202: Ich habe nach dem diesjährigen ASCO-Meeting noch einmal Studiendaten der verschiedenen anti-CD38-Antikörper verglichen. Dabei ist mir aufgefallen, dass bei MOR202 zwar deutlich weniger Infusionsreaktionen auftreten, dass aber - was aber viel kritischer ist - viel häufiger dann im Therapieverlauf schwere Nebenwirkungen Grad 3 und 4 zu beobachten sind. Aus dem Kopf möchte ich dies aber lieber nicht quantifizieren. Wird nachgeholt, wenn ich mehr Zeit habe.
Zitat von Ville7: Hi Joschka,
vielen Dank fuer deine analytischen Beitraege. Wie immer sehr erhellend...
Du haeltst Risankizumab fuer ueberlegen zu guselkumab. Meines Wissens gab es bisher erst phase 2 ergebnisse von Risankizumab, bitte korrigieren falls nicht korrekt.
Jnj hat m.e. drei Vorteile gegenueber diesem kuenftigen Mitbewerber:
1. Man ist ein gewaltiger platzhirsch in ra und psoriasis. Der Marketingmuskel wird das vielleicht schlechtere produkt mglw besser verkaufen
2. Bietet jnj eine klare wechselstrategie an mit NAVIGATE um die stelara "ausscheider" gleich auf guselkumab zu "navigieren" -sofern NAVIGATE erfolgreich ist
3. Man hat einen zeitlichen vorsprung zu Risankizumab
Ich bin zudem sehr gespannt darauf deine aktuelle Einschaetzung zu MOR 202 zu lesen. Fuer mich ist der AK ja ein sehr wahrscheinlicher Einstellungskandidat wegen mangelnder Wirksamkeit. Die geringeren infusion reactions und die schonung von nk zellen sind fuer mich nebelbomben um den kapitalmarkt bei laune zu halten.
Vielen Dank, das Kompliment gebe ich gerne zurück. Da mir aktuell etwas die Arbeit über den Kopf wächst, nur schnell etwas ins Unreine.
Im Grundsatz sehen wir die Dinge genauso, nur im Detail differiert der Blickwinkel:
zu Punkt 1: Das stimmt im Vergleich zu fast allen anderen Wettbewerbern, bzgl. Risankizumab nicht unbedingt, weil AbbVie im Autoimmunsektor der größte Platzhirsch ist.
zu Punkt 2: Zustimmung
zu Punkt 3: Zustimmung
Zur Frage der Überlegenheit. Bzgl. Risankizumab liegen in der Tat nur wenige Studiendaten vor, die es allerdings in sich haben: Mit nur 2 Injektionen packt Risankizumab nach 12 Wochen 81 % PASI 90 und 50 % PASI 100.
Guselkumab schafft mit 3 Injektionen (in die VOYAGE-1-Studie ist extra eine zusätzliche Zwischendosis integriert worden) nach 16 Wochen 73 % PASI 90. Die zusätzlichen vier Wochen Beobachtungszeit führen zu Verzerrungen!
Ich habe mir extra noch einmal den Anhang der Originalpublikation zur Guselkumab-Phase-II-Studie angesehen. Dort sieht man, dass durch die vier zusätzlichen Beobachtungswochen (Wochen 12 bis 16) der PASI 90-Wert um 12 % größer ausfällt, als dies bei einer 12-Wochen-Auswertung der Fall gewesen wäre. Man kann dies natürlich nicht 1:1 auf die VOYAGE 1-Studie übertragen, die Tendenz ist jedoch kalr: Die Ergebnisse der VOYAGE 1-Studie sind positiv überzeichnet.
Vor diesem Hintergrund kann man - natürlich unter dem Vorbehalt einer späteren Bestätigung in P3 - schon sagen, dass Risankizumab bei der Drei- oder Viermonatsbewertung ein ganzes Stück besser abschneidet als Guselkumab. Hohe PASI-Werte sind insoweit wichtig, als dadurch der teure Einsatz von Biotherapeutika optimiert werden kann (hoher Prozentwert verspricht höhere Erfolgswahrscheinlichkeit).
Richtig ist aber auch, dass es für den Patienten letztlich entscheidend ist, nachhaltig therapiert zu werden. Von einem Verschwinden der Symptomatik für einen Zeitraum von einigen Monaten mit anschließendem Wiederaufflackern der Symptome kann sich niemand etwas kaufen. Entscheidend ist die Nachhaltigkeit der Heilung ... und unter diesem Aspekt ist die Datenlage bei beiden Präparaten, insbesondere aber bei Risankizumab (bei Guselkumab gibt es wenigstens 24 Wochen-Daten) bislang einfach zu dürftig, um sich ein Urteil bilden zu können.
Per Saldo besitzt Risankizumab auf Basis der aktuellen Datenlage eine Favoritenstellung, für eine endgültige Beurteilung bedarf es aber deutlich mehr Daten aus längeren Beobachtungszeiträumen. Wäre diese Zusammenfassung für Dich ok?
Zu MOR202: Ich habe nach dem diesjährigen ASCO-Meeting noch einmal Studiendaten der verschiedenen anti-CD38-Antikörper verglichen. Dabei ist mir aufgefallen, dass bei MOR202 zwar deutlich weniger Infusionsreaktionen auftreten, dass aber - was aber viel kritischer ist - viel häufiger dann im Therapieverlauf schwere Nebenwirkungen Grad 3 und 4 zu beobachten sind. Aus dem Kopf möchte ich dies aber lieber nicht quantifizieren. Wird nachgeholt, wenn ich mehr Zeit habe.
Antwort auf Beitrag Nr.: 53.408.469 von Ville7 am 04.10.16 20:39:00
Ein wesentlicher Grund, wieso J&J quasi gezwungen ist, Guselkumab gegen Stelara zu testen, liegt darin, dass sowohl Risankizumab als auch Brodalumab und Secukinumab in head-to-head-Studien Stelara deutlich geschlagen haben. Sobald die Gusel-Stelara-Vergleichstudie vorliegt, werden Quervergleiche weitere Informationen darüber liefern, wie in etwa Guselkumab im Konkurrenzkampf positioniert sein wird. Fakt ist, dass der Wettbewerb im Psoriasismarkt sehr hart werden wird, wozu u.a. auch die von StefanR erwähnten Umstände beitragen werden.
Zitat von Ville7: 2. Bietet jnj eine klare wechselstrategie an mit NAVIGATE um die stelara "ausscheider" gleich auf guselkumab zu "navigieren" -sofern NAVIGATE erfolgreich ist
Ein wesentlicher Grund, wieso J&J quasi gezwungen ist, Guselkumab gegen Stelara zu testen, liegt darin, dass sowohl Risankizumab als auch Brodalumab und Secukinumab in head-to-head-Studien Stelara deutlich geschlagen haben. Sobald die Gusel-Stelara-Vergleichstudie vorliegt, werden Quervergleiche weitere Informationen darüber liefern, wie in etwa Guselkumab im Konkurrenzkampf positioniert sein wird. Fakt ist, dass der Wettbewerb im Psoriasismarkt sehr hart werden wird, wozu u.a. auch die von StefanR erwähnten Umstände beitragen werden.
Die aktuellen MOR202-Daten bieten wenig Neues. Im Vergleich zu den beim ASCO-Meeting präsentierten Daten hat es in der 8 mg/kg-POM/DEX-Gruppe eine Veschiebung von MR zu PR gegeben und in der 8 mg/lg-LEN/DEX-Gruppe eine Verschiebung von SD zu MR.
Sorge bereiten die ausgeprägten Nebenwirkungen größer gleich Grad 3 (in den Kombigruppen jeweils bei 100 % der Patienten). Da schneidet beispielsweise Daratumumab viel besser ab. Ein Vergleich: In Monotherapie sind bei 84 % der mit MOR202 behandelten Patienten derart schwere NW aufgetreten, im Daratumumab-Kollektiv nur bei ca. 46 % (davon fast alle Grad 3), obwohl die Dara-Patienten stärker vorbehandelt (= kranker) waren und die MOR202-Patienten zusätzlich zur Symptomlinderung DEX erhalten haben! Da hilft es wenig, dass der Infusionsvorgang bei MOR202 offenbar besser verträglich ist, was von Morphosys immer etwas einseitig herausgestellt wird (hier wäre allerdings auch noch zu klären, in welchem Umfang die DEX-Gabe zur besseren Verträglichkeit beitgetragen hat).
Sorge bereiten die ausgeprägten Nebenwirkungen größer gleich Grad 3 (in den Kombigruppen jeweils bei 100 % der Patienten). Da schneidet beispielsweise Daratumumab viel besser ab. Ein Vergleich: In Monotherapie sind bei 84 % der mit MOR202 behandelten Patienten derart schwere NW aufgetreten, im Daratumumab-Kollektiv nur bei ca. 46 % (davon fast alle Grad 3), obwohl die Dara-Patienten stärker vorbehandelt (= kranker) waren und die MOR202-Patienten zusätzlich zur Symptomlinderung DEX erhalten haben! Da hilft es wenig, dass der Infusionsvorgang bei MOR202 offenbar besser verträglich ist, was von Morphosys immer etwas einseitig herausgestellt wird (hier wäre allerdings auch noch zu klären, in welchem Umfang die DEX-Gabe zur besseren Verträglichkeit beitgetragen hat).
Antwort auf Beitrag Nr.: 53.491.312 von Joschka Schröder am 17.10.16 10:39:11Danke für eine Einschätzung in deinem Thread. Die Wirksamkeit scheint inzwischen nahezu auf Augenhöhe zu sein, nur die Sicherheit könnte in der Tat ein Problem darstellen. Nebenwirkungen, die ursächlich in MOR202 zu suchen sind...
Antwort auf Beitrag Nr.: 53.491.594 von Ville7 am 17.10.16 11:43:39Chance wäre einzig noch in der Duration of response zu finden, aber hier ist Dara in Kombi bereits hervorragend unterwegs... und wenn die Patienten wegen Nebenwirkungen vorher von der Studie genommen werden (viele SD wurden vor PD abgebrochen, teils auch PRs) wird das nichts: komisch z.B., wenn ein Patient direkt nach einer PR aus der Studie ausscheidet, das deutet auf derartige Probleme hin:

Antwort auf Beitrag Nr.: 53.410.197 von Joschka Schröder am 05.10.16 02:50:30
Ne direkte Vergleichsstudie (head-to-head) ist das meines Wissens aber nicht. In NAVIGATE soll lediglich geprüft werden, wie weit die Stelara-Non- oder low-responders auf Gusel ansprechen... oder hab ich das falsch in Erinnerung?
Zitat von Joschka Schröder: Ein wesentlicher Grund, wieso J&J quasi gezwungen ist, Guselkumab gegen Stelara zu testen, liegt darin, dass sowohl Risankizumab als auch Brodalumab und Secukinumab in head-to-head-Studien Stelara deutlich geschlagen haben. Sobald die Gusel-Stelara-Vergleichstudie vorliegt, werden Quervergleiche weitere Informationen darüber liefern, wie in etwa Guselkumab im Konkurrenzkampf positioniert sein wird. Fakt ist, dass der Wettbewerb im Psoriasismarkt sehr hart werden wird, wozu u.a. auch die von StefanR erwähnten Umstände beitragen werden.
Ne direkte Vergleichsstudie (head-to-head) ist das meines Wissens aber nicht. In NAVIGATE soll lediglich geprüft werden, wie weit die Stelara-Non- oder low-responders auf Gusel ansprechen... oder hab ich das falsch in Erinnerung?
Die Art, wie die Psoriasis-Vergleichsstudien konzipiert wurden, sagt vor allem etwas über die Pharmaindustrie aus. JnJ testet gegen den Blockbuster von AbbVie (Adalimumab), die anderen gegen den von JnJ (Ustekinumab). JnJ testet Guselkumab jetzt noch für die Patienten, bei denen ihr Ustekinumab nicht funktioniert. Es geht natürlich bei dieser Konzeption sehr viel um Marketing.
Interessant finde ich übrigens auch, was derzeit so verschrieben wird, offenbar sogar noch viel Etanercept, was von allen Biologicals bei Psoriasis am schlechtesten wirkt. Ich glaube, wir können hier lange analysieren, bei welchem Medikament der PASI-90 am höchsten ist und ob Risankizumab nicht noch ein bisschen besser ist als Guselkumab. Ich glaube nicht, dass das für die Marktaufteilung von allerhöchster Relevanz ist. Mal abgesehen davon, dass es keine Head to Head Vergleiche gibt (die Diskussion über die Quervergleiche möchte ich nicht wieder aufwärmen, aber man kann es ja an z.T. recht großen Unterschieden im Abschneiden des gleichen Medikamentes sehen, Bsp Secukinumab). Verträglichkeit, Art und Häufigkeit der Verabreichung und nicht zuletzt das Marketing (traurig, aber wahr) sind m.E. mindestens genauso wichtig. Die IL 17 A Antikörper und die IL 23p19 Antikörper sind halt alle sehr gut wirksam. Ich hab vor allem wegen der starken Position von JnJ keine größeren Zweifel, dass Guselkuman nach Zulassung sehr schnell zum Blockbuster wird. Und da nehme ich gerne Wetten drauf an.
Interessant finde ich übrigens auch, was derzeit so verschrieben wird, offenbar sogar noch viel Etanercept, was von allen Biologicals bei Psoriasis am schlechtesten wirkt. Ich glaube, wir können hier lange analysieren, bei welchem Medikament der PASI-90 am höchsten ist und ob Risankizumab nicht noch ein bisschen besser ist als Guselkumab. Ich glaube nicht, dass das für die Marktaufteilung von allerhöchster Relevanz ist. Mal abgesehen davon, dass es keine Head to Head Vergleiche gibt (die Diskussion über die Quervergleiche möchte ich nicht wieder aufwärmen, aber man kann es ja an z.T. recht großen Unterschieden im Abschneiden des gleichen Medikamentes sehen, Bsp Secukinumab). Verträglichkeit, Art und Häufigkeit der Verabreichung und nicht zuletzt das Marketing (traurig, aber wahr) sind m.E. mindestens genauso wichtig. Die IL 17 A Antikörper und die IL 23p19 Antikörper sind halt alle sehr gut wirksam. Ich hab vor allem wegen der starken Position von JnJ keine größeren Zweifel, dass Guselkuman nach Zulassung sehr schnell zum Blockbuster wird. Und da nehme ich gerne Wetten drauf an.
Auf MOR 202 ist manches davon übertragbar, bloß halt eher im negativen Sinne. Kann sein, dass es in der Kombination genauso gut wie Dara wirkt. Aber Dara ist halt schon im Markt, das ist schwer aufzuholen. Man bräuchte 1. mindestens vergleichbare Wirksamkeit 2. gute Verträglichkeit (hab die Daten noch nicht angeschaut, aber Joschka scheint skeptisch) und 3. einen Partner mit einer sehr starken Position für die Vermarktung. Momentan scheint keiner der Punkte eindeutig erfüllt, a.e. noch Punkt 1 (Wer hätte das gedacht?). Für Punkt 3 wäre Celgene optimal gewesen. Alleine hat man keine Chance.
Antwort auf Beitrag Nr.: 53.491.718 von Ville7 am 17.10.16 12:03:58@Ville zu Beitrag 568: Vielen Dank für den Hinweis, Du hast völlig recht. Ich hatte das falsch in Erinnerung. Zunächst erfolgt eine open-label-Periode mit Stelara. Switch zu Guselkumab nur bei inadaequater Antwort auf Stelara.
@deadflowers zu Beitrag 569: Auch hier volle Zustimmung. Letztlich werden wohl die Vertriebsmannschaften über den Erfolg entscheiden. Und dort sind J&J, AbbVie und Novartis mit am besten positioniert.
Schade, dass die Zulassungsbehörden (koordiniert auf internationaler Ebene) keine großangelegten head-to-head-Studien fordern.
@deadflowers zu Beitrag 569: Auch hier volle Zustimmung. Letztlich werden wohl die Vertriebsmannschaften über den Erfolg entscheiden. Und dort sind J&J, AbbVie und Novartis mit am besten positioniert.
Schade, dass die Zulassungsbehörden (koordiniert auf internationaler Ebene) keine großangelegten head-to-head-Studien fordern.
Antwort auf Beitrag Nr.: 53.491.312 von Joschka Schröder am 17.10.16 10:39:11
Mein Gott, das habe ich schlampig formuliert. Gemeint war, dass in beiden Dosisgruppen jeweils ein Patient eine bessere Response erzielt hat als bislang bekannt.
Zitat von Joschka Schröder: Die aktuellen MOR202-Daten bieten wenig Neues. Im Vergleich zu den beim ASCO-Meeting präsentierten Daten hat es in der 8 mg/kg-POM/DEX-Gruppe eine Veschiebung von MR zu PR gegeben und in der 8 mg/lg-LEN/DEX-Gruppe eine Verschiebung von SD zu MR.
Mein Gott, das habe ich schlampig formuliert. Gemeint war, dass in beiden Dosisgruppen jeweils ein Patient eine bessere Response erzielt hat als bislang bekannt.
Biogen hat ja heute gute Zahlen vorgelegt. Beim lesen der nachfolgen Veröffentlichung war ich doch ein bisschen überrascht, dass Biogen sich mit ihrer Entwicklung bei Aducanumab anscheinend sehr gute Chancen auf eine baldige Zulassung ausrechnet bzw. darauf hofft.
CAMBRIDGE (dpa-AFX) - Gute Verkäufe mit Multiple-Sklerose-Mitteln verleihen dem Biotech-Konzern Biogen weiterhin Schwung. Im dritten Quartal zogen die Umsätze um 6 Prozent auf rund 3 Milliarden US-Dollar an, wie der US-Konzern am Mittwoch mitteilte. Der Gewinn legte um 7 Prozent auf eine Milliarde Dollar zu. Dies war mehr als Analysten erwartet hatten.
Der Kassenschlager Tecfidera brachte im Quartal zehn Prozent mehr Umsatz ein. Damit schlug sich das Medikament besser als von Experten gedacht. Allerdings gingen die Erlöse des MS-Mittels außerhalb der USA im Vergleich mit dem Vorquartal zurück. Tecfidera ist der wichtigste Umsatzbringer von Biogen, er steht allerdings wegen starker Konkurrenzprodukte unter Druck. Der Konzern steckt daher viel Geld in die Erforschung anderer Krankheiten wie etwa Alzheimer und hofft auf eine baldige Zulassung in diesem Bereich.[/red] Offen ist weiterhin, wer Biogen in Zukunft führen wird. Noch-Chef George Scangos hatte im Sommer angekündigt, er bleibe nur noch so lange an Bord, bis ein geeigneter Nachfolger für ihn gefunden ist./she/men/stb
Quelle: http://www.finanznachrichten.de/nachrichten-2016-10/38972794…
CAMBRIDGE (dpa-AFX) - Gute Verkäufe mit Multiple-Sklerose-Mitteln verleihen dem Biotech-Konzern Biogen weiterhin Schwung. Im dritten Quartal zogen die Umsätze um 6 Prozent auf rund 3 Milliarden US-Dollar an, wie der US-Konzern am Mittwoch mitteilte. Der Gewinn legte um 7 Prozent auf eine Milliarde Dollar zu. Dies war mehr als Analysten erwartet hatten.
Der Kassenschlager Tecfidera brachte im Quartal zehn Prozent mehr Umsatz ein. Damit schlug sich das Medikament besser als von Experten gedacht. Allerdings gingen die Erlöse des MS-Mittels außerhalb der USA im Vergleich mit dem Vorquartal zurück. Tecfidera ist der wichtigste Umsatzbringer von Biogen, er steht allerdings wegen starker Konkurrenzprodukte unter Druck. Der Konzern steckt daher viel Geld in die Erforschung anderer Krankheiten wie etwa Alzheimer und hofft auf eine baldige Zulassung in diesem Bereich.[/red] Offen ist weiterhin, wer Biogen in Zukunft führen wird. Noch-Chef George Scangos hatte im Sommer angekündigt, er bleibe nur noch so lange an Bord, bis ein geeigneter Nachfolger für ihn gefunden ist./she/men/stb
Quelle: http://www.finanznachrichten.de/nachrichten-2016-10/38972794…
Hallo zusammen,
http://www.fool.com/investing/2016/10/30/will-this-trend-cos…
Vielleicht ein interessanter Artikel über die aktuelle und zukünftige Bezahlung
der Pharma- und Biotechbranche...
Tenor: Nur was wirklich wirkt, wird auch sehr gut bezahlt.
Gruß
http://www.fool.com/investing/2016/10/30/will-this-trend-cos…
Vielleicht ein interessanter Artikel über die aktuelle und zukünftige Bezahlung
der Pharma- und Biotechbranche...
Tenor: Nur was wirklich wirkt, wird auch sehr gut bezahlt.
Gruß
MorphoSys AG: MorphoSys und LEO Pharma vereinbaren strategische Allianz zur Entwicklung therapeutischer Antikörper gegen Hauterkrankungen
Die MorphoSys AG (Frankfurt: MOR; Prime Standard Segment, TecDAX, OTC: MPSYY) und LEO Pharma gaben heute den Start einer strategischen Allianz für die Erforschung und Entwicklung therapeutischer Antikörper zur Behandlung von Hautkrankheiten bekannt. Das Ziel der Partnerschaft ist es, neue antikörperbasierte Therapeutika zur Behandlung von Krankheiten mit bislang ungedecktem medizinischen Bedarf zu identifizieren, die eine wertvolle Ergänzung für die Entwicklungspipelines beider Unternehmen darstellen sollen.
Die beiden Partner werden gemeinsam neue therapeutische Antikörper gegen mehrere, von LEO Pharma ausgewählte, Zielmoleküle identifizieren, validieren und entwickeln. MorphoSys wird seine firmeneigene Ylanthia-Technologieplattform einsetzen, um vollständig humane Antikörperkandidaten gegen die ausgewählten Zielmoleküle zu erzeugen. Zudem wird MorphoSys die Entwicklungsaktivitäten bis zum Beginn der klinischen Prüfung durchführen. LEO Pharma wird für die klinische Entwicklung und Vermarktung der entstandenen Wirkstoffe in allen Indikationen, ausgenommen Krebs, verantwortlich sein. Bei Hautkrebsindikationen hat MorphoSys die Möglichkeit, die Antikörperwirkstoffe klinisch mit zu entwickeln und in Europa mit zu vermarkten. Darüber hinaus wird MorphoSys die Möglichkeit haben, therapeutische Programme aus dieser Zusammenarbeit in anderen Krebsindikationen zu entwickeln und zu vermarkten.
MorphoSys wird Zahlungen für Forschung und Entwicklung erhalten sowie erfolgsbasierte entwicklungs-, zulassungs- und vermarktungsbezogene Meilensteinzahlungen. Zudem wird MorphoSys aus der späteren Vermarktung der Medikamente Tantiemen auf Basis der Nettoverkaufserlöse erhalten. Vorausgesetzt alle Entwicklungs-, Zulassungs- und Umsatzziele werden erreicht, könnten die Meilensteinzahlungen bis zu insgesamt 111,5 Mio. Euro pro Antikörperprogramm erreichen.
"Wir freuen uns sehr über diese strategische Allianz mit LEO Pharma, um gemeinsam neue antikörperbasierte Medikamente zu entwickeln", kommentierte Dr. Simon Moroney, Vorstandsvorsitzender der MorphoSys AG. "Die Kombination von LEO Pharmas umfassender Kenntnis in der Dermatologie und unserer Antikörperexpertise ermöglicht die Entwicklung für neue Produkte in einem Bereich mit einem hohen ungedeckten medizinischen Bedarf. Diese neue Partnerschaft ist ein weiterer wichtiger Schritt in der Umsetzung unserer Strategie, auf Basis unserer Expertise eine starke Pipeline innovativer therapeutischer Antikörper aufzubauen."
"Wir sehen spannende Möglichkeiten, um auf Basis der strategischen Allianz mit MorphoSys neue Therapien zur Behandlung von Menschen mit Hauterkrankungen zu entwickeln", sagte Gitte Aabo, CEO und President von LEO Pharma. "Durch die gemeinsame Erforschung und Entwicklung neuer Antikörperwirkstoffe wollen wir für Menschen mit Hauterkrankungen Bereiche mit einem hohen ungedeckten medizinischen Bedarf erschließen. Diese Partnerschaft untermauert unser Ziel, unser therapeutisches Angebot zu erweitern und unsere Position bei Wirkstoffen auf biologischer Basis innerhalb der Dermatologie zu stärken."
...
http://www.finanznachrichten.de/nachrichten-2016-11/39045233…
Die MorphoSys AG (Frankfurt: MOR; Prime Standard Segment, TecDAX, OTC: MPSYY) und LEO Pharma gaben heute den Start einer strategischen Allianz für die Erforschung und Entwicklung therapeutischer Antikörper zur Behandlung von Hautkrankheiten bekannt. Das Ziel der Partnerschaft ist es, neue antikörperbasierte Therapeutika zur Behandlung von Krankheiten mit bislang ungedecktem medizinischen Bedarf zu identifizieren, die eine wertvolle Ergänzung für die Entwicklungspipelines beider Unternehmen darstellen sollen.
Die beiden Partner werden gemeinsam neue therapeutische Antikörper gegen mehrere, von LEO Pharma ausgewählte, Zielmoleküle identifizieren, validieren und entwickeln. MorphoSys wird seine firmeneigene Ylanthia-Technologieplattform einsetzen, um vollständig humane Antikörperkandidaten gegen die ausgewählten Zielmoleküle zu erzeugen. Zudem wird MorphoSys die Entwicklungsaktivitäten bis zum Beginn der klinischen Prüfung durchführen. LEO Pharma wird für die klinische Entwicklung und Vermarktung der entstandenen Wirkstoffe in allen Indikationen, ausgenommen Krebs, verantwortlich sein. Bei Hautkrebsindikationen hat MorphoSys die Möglichkeit, die Antikörperwirkstoffe klinisch mit zu entwickeln und in Europa mit zu vermarkten. Darüber hinaus wird MorphoSys die Möglichkeit haben, therapeutische Programme aus dieser Zusammenarbeit in anderen Krebsindikationen zu entwickeln und zu vermarkten.
MorphoSys wird Zahlungen für Forschung und Entwicklung erhalten sowie erfolgsbasierte entwicklungs-, zulassungs- und vermarktungsbezogene Meilensteinzahlungen. Zudem wird MorphoSys aus der späteren Vermarktung der Medikamente Tantiemen auf Basis der Nettoverkaufserlöse erhalten. Vorausgesetzt alle Entwicklungs-, Zulassungs- und Umsatzziele werden erreicht, könnten die Meilensteinzahlungen bis zu insgesamt 111,5 Mio. Euro pro Antikörperprogramm erreichen.
"Wir freuen uns sehr über diese strategische Allianz mit LEO Pharma, um gemeinsam neue antikörperbasierte Medikamente zu entwickeln", kommentierte Dr. Simon Moroney, Vorstandsvorsitzender der MorphoSys AG. "Die Kombination von LEO Pharmas umfassender Kenntnis in der Dermatologie und unserer Antikörperexpertise ermöglicht die Entwicklung für neue Produkte in einem Bereich mit einem hohen ungedeckten medizinischen Bedarf. Diese neue Partnerschaft ist ein weiterer wichtiger Schritt in der Umsetzung unserer Strategie, auf Basis unserer Expertise eine starke Pipeline innovativer therapeutischer Antikörper aufzubauen."
"Wir sehen spannende Möglichkeiten, um auf Basis der strategischen Allianz mit MorphoSys neue Therapien zur Behandlung von Menschen mit Hauterkrankungen zu entwickeln", sagte Gitte Aabo, CEO und President von LEO Pharma. "Durch die gemeinsame Erforschung und Entwicklung neuer Antikörperwirkstoffe wollen wir für Menschen mit Hauterkrankungen Bereiche mit einem hohen ungedeckten medizinischen Bedarf erschließen. Diese Partnerschaft untermauert unser Ziel, unser therapeutisches Angebot zu erweitern und unsere Position bei Wirkstoffen auf biologischer Basis innerhalb der Dermatologie zu stärken."
...
http://www.finanznachrichten.de/nachrichten-2016-11/39045233…
Leo Pharma, nie gehört. Obwohl anscheinend 5000 Mitarbeiter weltweit und machen nur etwas über 1 Mrd Umsatz. Sortiere ich mal in die dritte Reihe Pharmas ein.
Was aber umso erstaunlicher ist, ist der Strategiewechsel bei Morphosys. Solche Verträge wollte man doch eigentlich nicht mehr machen, da hatte man die Nase ganz oben. Meine Lesung dazu war stets eine andere und die sehe ich bestätigt: Man hat einfach keinen top-pharma mehr mit Ylanthia an Land ziehen können und nun, nachdem man finanziell doch nicht so auf sicheren Beinen steht (nachdem bimagrumab in SBIM gescheitert ist) rudert man wieder zurück und nimmt auch die Kleineren Dinge wieder mit.
Was aber umso erstaunlicher ist, ist der Strategiewechsel bei Morphosys. Solche Verträge wollte man doch eigentlich nicht mehr machen, da hatte man die Nase ganz oben. Meine Lesung dazu war stets eine andere und die sehe ich bestätigt: Man hat einfach keinen top-pharma mehr mit Ylanthia an Land ziehen können und nun, nachdem man finanziell doch nicht so auf sicheren Beinen steht (nachdem bimagrumab in SBIM gescheitert ist) rudert man wieder zurück und nimmt auch die Kleineren Dinge wieder mit.
Antwort auf Beitrag Nr.: 53.601.657 von Ville7 am 02.11.16 08:25:42So ähnlich war auch mein erster Gedanke. Aber erstaunlich ist doch, dass Morphosys es geschafft hat, mit einer Technologie, die "keiner mehr will", seine üblichen Milestones mehr als zu verzehnfachen!
Entwicklung des Nasdaq Biotech Index nach den US-Wahlen
Antwort auf Beitrag Nr.: 53.601.954 von HK12 am 02.11.16 08:48:16Interessantes Muster. Sollte dann aber eher heißen "Entwicklung des Nasdaq Biotech Index vor den US-Wahlen", denn die Wahlen finden m.E. immer im November statt.
Antwort auf Beitrag Nr.: 53.601.789 von Milestones am 02.11.16 08:35:06Dafür ist davon auszugehen, dass die künftigen Tantiemen auch deutlich geringer sein werden. Ingesamt gehe ich davon aus, dass der Deal pro Anikörper nicht annähernd an die Konditionen heranreicht, die früher mit big pharma abgeschlossen werden konnten.
Antwort auf Beitrag Nr.: 53.601.657 von Ville7 am 02.11.16 08:25:42
Ja, das war mir auch durch den Kopf gegangen. Grundsätzlich ist der (abermalige) Strategiewechsel aber zu begrüßen. Eine kleine Änderung im Vergleich zu früher scheint es ja zu geben: "Darüber hinaus wird MorphoSys die Möglichkeit haben, therapeutische Programme aus dieser Zusammenarbeit in anderen Krebsindikationen zu entwickeln und zu vermarkten".
Diese Passage war/ist mir allerdings etwas unklar. Die Allianz bezieht sich explizit auf Hauterkrankungen und bei Hautkrebsindikationen hat MorphoSys die Möglichkeit, die Antikörperwirkstoffe klinisch mit zu entwickeln und in Europa mit zu vermarkten. Welche darüber hinaus gehende Krebsformen können im Kontext von Hauterkrankungen gemeint sein?
Zitat von Ville7: Was aber umso erstaunlicher ist, ist der Strategiewechsel bei Morphosys. Solche Verträge wollte man doch eigentlich nicht mehr machen, da hatte man die Nase ganz oben. Meine Lesung dazu war stets eine andere und die sehe ich bestätigt: Man hat einfach keinen top-pharma mehr mit Ylanthia an Land ziehen können und nun, nachdem man finanziell doch nicht so auf sicheren Beinen steht (nachdem bimagrumab in SBIM gescheitert ist) rudert man wieder zurück und nimmt auch die Kleineren Dinge wieder mit.
Ja, das war mir auch durch den Kopf gegangen. Grundsätzlich ist der (abermalige) Strategiewechsel aber zu begrüßen. Eine kleine Änderung im Vergleich zu früher scheint es ja zu geben: "Darüber hinaus wird MorphoSys die Möglichkeit haben, therapeutische Programme aus dieser Zusammenarbeit in anderen Krebsindikationen zu entwickeln und zu vermarkten".
Diese Passage war/ist mir allerdings etwas unklar. Die Allianz bezieht sich explizit auf Hauterkrankungen und bei Hautkrebsindikationen hat MorphoSys die Möglichkeit, die Antikörperwirkstoffe klinisch mit zu entwickeln und in Europa mit zu vermarkten. Welche darüber hinaus gehende Krebsformen können im Kontext von Hauterkrankungen gemeint sein?
Antwort auf Beitrag Nr.: 53.603.355 von Joschka Schröder am 02.11.16 11:27:06Also ich verstehe das so, dass Morphosys
1. bei Hautkrebsindikationen entscheiden darf, ob sie mit entwickeln und in Europa vermarkten.
2. Wenn der Antikörper auch gegen andere Krebsarten zum Beispiel Lungen-, Prostatakrebs oder was auch immer taugt, dann darf Morphosys auch komplett selbstständig entwickeln und vermarkten.
Und 3. wenn der Antikörper gegen Hautkrankheiten, die nicht Krebs sind, also zum Beispiel gegen Schuppenflechte hilft, dann entwickelt und vermarktet LEO alleine.
Ich finds auch gut, mal wieder so eine Allianz einzugehen, sollten Morphosys wieder mehr von eingehen.
1. bei Hautkrebsindikationen entscheiden darf, ob sie mit entwickeln und in Europa vermarkten.
2. Wenn der Antikörper auch gegen andere Krebsarten zum Beispiel Lungen-, Prostatakrebs oder was auch immer taugt, dann darf Morphosys auch komplett selbstständig entwickeln und vermarkten.
Und 3. wenn der Antikörper gegen Hautkrankheiten, die nicht Krebs sind, also zum Beispiel gegen Schuppenflechte hilft, dann entwickelt und vermarktet LEO alleine.
Ich finds auch gut, mal wieder so eine Allianz einzugehen, sollten Morphosys wieder mehr von eingehen.
Antwort auf Beitrag Nr.: 53.603.355 von Joschka Schröder am 02.11.16 11:27:06Ich verstehe das so, dass Morphosys Hautkrebsindikationen mit entwickeln und in Europa vermarkten darf. Sollte es Targets für diese Hautkrebsindikationen geben, die auch in nicht-Hautkrebsindikationen greifen, dann darf Morphosys diese entwickeln und vermarkten.
Unklar ist mir aber bei diesem Strategiewechsel die Target-Exklusivität, falls es weitere Deals geben wird. Wenn Leo Pharma Target XY angeht und ein weiterer Partner (okay gibt es bisher noch nicht) dann dieses Target auch haben möchte, was dann?
Unklar ist mir aber bei diesem Strategiewechsel die Target-Exklusivität, falls es weitere Deals geben wird. Wenn Leo Pharma Target XY angeht und ein weiterer Partner (okay gibt es bisher noch nicht) dann dieses Target auch haben möchte, was dann?
Antwort auf Beitrag Nr.: 53.603.481 von hinz12 am 02.11.16 11:43:47Hi, habe deinen post zu spät gesehen, deckt sich mit meinem verständnis... grüße
Antwort auf Beitrag Nr.: 53.603.481 von hinz12 am 02.11.16 11:43:47Ja, das ist richtig, so muss man es wohl verstehen.
Also das mit Hautkrebs verstehe ich gar nicht in der Meldung. Entweder geht es um harmlose lokale Tumore (LEO schreiben "non-melanoma skin cancer" als Indikationsgebiet auf Ihrer HP). Die soll man bitte schön rausschneiden, liegt an der Oberfläche, kleiner Eingriff, kaum Folgen für den Patienten und das einzig wirklich kurative Vorgehen bei fast allen Krebsarten. Oder es geht um metastasierte Tumore, dann sprechen wir von einer systemischen Onkologie-Behandlung. Nur für zweiteres macht es Sinn, einen Antikörper zu verwenden - der Ansatz von LEO scheint mir das aber nicht einzuschliessen, das klingt eher nach rundum gesunder Haut als Zielvorgabe.
Via Twitter:
Trinity Delta @trinity_delta 7 Std.Vor 7 Stunden
Positive news for @MorphoSys, MOR208 loses a major competitor in DLBCL as @AstraZeneca focuses MED-551 on neuromyelitis; $MOR shares up 1.4%
Trinity Delta @trinity_delta 7 Std.Vor 7 Stunden
Positive news for @MorphoSys, MOR208 loses a major competitor in DLBCL as @AstraZeneca focuses MED-551 on neuromyelitis; $MOR shares up 1.4%
Was sagen die Experten zu den gestrigen Gusel-Daten?
Antwort auf Beitrag Nr.: 53.123.550 von Joschka Schröder am 24.08.16 08:18:31Richtig ist nun auf Recruiting.
Könnte RTH258 Brolucizumab
evt. NOV 7 oder 9 sein ?
https://clinicaltrials.gov/ct2/results?term=RTH258&pg=1
http://adisinsight.springer.com/drugs/800033756
evt. NOV 7 oder 9 sein ?
https://clinicaltrials.gov/ct2/results?term=RTH258&pg=1
http://adisinsight.springer.com/drugs/800033756
Antwort auf Beitrag Nr.: 53.816.820 von schnappi am 02.12.16 15:13:33
Nur dann, wenn Morphosys und Novartis zufällig entgangen ist, dass sich die Substanz bereits in Phase 3 befindet...
Zitat von schnappi: Könnte RTH258 Brolucizumab
evt. NOV 7 oder 9 sein ?
https://clinicaltrials.gov/ct2/results?term=RTH258&pg=1
http://adisinsight.springer.com/drugs/800033756
Nur dann, wenn Morphosys und Novartis zufällig entgangen ist, dass sich die Substanz bereits in Phase 3 befindet...

Antwort auf Beitrag Nr.: 53.820.384 von Milestones am 03.12.16 01:24:19Von Seiten Morphosys heißt es immer das Novartis bestimmt was sie melden dürfen, es ist auch kaum vorstellbar das von den nicht bekannten Projeketen bis jetzt keines eine weitere Phase erreicht haben soll wie auf der Hompage angegeben.
Infos über den AK findet man eine Menge nur aus welcher Quelle er stammt habe ich nicht gefunden.
Ich finde die recherche nach solchen Dingen aber viel interessanter als das ständige gejammer über den Kurs zu lesen.
Infos über den AK findet man eine Menge nur aus welcher Quelle er stammt habe ich nicht gefunden.
Ich finde die recherche nach solchen Dingen aber viel interessanter als das ständige gejammer über den Kurs zu lesen.
Antwort auf Beitrag Nr.: 53.816.820 von schnappi am 02.12.16 15:13:33Quelle gefunden http://www.esbatech.com/.
zu Mor208 von heute
https://www.morphosys.de/sites/default/files/downloads/16120…
https://www.morphosys.de/sites/default/files/downloads/16120…
Antwort auf Beitrag Nr.: 53.832.697 von schnappi am 05.12.16 23:12:25Zu viele Balken, Striche und Abkürzungen für mich Kann das jemand "lesen" oder "übersetzen"? 



MOR208 scheint in iNHL als single agent doch besser zu sein als in DLBCL. Trotzdem ist man in der pivotal Studie in DLBCL (in Kombi) unterwegs.
Was mich am meisten beeindruckt, ist die klare Signifikanz für besseres Ansprechen mit höherem NK cell count (und noch einen Tick besser für CD16). Das untermauert zum einen den mode of action und bietet vor allem ein sehr einfach durchzuführendes Stratifizierungsverfahren.
Ich schaue mir gerade die Präsentationen an und bin abseits des Fachlichen auf eine äußerst spannende Sache gestoßen:
Wenn man mit dem Cursor in der ASH-Präsentation der Morphosys AG über einzelne Begriffe fährt, z.B. um sie zu markieren, taucht regelmäßig der Link zu einem User namens ekki auf.
Beispiel: Cursor auf den Begriff "Biomarker" auf S.22 gesetzt, führt zum Link
C:UsersekkiDocumentsProPubMOREKKI208EKKI208MOR208_F16u18_Bars.png.
Zieht man den Cursor auf S.21 über die Abszisse der Graphik, so erscheint der Link
C:UsersekkiDocumentsProPubMOREKKI208EKKI208MOR208_F15_Subtypes.png.
Usw. usw.
Die Präsentation muss auf einem Computer erstellt worden sein, dessen user ein Ekki ist bzw. zu dessen Usern ein Ekki gehört.
Wie geht es eigentlich dem Wallstreet-Online-User Ekki (eck64), der viele Jahre lang in diesem Forum akribisch Morphosys besprochen und durchaus wertvolle (eigene) Graphiken zu/über Morphosys ins Netz gestellt hat? Seit langem hat man nichts mehr von ihm gehört. Hat Ekki jetzt auf anderem Weg mit Morphosys zu tun?
Wenn man mit dem Cursor in der ASH-Präsentation der Morphosys AG über einzelne Begriffe fährt, z.B. um sie zu markieren, taucht regelmäßig der Link zu einem User namens ekki auf.
Beispiel: Cursor auf den Begriff "Biomarker" auf S.22 gesetzt, führt zum Link
C:UsersekkiDocumentsProPubMOREKKI208EKKI208MOR208_F16u18_Bars.png.
Zieht man den Cursor auf S.21 über die Abszisse der Graphik, so erscheint der Link
C:UsersekkiDocumentsProPubMOREKKI208EKKI208MOR208_F15_Subtypes.png.
Usw. usw.
Die Präsentation muss auf einem Computer erstellt worden sein, dessen user ein Ekki ist bzw. zu dessen Usern ein Ekki gehört.

Wie geht es eigentlich dem Wallstreet-Online-User Ekki (eck64), der viele Jahre lang in diesem Forum akribisch Morphosys besprochen und durchaus wertvolle (eigene) Graphiken zu/über Morphosys ins Netz gestellt hat? Seit langem hat man nichts mehr von ihm gehört. Hat Ekki jetzt auf anderem Weg mit Morphosys zu tun?

Antwort auf Beitrag Nr.: 53.836.958 von Joschka Schröder am 06.12.16 15:53:10Bei den Links sind jeweils die Backslashes zu ergänzen. Die hat der Wallstreet-Online-Editor verschluckt.
Antwort auf Beitrag Nr.: 53.836.958 von Joschka Schröder am 06.12.16 15:53:10Moin,
eck64 hatte sich ja vor gar nicht so langer Zeit mal wieder gemeldet und liest bestimmt alles hier mit. Vielleicht finden wir ja am 02.01.17 wieder einen neuen Thread von ihm vor. Der Titel lautet dann in etwa:
Morphosys: Erste Zulassungen bringen Tantiemen
Gruß q.
eck64 hatte sich ja vor gar nicht so langer Zeit mal wieder gemeldet und liest bestimmt alles hier mit. Vielleicht finden wir ja am 02.01.17 wieder einen neuen Thread von ihm vor. Der Titel lautet dann in etwa:
Morphosys: Erste Zulassungen bringen Tantiemen
Gruß q.
Antwort auf Beitrag Nr.: 53.836.958 von Joschka Schröder am 06.12.16 15:53:10Schon interessant, aber da steht ja genaugenommen "Usersekki" . Wäre das dann nicht der -User sekki- ?
Von der Schreibweise mit "kk" mal ganz abgesehen.
Als "UsersekkiDocumentsProPubMOREKKI208.." Users Ekki Documents... könnte es ja mit dem s passen, aber ich glaub da eher an einen Zufall
-
Der Ak... ist weiter am Ball;
http://www.deraktionaer.de/aktie/morphosys-aktie-mit-kraefti…
Von der Schreibweise mit "kk" mal ganz abgesehen.
Als "UsersekkiDocumentsProPubMOREKKI208.." Users Ekki Documents... könnte es ja mit dem s passen, aber ich glaub da eher an einen Zufall
-
Der Ak... ist weiter am Ball;
http://www.deraktionaer.de/aktie/morphosys-aktie-mit-kraefti…
Antwort auf Beitrag Nr.: 53.837.027 von Joschka Schröder am 06.12.16 15:57:50Das hat absolut nichts mit dem User Eck64 zu tun. Reiner Zufall. Aber wenn dir sonst nichts zur Präsentation einfällt, dann kann es ja nichts besonderes gewesen sein!!?? Ich persönlich habe es mir noch nicht anhören können, da der Webcast noch nicht zum erneuten Abspielen archiviert war....
Antwort auf Beitrag Nr.: 53.838.053 von RichyBerlin am 06.12.16 17:40:44Nein, das ist ein ganz gewöhnlicher windows-Path
c: = Laufwerk
Users = Nutzer-Directory
ekki = Nutzername
Jetzt zum Fachlichen:
Die MOR208-Daten sind wirklich gut.
DoR in der Indikation r/r DLBCL 20,1 Monate (zum Vergleich: Venetoclax packt bei ähnlichem Patientenkollektiv gerade mal 3,3 Monate!), zwei ausgezeichnete, aussagekräftige Biomarker (CD16 und NK), gutes Ansprechen Retuximab-refraktärer Patienten, die allgemeinen Ansprechraten waren bereits bekannt.
Die MOR202-Wirkungsdaten haben sich weiter verbessert, in der POM/DEX-Kombi mit 91 % ORR, das ist ein unerwarteter Spitzenwert. Daratumumab bei ähnlich vorbehandeltem Patientenkollektiv mit 71 %. Negativ weiterhin das Nebenwirkungsprofil (Grad 3 und höher).
Auch wenn es für ein endgültiges Urteil natürlich zu früh ist, so kann man zum jetzigen Zeitpunkt schon feststellen, dass sich die Entscheidung, MOR202 alleine weiterzuentwickeln, als richtig erwiesen hat. Bzgl. MOR202 war ich, das muss ich klar zugeben, zu skeptisch.
Insgesamt eine sehr positive Entwicklung!
c: = Laufwerk
Users = Nutzer-Directory
ekki = Nutzername
Jetzt zum Fachlichen:
Die MOR208-Daten sind wirklich gut.
DoR in der Indikation r/r DLBCL 20,1 Monate (zum Vergleich: Venetoclax packt bei ähnlichem Patientenkollektiv gerade mal 3,3 Monate!), zwei ausgezeichnete, aussagekräftige Biomarker (CD16 und NK), gutes Ansprechen Retuximab-refraktärer Patienten, die allgemeinen Ansprechraten waren bereits bekannt.
Die MOR202-Wirkungsdaten haben sich weiter verbessert, in der POM/DEX-Kombi mit 91 % ORR, das ist ein unerwarteter Spitzenwert. Daratumumab bei ähnlich vorbehandeltem Patientenkollektiv mit 71 %. Negativ weiterhin das Nebenwirkungsprofil (Grad 3 und höher).
Auch wenn es für ein endgültiges Urteil natürlich zu früh ist, so kann man zum jetzigen Zeitpunkt schon feststellen, dass sich die Entscheidung, MOR202 alleine weiterzuentwickeln, als richtig erwiesen hat. Bzgl. MOR202 war ich, das muss ich klar zugeben, zu skeptisch.
Insgesamt eine sehr positive Entwicklung!
Antwort auf Beitrag Nr.: 53.840.465 von Joschka Schröder am 06.12.16 21:21:26Danke für deine Einschätzung zu den Daten!
Was zuletzt etwas untergegangen ist: Morphosys entwickelt neuerdings auch bispezifische Antikörper. Eine späte, aber gute Entscheidung. In den üblichen Patentdatenbanken war dazu in der vergangenen Woche allerdings noch nichts zu finden.
Antwort auf Beitrag Nr.: 53.840.465 von Joschka Schröder am 06.12.16 21:21:26
"Der Markt" sieht´s per Saldo wohl ähnlich, Xencor in Reaktion auf die MOR208-Daten aktuell +7 %.
Zitat von Joschka Schröder: Die MOR208-Daten sind wirklich gut.
DoR in der Indikation r/r DLBCL 20,1 Monate (zum Vergleich: Venetoclax packt bei ähnlichem Patientenkollektiv gerade mal 3,3 Monate!), zwei ausgezeichnete, aussagekräftige Biomarker (CD16 und NK), gutes Ansprechen Retuximab-refraktärer Patienten, die allgemeinen Ansprechraten waren bereits bekannt.
"Der Markt" sieht´s per Saldo wohl ähnlich, Xencor in Reaktion auf die MOR208-Daten aktuell +7 %.
Antwort auf Beitrag Nr.: 53.840.663 von Joschka Schröder am 06.12.16 21:49:31Danke für deine Einschätzung. MOR208 wird in der pivotal Studie die Zulassung locker schaffen. Nur kann ich da überhaupt nicht das Marktpotential (dann) in 2020ff einschätzen. Wie viel Kuchen-Anteil bleibt bei all den neuen Therapien überhaupt übrig für diese Kombi-Therapie? Und MOR202, die RR erscheinen schon gut, nur das NW Profil und dass man dann sehr spät am Markt wäre ist das Problem. Zudem würde ich mir einen Partner wünschen, aber ich befürchte auch MOR202 versuchen sie alleine durch die Phase 3 zu bringen...
Antwort auf Beitrag Nr.: 53.841.653 von Ville7 am 07.12.16 07:21:56
Da kannst du denke ich beruhigt sein. Wenn ich mich recht erinnere, hat Schottelius oder Holstein eine eigenständige Phase 3 in einem Interview vor kurzem ausgeschlossen. Ich weiß nicht mehr wann und in welchem Zusammenhang und habe jetzt auch nicht die Zeit zu suchen. Vielleicht findet es ja jemand (oder erinnert sich auch).
Zitat von Ville7: Und MOR202, die RR erscheinen schon gut, nur das NW Profil und dass man dann sehr spät am Markt wäre ist das Problem. Zudem würde ich mir einen Partner wünschen, aber ich befürchte auch MOR202 versuchen sie alleine durch die Phase 3 zu bringen...
Da kannst du denke ich beruhigt sein. Wenn ich mich recht erinnere, hat Schottelius oder Holstein eine eigenständige Phase 3 in einem Interview vor kurzem ausgeschlossen. Ich weiß nicht mehr wann und in welchem Zusammenhang und habe jetzt auch nicht die Zeit zu suchen. Vielleicht findet es ja jemand (oder erinnert sich auch).
Antwort auf Beitrag Nr.: 53.842.382 von Milestones am 07.12.16 08:54:54So habe ich es auch in Erinnerung, daß man alleine ein Phase 3 nicht stämmen kann.
Antwort auf Beitrag Nr.: 53.844.143 von schnappi am 07.12.16 11:21:01Danke für Euer Feedback...
Ob nach der jüngsten KE diese Aussage noch gilt?
Ich hoffe es sehr..
Ob nach der jüngsten KE diese Aussage noch gilt?
Ich hoffe es sehr..
Antwort auf Beitrag Nr.: 53.844.398 von Ville7 am 07.12.16 11:46:22Das ist richtig aber bis zu einer Phase 3 ist es ja auch noch Zeit.
Wo du gerade KE schreibst die war ja ein Armutszeugnis der begleiteten Banken den Kurs so abkacken zu lassen sollten sich mal ein Beispiel bei XNCR holen.
Wo du gerade KE schreibst die war ja ein Armutszeugnis der begleiteten Banken den Kurs so abkacken zu lassen sollten sich mal ein Beispiel bei XNCR holen.
Die vorgestellten Daten sind insgesamt überzeugend. MOR208 wird es zur Zulassung schaffen und seinen Platz finden. Es geht mir allerdings ähnlich wie ville: Mir ist überhaupt nicht klar, wie man sich positionieren will und was das an Umsatzpotential mit sich bringt. Grundsätzlich sind alle Indikationen denkbar, in denen CD20-Antikörper eingesetzt werden. Allerdings bräuchte man dann viel mehr Studien und vermutlich auch Head-to-head-Vergleiche, um sich möglicherweise als eine Art "besseres Rituximab" zu postionieren. Wenn man über die Nische hinaus will, geht das aber definitiv nicht ohne einen starken Partner.
Was MOR202 betrifft, bin ich nach wie vor etwas skeptisch, obwohl die Daten gut sind. Aber irgendwie muss man sich ja gegen die anderen CD38-Antikörper positionieren. Mit Celgene und deren Marktmacht hätte das eine interessante Sache werden können. Mir ist immer weniger klar, warum die sich zurückgezogen haben. Ich kann mir mittlerweile schon vorstellen, dass man die P3 alleine machen will. Es war ja schon mal angekündigt, dass 2017 eine P2/3 beginnen soll (Link Seite 13).
https://www.morphosys.de/sites/default/files/presentations/1… Es muss ja insgesamt schon schnell gehen, wenn man noch was reißen will. Lieber wäre es mir allemal, wenn man das nicht alleine macht.
Fakt ist: CD38 + Lenalidomid oder CD38 + Pomalidomid, jeweils + Dex, könnte eine Art Standard werden. Allerdings ist man nirgendwo der erste.
Vorerst ist das der Maßstab (Dara + Lenalidomid+Dex)
http://www.nejm.org/doi/full/10.1056/NEJMoa1607751
Sanofi macht die analoge Studie mit Pomalidomid, das hatte ville schon mal verlinkt
http://www.businesswire.com/news/home/20161205006021/en/Sano…
Ich bin gespannt, wie die P2/3 mit MOR202 konzipiert wird.
Insgesamt habe ich den Eindruck, dass das Sentiment für Morphosys sich gerade zum Positiven ändert und man vielleicht wieder eher die Chancen als die Risken sieht. Die Eigenpipeline sieht für mich jedenfalls alles andere als schlecht aus. Ich würde mir wünschen, dass man 2017/2018 starke Partner für MOR 208 und MOR 202 findet. Das könnte den Kurs ordentlich beflüglen.
Was MOR202 betrifft, bin ich nach wie vor etwas skeptisch, obwohl die Daten gut sind. Aber irgendwie muss man sich ja gegen die anderen CD38-Antikörper positionieren. Mit Celgene und deren Marktmacht hätte das eine interessante Sache werden können. Mir ist immer weniger klar, warum die sich zurückgezogen haben. Ich kann mir mittlerweile schon vorstellen, dass man die P3 alleine machen will. Es war ja schon mal angekündigt, dass 2017 eine P2/3 beginnen soll (Link Seite 13).
https://www.morphosys.de/sites/default/files/presentations/1… Es muss ja insgesamt schon schnell gehen, wenn man noch was reißen will. Lieber wäre es mir allemal, wenn man das nicht alleine macht.
Fakt ist: CD38 + Lenalidomid oder CD38 + Pomalidomid, jeweils + Dex, könnte eine Art Standard werden. Allerdings ist man nirgendwo der erste.
Vorerst ist das der Maßstab (Dara + Lenalidomid+Dex)
http://www.nejm.org/doi/full/10.1056/NEJMoa1607751
Sanofi macht die analoge Studie mit Pomalidomid, das hatte ville schon mal verlinkt
http://www.businesswire.com/news/home/20161205006021/en/Sano…
Ich bin gespannt, wie die P2/3 mit MOR202 konzipiert wird.
Insgesamt habe ich den Eindruck, dass das Sentiment für Morphosys sich gerade zum Positiven ändert und man vielleicht wieder eher die Chancen als die Risken sieht. Die Eigenpipeline sieht für mich jedenfalls alles andere als schlecht aus. Ich würde mir wünschen, dass man 2017/2018 starke Partner für MOR 208 und MOR 202 findet. Das könnte den Kurs ordentlich beflüglen.
Ich glaube, ihr macht euch um Verpartnerung zu viele Sorgen. Wenn die klinischen Daten wirklich gut sind, dann klopfen die Pharmas an und dann gibt es irgendwann Verträge. Auch wenn man nicht first-in-class ist, solange die Hoffnung besteht, dass man zumindest so gut oder besser als der Platzhirsch ist.
Joschka, ich glaube auch an keine Zufälle, vielleicht erzählt uns Eck ja irgendwann, ob er schon immer die Folien für MOR gemacht hat oder ob er sich für den Job erst durch sein WO-Engagement verdient hat ;-)
Joschka, ich glaube auch an keine Zufälle, vielleicht erzählt uns Eck ja irgendwann, ob er schon immer die Folien für MOR gemacht hat oder ob er sich für den Job erst durch sein WO-Engagement verdient hat ;-)
Antwort auf Beitrag Nr.: 53.859.029 von yok am 08.12.16 22:41:41
Das ist eine der wenigen schlüssigen Erklärungen für das Verbleiben des Users eck64. Man ändert ja nicht so einfach sein Postingverhalten, das zeigen ja auch die Versuche anderer Leute, die schreiben, sie schreiben jetzt mal längere Zeit nichts, wenn sie denn vorher sehr aktiv waren, dann kommen die schnell wieder.
Immerhin ist er ja jetzt dann Insider, da darf man nicht so einfach weiterschreiben wie vorher
Zitat von yok: Ich glaube, ihr macht euch um Verpartnerung zu viele Sorgen. Wenn die klinischen Daten wirklich gut sind, dann klopfen die Pharmas an und dann gibt es irgendwann Verträge. Auch wenn man nicht first-in-class ist, solange die Hoffnung besteht, dass man zumindest so gut oder besser als der Platzhirsch ist.
Joschka, ich glaube auch an keine Zufälle, vielleicht erzählt uns Eck ja irgendwann, ob er schon immer die Folien für MOR gemacht hat oder ob er sich für den Job erst durch sein WO-Engagement verdient hat ;-)
Das ist eine der wenigen schlüssigen Erklärungen für das Verbleiben des Users eck64. Man ändert ja nicht so einfach sein Postingverhalten, das zeigen ja auch die Versuche anderer Leute, die schreiben, sie schreiben jetzt mal längere Zeit nichts, wenn sie denn vorher sehr aktiv waren, dann kommen die schnell wieder.
Immerhin ist er ja jetzt dann Insider, da darf man nicht so einfach weiterschreiben wie vorher
Antwort auf Beitrag Nr.: 53.859.473 von riverstar_de am 09.12.16 02:04:22Es gibt andere Gründe, dass er nicht mehr schreibt. Nochmal: Eck64 hat damit nichts zu tun.
aducanumab zeigt, dass die a-beta hypothese nicht tot ist. aducanumab hat einen anderen angriffspunkt wie soleneuzumab und entfernt dadurch weit mehr plaque. Hinweise auf kognitive Verbesserungen im Vgl zu Plazebo inklusive. Moroney wies juengst darauf hin, dass gantenerumab denselben angriffspunkt hat. Fuer Roche, Biogen, ja selbst fuer Lilly ist diese These noch nicht tot. Die aducanumab phase 3 Ergebnisse werden es dann zeigen.
Das war heute auch noch was neues in der Meldung.
Darüber hinaus beabsichtigen wir, MOR208 in Kombination mit einem weiteren Krebsmedikament in dieser Patientengruppe zu erproben und hoffen, dazu in Kürze weitere Informationen veröffentlichen zu können.
Darüber hinaus beabsichtigen wir, MOR208 in Kombination mit einem weiteren Krebsmedikament in dieser Patientengruppe zu erproben und hoffen, dazu in Kürze weitere Informationen veröffentlichen zu können.
UPDATED: Eli Lilly shutters the last PhIII sola study, certain of failure
https://endpts.com/eli-lilly-shutters-the-last-phiii-sola-st…
https://endpts.com/eli-lilly-shutters-the-last-phiii-sola-st…
Merck’s leading PhIII BACE drug implodes in latest Alzheimer’s disaster
https://endpts.com/mercks-leading-phiii-bace-drug-implodes-i…
https://endpts.com/mercks-leading-phiii-bace-drug-implodes-i…
Roche confirms patient death in ACE910 PhIII hemophilia trial, spurring new questions about top blockbuster hopeful
https://endpts.com/roche-confirms-patient-death-in-ace910-ph…
https://endpts.com/roche-confirms-patient-death-in-ace910-ph…
Sugarbaker: Mesothelioma Clinical Trial May Be ‘Home Run’
https://www.asbestos.com/news/2016/10/05/david-sugarbaker-me…
https://www.asbestos.com/news/2016/10/05/david-sugarbaker-me…
aus dem Q4 Call von Bayer - die Rekrutierung von Anetumab ravtansine läuft viel schneller und besser:
Dieter Weinand - Bayer AG - Head, Pharmaceuticals
All right, so you know that I've been very excited about anetumab. And the much more rapid completion of the enrollment in the mesothelioma trial gives me confidence that the obvious physicians must be seeing something to speed up the enrollment so much faster.
Now you know we have initiated a multi-indication, a basket trial and six different tumor types that include certain non-small cell lung cancer, triple negative breast cancer, gastric cancer, thymic cancer, pancreatic cancer and cholangiocarcinoma. These trials, as you know you have to wait for some, first you have to see a signal.
The initiation for these trials is now underway.
And based on the signals we may see some of that signal in the time frame of 2018 that would allow us then to make some decisions on where to expand rapidly moving into a Phase II trial setting with registrational intent. So I would say we have to wait until we see these signals. That will probably be in the 2018 time frame before we can make further decisions on those.
You asked about the confirmation of the 2019 trial and I'm assuming that is the mesothelioma trial, the filing. My sense is that you are asking because we have completed enrollment so much faster than previously anticipated. This is an events-driven trial and we have to wait for the events to come in now and that will dictate our filing timeline.
So for now our timeline is unchanged for the first launch in 2019. Should that change we would update that. But we have to wait for the event.
Dieter Weinand - Bayer AG - Head, Pharmaceuticals
All right, so you know that I've been very excited about anetumab. And the much more rapid completion of the enrollment in the mesothelioma trial gives me confidence that the obvious physicians must be seeing something to speed up the enrollment so much faster.
Now you know we have initiated a multi-indication, a basket trial and six different tumor types that include certain non-small cell lung cancer, triple negative breast cancer, gastric cancer, thymic cancer, pancreatic cancer and cholangiocarcinoma. These trials, as you know you have to wait for some, first you have to see a signal.
The initiation for these trials is now underway.
And based on the signals we may see some of that signal in the time frame of 2018 that would allow us then to make some decisions on where to expand rapidly moving into a Phase II trial setting with registrational intent. So I would say we have to wait until we see these signals. That will probably be in the 2018 time frame before we can make further decisions on those.
You asked about the confirmation of the 2019 trial and I'm assuming that is the mesothelioma trial, the filing. My sense is that you are asking because we have completed enrollment so much faster than previously anticipated. This is an events-driven trial and we have to wait for the events to come in now and that will dictate our filing timeline.
So for now our timeline is unchanged for the first launch in 2019. Should that change we would update that. But we have to wait for the event.
Sollte am Montag wieder Richtung 53 Euro laufen.Hier passt Chart und Nachrichtenlage perfekt.
Kann eigentlich nur weiter nach oben laufen.......außer der Gesamtmarkt dreht.
Bei erfolgreicher Phase 3 sollte sich auch der Kurs 3 stellig entwickeln.
Kann eigentlich nur weiter nach oben laufen.......außer der Gesamtmarkt dreht.
Bei erfolgreicher Phase 3 sollte sich auch der Kurs 3 stellig entwickeln.
Neue Studie Anetumab Ravtansine plus Pembrolizumab (anti-PD-1 von Pfizer) in der Indikation "Mesotheliom" -> https://clinicaltrials.gov/ct2/show/NCT03126630?term=Anetuma…
Mir ist immer noch rätselhaft, wieso Novartis die Entwicklung des von Morphosys konstruierten anti-sclorostin-Antikörpers BPS804 eingestellt (bzw. für eine Nischenindikation auslizensiert) hat, wo doch die präklinischen Ergebnisse so hervorragend waren und auch in den ersten klinischen Untersuchungen (Einmalgabe) bestätigt worden sind.
Heute hatte ich endlich Gelegenheit, in der Novartis-Datenbank die Ergebnisse der Phase II-Studie mit BPS804 in der Indikation Osteoporose durchzusehen. Tatsächlich, auch hier finden sich in jeder Hinsicht absolut überzeugende Ergebnisse und keinerlei Nebenwirkungen, die Anlaß zur Besorgnis gäben. Ich bin so frei, die allgemein verständliche Zusammenfassung der Datenanalyse wörtlich wiederzugeben (hoffentlich schlägt jetzt nicht wieder der w:o-Löschalgorithmus zu ... Novartis hat die Daten freigegeben!):
This study was designed as a small, exploratory dosing regimen study and showed that BPS804 is well tolerated and effective in increasing bone mineral density in postmenopausal women with low bone mineral density. The short duration weekly dosing regimen showed a more rapid onset and greater increase in lumbar spine bone mineral density then the monthly and quarterly treatment. The study also showed that BPS804 is associated with rapid and substantial increases in serum levels of bone formation biomarkers and decreased serum levels of a bone resorption marker. It was also demonstrated that the target capture, measured as total sclerostin serum concentration, occurred with BPS804 dosing, an observation consistent with previous clinical studies. In sum, these results suggest that BPS804 has potential as a therapy for patients with a number of conditions involving low bone mass, such as for example postmenopausal osteoporosis.
Ich bin auf das anti-sclerostin-Thema am vergangenen Wochenende gestoßen, als bekannt wurde, dass während der P3-Evenity-Studie unter dem von Amgen und UCB entwickelten Konkurrenzantikörper romosozumab gehäuft kardiovaskuläre Probleme aufgetreten sind, die (zumindest) zu einer Verzögerung der Zulassung führen werden. Novartis´Datenanalyse entnehme ich, dass unter BPS804 keine derartigen Nebenwirkungen beobachtet worden sind. Um so bedauerlicher ist es, dass die klinische Entwicklung des BPS804 von Novartis nicht weiter fortgeführt worden ist.
Heute hatte ich endlich Gelegenheit, in der Novartis-Datenbank die Ergebnisse der Phase II-Studie mit BPS804 in der Indikation Osteoporose durchzusehen. Tatsächlich, auch hier finden sich in jeder Hinsicht absolut überzeugende Ergebnisse und keinerlei Nebenwirkungen, die Anlaß zur Besorgnis gäben. Ich bin so frei, die allgemein verständliche Zusammenfassung der Datenanalyse wörtlich wiederzugeben (hoffentlich schlägt jetzt nicht wieder der w:o-Löschalgorithmus zu ... Novartis hat die Daten freigegeben!):
This study was designed as a small, exploratory dosing regimen study and showed that BPS804 is well tolerated and effective in increasing bone mineral density in postmenopausal women with low bone mineral density. The short duration weekly dosing regimen showed a more rapid onset and greater increase in lumbar spine bone mineral density then the monthly and quarterly treatment. The study also showed that BPS804 is associated with rapid and substantial increases in serum levels of bone formation biomarkers and decreased serum levels of a bone resorption marker. It was also demonstrated that the target capture, measured as total sclerostin serum concentration, occurred with BPS804 dosing, an observation consistent with previous clinical studies. In sum, these results suggest that BPS804 has potential as a therapy for patients with a number of conditions involving low bone mass, such as for example postmenopausal osteoporosis.
Ich bin auf das anti-sclerostin-Thema am vergangenen Wochenende gestoßen, als bekannt wurde, dass während der P3-Evenity-Studie unter dem von Amgen und UCB entwickelten Konkurrenzantikörper romosozumab gehäuft kardiovaskuläre Probleme aufgetreten sind, die (zumindest) zu einer Verzögerung der Zulassung führen werden. Novartis´Datenanalyse entnehme ich, dass unter BPS804 keine derartigen Nebenwirkungen beobachtet worden sind. Um so bedauerlicher ist es, dass die klinische Entwicklung des BPS804 von Novartis nicht weiter fortgeführt worden ist.
Kurz meine Eindrücke zu den ASCO-Präsentationen. Aus Zeitmangel mache ich es kurz.
MOR202:
Wirksamkeitsdaten deutlich schlechter als z.B. Daratumumab, aber auch schlechter als bei anderen Präparaten wie z.B. anti-CD138-ADC von Biotest etc.
Auch im Vergleich zu SAR650984 fallen die MOR202-Daten ab, obwohl die SAR-Patienten weitaus stärker austherapiert (= kranker waren).
MOR202 betont immer wieder die bessere Infusionsvertäglichkeit. Mag ja sein, aber was hilft´s, wenn das Präparat weniger wirkt ... und wenn die Gesamtverträglichkeit miserabel ist -> deutlich mehr Nebenwirkungen Grad 3 und mehr als bei der Konkurrenz.
MOR202 habe ich für mich abgehakt. Kann mir nicht vorstellen, dass ein größerer Player diesen Antikörper einlizensieren wird.
MOR208:
ORR 56 % in der LEN-Kombi. Während der Präsentation wurde als Vergleichswert für die Obinutuzumab-LEN-Kombi ein Wert von 45 % angegeben. Verschwiegen wurde aber, dass in der Obinutuzumab-Kombi LEN niedrieger dosiert worden ist als in der L-Mind-Studie und dass die Patienten in der Obinutuz.-Kombi stärker vorbehandelt und kranker waren.
Zur Einschätzung der ORR: Vor eineinhalb Jahren meinte Moris/ImmunoGen im Conference Call, man werde die eigenen Präparate wie SAR3419 und IMGN529, die ja bereits als Monotherapeutika bzgl. ORR besser abgeschnitten hatten als MOR208, nur dann in die Phase 3 bringen, wenn die ORR in Kombistudien > 60 % sei.
Hinsichtlich der B-Mind-Studie fällt die schleppende Patientenrekrutierung auf. Im ersten Jahr sind gerade mal 27 Patienten rekrutiert worden, davon haben erst 25 Patienten mindestens eine Medikamentendosis erhalten. Geplant ist die Untersuchung von 330 Patienten. Wenn das in diesem Tempo weitergeht, rekrutiert Morphosys noch in 10 Jahren.
Insgesamt sind die diesjährigen ASCO-Daten ernüchternd.
MOR202:
Wirksamkeitsdaten deutlich schlechter als z.B. Daratumumab, aber auch schlechter als bei anderen Präparaten wie z.B. anti-CD138-ADC von Biotest etc.
Auch im Vergleich zu SAR650984 fallen die MOR202-Daten ab, obwohl die SAR-Patienten weitaus stärker austherapiert (= kranker waren).
MOR202 betont immer wieder die bessere Infusionsvertäglichkeit. Mag ja sein, aber was hilft´s, wenn das Präparat weniger wirkt ... und wenn die Gesamtverträglichkeit miserabel ist -> deutlich mehr Nebenwirkungen Grad 3 und mehr als bei der Konkurrenz.
MOR202 habe ich für mich abgehakt. Kann mir nicht vorstellen, dass ein größerer Player diesen Antikörper einlizensieren wird.
MOR208:
ORR 56 % in der LEN-Kombi. Während der Präsentation wurde als Vergleichswert für die Obinutuzumab-LEN-Kombi ein Wert von 45 % angegeben. Verschwiegen wurde aber, dass in der Obinutuzumab-Kombi LEN niedrieger dosiert worden ist als in der L-Mind-Studie und dass die Patienten in der Obinutuz.-Kombi stärker vorbehandelt und kranker waren.
Zur Einschätzung der ORR: Vor eineinhalb Jahren meinte Moris/ImmunoGen im Conference Call, man werde die eigenen Präparate wie SAR3419 und IMGN529, die ja bereits als Monotherapeutika bzgl. ORR besser abgeschnitten hatten als MOR208, nur dann in die Phase 3 bringen, wenn die ORR in Kombistudien > 60 % sei.
Hinsichtlich der B-Mind-Studie fällt die schleppende Patientenrekrutierung auf. Im ersten Jahr sind gerade mal 27 Patienten rekrutiert worden, davon haben erst 25 Patienten mindestens eine Medikamentendosis erhalten. Geplant ist die Untersuchung von 330 Patienten. Wenn das in diesem Tempo weitergeht, rekrutiert Morphosys noch in 10 Jahren.
Insgesamt sind die diesjährigen ASCO-Daten ernüchternd.
Zu Anetumab ravtansine: Starke Konkurrenz durch die Checkpoint-Inhibitoren (s. ASCO-Daten zur nivolumab/ipilimumab-Kombi!)
Meine Interpretation:
MOR202: Es kam mir schon früher so vor, als würde dieser vor allem deshalb weitergezogen werden, um Verhandlungsmasse mit Janssen auf den Tisch legen zu können und den Patentanspruch zu untermauern. Die Daten sind nicht grossartig, und das wording von MOR sagt alles: "further development of MOR202 as a representative of the CD38-class"... da steht nichts von best in class oder whatever, man glaubt selbst nicht daran, Dara schlagen zu könnnen.
MOR208: Hier sehe ich mehr Chancen als Joschka. Wenn man sich das slide "Time on Study and Duration of Response" anschaut, sieht man, dass eine Wirkung durchgehend erst nach 2 Monaten erzielt wird, die sich dann oft später noch verbessert. Die sieben untersten NE und PD würde ich also nicht als relevant betrachten, da zu diesem Zeitpunkt noch keine Wirkung erzielt werden kann.
Neutropenia grade 4 auf der anderen Seite ist allerdings sicher nichts zum Spassen. (Die ständige Erwähnung der geringen Infusionsreaktionen empfinde ich als unprofessionell, da das wirklich nicht die limitierende Nebenwirkung ist.)
Was ich überhaupt nicht verstehe, ist das MIND Studiendesign, vielleicht kann mir das einer erklären. Warum kombiniert man MOR208 mit Lenalidomide, wo dies nur für Myelom zugelassen ist und in B-cell malignacies eher experimentellen Charakter hat? (Weil es LEN grad umsonst von Celgene gab??) Warum führt man die erfolgreiche L-MIND Studie nicht in einer Ph 2 oder 3 weiter? Wo sind die Ph 1-Daten von B-MIND, die eine Weiterführung als Ph 2/3 rechtfertigen?
MOR202: Es kam mir schon früher so vor, als würde dieser vor allem deshalb weitergezogen werden, um Verhandlungsmasse mit Janssen auf den Tisch legen zu können und den Patentanspruch zu untermauern. Die Daten sind nicht grossartig, und das wording von MOR sagt alles: "further development of MOR202 as a representative of the CD38-class"... da steht nichts von best in class oder whatever, man glaubt selbst nicht daran, Dara schlagen zu könnnen.
MOR208: Hier sehe ich mehr Chancen als Joschka. Wenn man sich das slide "Time on Study and Duration of Response" anschaut, sieht man, dass eine Wirkung durchgehend erst nach 2 Monaten erzielt wird, die sich dann oft später noch verbessert. Die sieben untersten NE und PD würde ich also nicht als relevant betrachten, da zu diesem Zeitpunkt noch keine Wirkung erzielt werden kann.
Neutropenia grade 4 auf der anderen Seite ist allerdings sicher nichts zum Spassen. (Die ständige Erwähnung der geringen Infusionsreaktionen empfinde ich als unprofessionell, da das wirklich nicht die limitierende Nebenwirkung ist.)
Was ich überhaupt nicht verstehe, ist das MIND Studiendesign, vielleicht kann mir das einer erklären. Warum kombiniert man MOR208 mit Lenalidomide, wo dies nur für Myelom zugelassen ist und in B-cell malignacies eher experimentellen Charakter hat? (Weil es LEN grad umsonst von Celgene gab??) Warum führt man die erfolgreiche L-MIND Studie nicht in einer Ph 2 oder 3 weiter? Wo sind die Ph 1-Daten von B-MIND, die eine Weiterführung als Ph 2/3 rechtfertigen?
Gute Daten mit den checkpoint inhibitors nivo/ipi sind sicher kein Problem für den ADC Anetumab ravtansine. Es handelt sich um zwei ganz verschiedene Wirkungsmechanismen, die man auch bei anderen Targets/Indikationen diskutiert, miteinander zu kombinieren.
Antwort auf Beitrag Nr.: 55.094.095 von yok am 07.06.17 10:22:34Es wurden vorher nie Patienten mit der Kombi MOR 208 +
Bemdamustin behandelt, deswegen gab es ja den Safety Run-In. Ich glaub,man hat diese Kombi gewählt, weil es ein paar Daten zu Rituximab + Bendamustin in genau diesem zu untersuchenden Kollektiv R/R DLBCL gibt. Lenalidomid ist in der Indikation gar nicht zugelassen, würde ich vermuten.
Noch ein Wort zu den Checkpoint Inhibitoren beim Meotheliom. Das sind m.E. keine Daten, die einem bzgl. Anetumab Ravtansine Angst machen müssen. Glaube, dass die Checkpoint Inhibitoren da eher als Kombi in Frage kommen. Eine solche Studie läuft ja mit Pembrolizumab gerade an. Welche Gesundheitssysteme das alles bezahlen sollen, muss dann bloß noch geklärt werden. 🤔
Bemdamustin behandelt, deswegen gab es ja den Safety Run-In. Ich glaub,man hat diese Kombi gewählt, weil es ein paar Daten zu Rituximab + Bendamustin in genau diesem zu untersuchenden Kollektiv R/R DLBCL gibt. Lenalidomid ist in der Indikation gar nicht zugelassen, würde ich vermuten.
Noch ein Wort zu den Checkpoint Inhibitoren beim Meotheliom. Das sind m.E. keine Daten, die einem bzgl. Anetumab Ravtansine Angst machen müssen. Glaube, dass die Checkpoint Inhibitoren da eher als Kombi in Frage kommen. Eine solche Studie läuft ja mit Pembrolizumab gerade an. Welche Gesundheitssysteme das alles bezahlen sollen, muss dann bloß noch geklärt werden. 🤔
zu MOR208:
Heute Abend sind Zwischerergebnisse der JULIET-Studie (CTL019 von Novartis in relapsed or refractory DLBCL after ≥2 lines of chemotherapy including rituximab and anthracycline and either having failed autologous Hematopoietic stem cell transplantation (ASCT), or being ineligible for or not consenting to ASCT -> https://clinicaltrials.gov/ct2/show/NCT02445248?term=JULIET&… ) veröffentlicht worden.
Dabei wurde eine best ORR 59 % erzielt mit 43 % complete response.
Das übertrifft natürlich alles.
Heute Abend sind Zwischerergebnisse der JULIET-Studie (CTL019 von Novartis in relapsed or refractory DLBCL after ≥2 lines of chemotherapy including rituximab and anthracycline and either having failed autologous Hematopoietic stem cell transplantation (ASCT), or being ineligible for or not consenting to ASCT -> https://clinicaltrials.gov/ct2/show/NCT02445248?term=JULIET&… ) veröffentlicht worden.
Dabei wurde eine best ORR 59 % erzielt mit 43 % complete response.
Das übertrifft natürlich alles.
Antwort auf Beitrag Nr.: 55.100.428 von Joschka Schröder am 07.06.17 23:43:23Die Patienten waren zuvor tatsächlich weitgehend "austherapiert": Sixty percent of the patients had three or more lines of chemotherapy and 51% had a prior autoSCT.
Für CAR-T war das sicher ne gute Woche. Alle Zweifel sind da aber nicht beseitigt. Im JULIET Trial waren übrigens 141 Parienten, ausgewertet wurden nur 51 und auf diese 51 beziehen sich die Ansprechraten. Da würden mich die Details schon mal interessieren. 57 % CRS sind auch erwähnenswert, waren hier aber beherrschbar. Das ganze Verfahren ist natürlich aufwendig und wird vermutlich wirklich erst mal nur für austherapierte Patienten in Frage kommen.
Antwort auf Beitrag Nr.: 55.101.880 von deadflowers am 08.06.17 10:05:59
Das Patientenkollektiv war so krank, dass sehr viele Patienten bereits vor der ersten Behandlung/Infusion schon wieder aus der Studie ausgeschieden sind.
"In the JULIET trial, 43 patients discontinued before infusion and the majority did so due to rapid progression of their disease or deterioration in their clinical status. This reflects the acute and progressive nature of the disease of the patients. Only nine of 141 (6%) enrolled patients could not be infused due to inability to manufacture an adequate dose of CAR-T cells. Over the course of JULIET, with continuous process improvements, manufacturing success rate improved to 97% for the last 30 patients."
Sollten die Studienergebnisse weiter bestätigt weerde, wird sich CAR-T wohl mit der Zeit als bevorzugte Therapie etablieren.
Das CRS hat man mittlerweile gut im Griff, tritt ja bei recht vielen neueren immunonkol. Behandlungsmethoden auf, so dass inzwischen ausreichende Erfahrungen vorliegen.
Zitat von deadflowers: Für CAR-T war das sicher ne gute Woche. Alle Zweifel sind da aber nicht beseitigt. Im JULIET Trial waren übrigens 141 Parienten, ausgewertet wurden nur 51 und auf diese 51 beziehen sich die Ansprechraten. Da würden mich die Details schon mal interessieren.
Das Patientenkollektiv war so krank, dass sehr viele Patienten bereits vor der ersten Behandlung/Infusion schon wieder aus der Studie ausgeschieden sind.
"In the JULIET trial, 43 patients discontinued before infusion and the majority did so due to rapid progression of their disease or deterioration in their clinical status. This reflects the acute and progressive nature of the disease of the patients. Only nine of 141 (6%) enrolled patients could not be infused due to inability to manufacture an adequate dose of CAR-T cells. Over the course of JULIET, with continuous process improvements, manufacturing success rate improved to 97% for the last 30 patients."
Sollten die Studienergebnisse weiter bestätigt weerde, wird sich CAR-T wohl mit der Zeit als bevorzugte Therapie etablieren.
Das CRS hat man mittlerweile gut im Griff, tritt ja bei recht vielen neueren immunonkol. Behandlungsmethoden auf, so dass inzwischen ausreichende Erfahrungen vorliegen.
Antwort auf Beitrag Nr.: 55.102.324 von Joschka Schröder am 08.06.17 10:54:58Ergänzend zu den Evaluationskriterien der Zwischenanalyse:
"Among 51 patients with three months or more of follow-up or earlier discontinuation, best ORR was 59% (95% CI, 44.2-72.4; p<0.0001), with 43% achieving CR and 16% achieving PR. The full JULIET primary analysis is expected to be available later this year and will serve as the basis for US and EU regulatory submissions."
"Among 51 patients with three months or more of follow-up or earlier discontinuation, best ORR was 59% (95% CI, 44.2-72.4; p<0.0001), with 43% achieving CR and 16% achieving PR. The full JULIET primary analysis is expected to be available later this year and will serve as the basis for US and EU regulatory submissions."
Antwort auf Beitrag Nr.: 55.102.417 von Joschka Schröder am 08.06.17 11:04:57...und was schließt ihr daraus für das Unternehmen?
Cart wird nichts für den "massenmarkt". Habe heute eine schätzung von 500 k für die Behandlung gelesen... Und selbst wenn es nur 300 k werden, werden sich das nur sehr privilegierte Patienten leisten können. Plus lebenslange ivig- Behandlung...
Antwort auf Beitrag Nr.: 55.107.571 von yok am 08.06.17 22:40:10
Wieso eigentlich? Die CAR-T-Zellen haben doch eine begrenzte Lebensdauer, entsprechend sollte die therapieinduzierte B-Zell-Aplasie nach einigen Jahren wieder verschwinden (die haematopoetische Stammzelle bleibt ja während der Therapie erhalten, erst ab der Pro-B-Zelle aufwärts wird CD19 exprimiert).
Zitat von yok: Plus lebenslange ivig- Behandlung...
Wieso eigentlich? Die CAR-T-Zellen haben doch eine begrenzte Lebensdauer, entsprechend sollte die therapieinduzierte B-Zell-Aplasie nach einigen Jahren wieder verschwinden (die haematopoetische Stammzelle bleibt ja während der Therapie erhalten, erst ab der Pro-B-Zelle aufwärts wird CD19 exprimiert).
Antwort auf Beitrag Nr.: 55.108.105 von Joschka Schröder am 09.06.17 01:35:36Also 100%ig kann ich es dir auch nicht erklären. Es werden auf jeden Fall nicht die HSC gekillt, dann wären ja ausser den B-Zellen auch noch andere Zellpopulationen betroffen. Aber die CAR-T-Zellen sollen wohl lebenslang überleben (reale Langzeitdaten bleiben natürlich abzuwarten). Sie vermehren sich auf jeden Fall im Körper, daher auch das Problem mit dem CRS relativ unabhängig von der dosierten Zellzahl. Ob das langfristige Überleben über memory cells geht oder über die permanente Stimulation durch neu entstehende B-Zellen, weiss ich nicht.
Antwort auf Beitrag Nr.: 55.119.346 von yok am 11.06.17 10:24:27Ehrlich gesagt kann ich Dir nicht folgen.
1. Woher hast Du, dass CAR-T-Zellen lebenslang im Körper überleben sollen?
2. Und wie stellst Du Dir die Vermehrung einer reifen (modizifierten) T-Zelle vor bzw. wo wird so etwas beschrieben?
3. Und welcher Wissenschaftler behauptet, das CRS beruhe u.a. auf einer Vermehrung infundierter CAR-T-Zellen im menschlichen Körper?
Allgemein verständlich werden die Zusammenhäng (inkl. begrenzter Lebensdauer der CAR-T-Zellen) z.B. hier beschrieben -> http://www.wissensschau.de/krebs_tumor/car-t-zellen_nebenwir…
1. Woher hast Du, dass CAR-T-Zellen lebenslang im Körper überleben sollen?
2. Und wie stellst Du Dir die Vermehrung einer reifen (modizifierten) T-Zelle vor bzw. wo wird so etwas beschrieben?
3. Und welcher Wissenschaftler behauptet, das CRS beruhe u.a. auf einer Vermehrung infundierter CAR-T-Zellen im menschlichen Körper?
Allgemein verständlich werden die Zusammenhäng (inkl. begrenzter Lebensdauer der CAR-T-Zellen) z.B. hier beschrieben -> http://www.wissensschau.de/krebs_tumor/car-t-zellen_nebenwir…
Antwort auf Beitrag Nr.: 55.119.784 von Joschka Schröder am 11.06.17 12:17:25Nein, Du könntest doch recht haben. Die CAR-T-Zelle verliert durch die Umprogrammierung ja nicht ihre Teilungsfähigkeit. Theoretisch ist also wirklich vorstellbar, dass eine lebenslange Substitution Immunoglobulin-Substitution notwendig werden könnte.
Andererseits habe ich schon Wissenschaftler über die limitierte Lebensdauer von CAR-T klagen hören.
Es führt wohl leider kein Weg daran vorbei, etwas fundiertere Literatur zum Thema zu lesen.
Andererseits habe ich schon Wissenschaftler über die limitierte Lebensdauer von CAR-T klagen hören.
Es führt wohl leider kein Weg daran vorbei, etwas fundiertere Literatur zum Thema zu lesen.
Antwort auf Beitrag Nr.: 55.119.814 von Joschka Schröder am 11.06.17 12:40:22yok, was die Proliferation post inject. betrifft, auch mit Bezug auf das CRS, hast Du hast vollumfänglich recht. Da reicht die Durchsicht eines Interviews mit Stephan Grupp (-> http://www.chop.edu/doctors/grupp-stephan-a) aus, der in der CAR-T-Zelltherapie mit am erfahrensten ist.
http://www.trillium.de/heft-22016/interview-mit-prof-dr-step…
http://www.trillium.de/heft-22016/interview-mit-prof-dr-step…
Extrem starke Daten des anti-CD19-ADCs ADCT-402 bei relaps./refrakt. DLBCL, Median 3 Vorbehandlungen, Dosiseskalation, Dosis größer gleich 120 Mikrogramm/kg: 41 % CR, 17 % PR, 25 % SD -> http://onlinelibrary.wiley.com/doi/10.1002/hon.2437_33/epdf
Das Wettbewerbsumfeld für MOR208 ist hart.
Das Wettbewerbsumfeld für MOR208 ist hart.
Antwort auf Beitrag Nr.: 55.130.315 von Joschka Schröder am 13.06.17 10:56:04Das sieht mau aus für MOR 208.
ADC Th. ist ein interessantes Unternehmen, schade, dass es nicht an der Börse gehandelt wird.
Persönlich hoffe ich auf eine Zulassung von Guselkumab und werde dann erstmal Gewinne mitnehmen... so rosig sieht es für Morphosys derzeit dann doch nicht aus.
ADC Th. ist ein interessantes Unternehmen, schade, dass es nicht an der Börse gehandelt wird.
Persönlich hoffe ich auf eine Zulassung von Guselkumab und werde dann erstmal Gewinne mitnehmen... so rosig sieht es für Morphosys derzeit dann doch nicht aus.
Antwort auf Beitrag Nr.: 55.130.315 von Joschka Schröder am 13.06.17 10:56:04Wie aussagekräftig sind solche Ergebnisse (jetzt schon) ?
Bei ADCT-402 waren es 12 Patienten in der Auswertung und bei MOR208 34.
Bei ADCT-402 waren es 12 Patienten in der Auswertung und bei MOR208 34.
Antwort auf Beitrag Nr.: 55.131.302 von -weitblick- am 13.06.17 13:46:43Sämtliche Daten aus derartigen Studien sind wegen der geringen Fallzahlen mit Vorsicht zu genießen. Dennoch geben sie eine gewisse Tendenz vor.
Und so sehen die Ergebnisse von ADCT402 im Wasserfall-Diagramm aus:

Relevant für MOR202 und die etwaigen Darzalex-Royalties evtl. ja auch die ASCO Ergebnisse von BLUE/CELG. Und die waren wirklich stark!
Hier die journalistische Aufarbeitung:
https://www.thestreet.com/story/14162047/1/bluebird-celgene-…
https://endpts.com/bluebird-has-a-promising-new-update-on-it…" target="_blank" rel="nofollow ugc noopener">https://endpts.com/bluebird-has-a-promising-new-update-on-it…

Diese Folie stellte der Arzt in der ASCO-Präsentation vor mit den Worten "And this is the money slide i guess!"
100% Ansprechrate (wenn man die offensichtlich zu niedrige Dosis weglässt) und alle anhaltend. Auch hier handelt es sich um stark austherapierte Patienten. Im Schnitt hatten sie 7 vorherige Therapien. Ein guter Indikator sind hier auch die 3 "Armen" Teilnehmer in der wohl zu geringen Dosis. Diese sind alle recht schnell verstorben.
Es werden erste Rufe laut diese Technologie auch in früheren Therapielinien zu testen...
Relevant für MOR202 und die etwaigen Darzalex-Royalties evtl. ja auch die ASCO Ergebnisse von BLUE/CELG. Und die waren wirklich stark!
Hier die journalistische Aufarbeitung:
https://www.thestreet.com/story/14162047/1/bluebird-celgene-…
https://endpts.com/bluebird-has-a-promising-new-update-on-it…" target="_blank" rel="nofollow ugc noopener">https://endpts.com/bluebird-has-a-promising-new-update-on-it…
Diese Folie stellte der Arzt in der ASCO-Präsentation vor mit den Worten "And this is the money slide i guess!"
100% Ansprechrate (wenn man die offensichtlich zu niedrige Dosis weglässt) und alle anhaltend. Auch hier handelt es sich um stark austherapierte Patienten. Im Schnitt hatten sie 7 vorherige Therapien. Ein guter Indikator sind hier auch die 3 "Armen" Teilnehmer in der wohl zu geringen Dosis. Diese sind alle recht schnell verstorben.
Es werden erste Rufe laut diese Technologie auch in früheren Therapielinien zu testen...
Antwort auf Beitrag Nr.: 55.140.398 von kmastra am 14.06.17 19:43:19Ich habe mir zwischenzeitlich die letzte Bluebird-Präsentation im Detail angesehen. Angesichts der mit bb2121 (= anti-BCMA CAR) erzielten Wahnsinnsdaten hätte es für Celgene wirklich keinen Sinn gemacht, dass MOR202-Projekt fortzusetzen (zumal sich MOR202 bislang als schwächster Kandidat unter den anti-CD38-Mabs entpuppt hat).
Die Ergebnisdaten kurz zusammengefaßt:
im Mittel 6 (!) Vorbehandlungen, dennoch
ORR 100 %
CR 27 %
VGPR 46 %
PR 27 %
Lang anhaltender Therapieeffekt ... MM-Patienten haben endlich eine richtig gute Perspektive, auch wenn sie schwerstkrank und "austherapiert" sind. Es ist einfach eine große Freude, solche Studiendaten zu sehen.
Die Ergebnisdaten kurz zusammengefaßt:
im Mittel 6 (!) Vorbehandlungen, dennoch
ORR 100 %
CR 27 %
VGPR 46 %
PR 27 %
Lang anhaltender Therapieeffekt ... MM-Patienten haben endlich eine richtig gute Perspektive, auch wenn sie schwerstkrank und "austherapiert" sind. Es ist einfach eine große Freude, solche Studiendaten zu sehen.
Eine der großen offenen Fragen zu CAR-T ist allerdings, wie das alles finanzierbar sein soll. Das betrifft die Therapie selbst und die sehr teure lebenlange IVIG Gabe. Wenn man das in der Breite einsetzt, läuft das ganzes System aus dem Ruder. M.E. unbezahlbar, und eigentlich nicht vorstellbar, dass es bei Patienten zum Einsatz kommt, wo es noch andere Therapiemöglichkeiten gibt.
Antwort auf Beitrag Nr.: 55.149.869 von deadflowers am 16.06.17 09:14:36Das ist in der Tat ein nicht zu unterschätzendes Argument. Ich kann mich an die Preisdiskussion mit Gilead bzgl. Sovaldi erinnern. Da ich bei einer der größten Krankenkassen arbeite, weiß ich, das die Kosten alleine für dieses Medikament (Sovaldi) ein Ausmaß erreicht haben, dass budgetrelavant wurde.
Es ist einerseits natürlich ein großer und sehr erfreulicher Wurf für die betroffenen Patienten, andererseits natürlich auch eine Frage der (begrenzten) Finanzierungsmöglichkeiten. Ich kann mir auch vorstellen, dass das noch ein Thema werden wird.
Es ist einerseits natürlich ein großer und sehr erfreulicher Wurf für die betroffenen Patienten, andererseits natürlich auch eine Frage der (begrenzten) Finanzierungsmöglichkeiten. Ich kann mir auch vorstellen, dass das noch ein Thema werden wird.
Antwort auf Beitrag Nr.: 55.148.906 von Joschka Schröder am 16.06.17 00:31:56In der Tat eine Freude! Die Ergebnisse von BLUE/CELG werden ja auch durch die Ergebnisse der etwas merkwürdig anmutenden Nanjing Legend Biotech untermauert:
http://www.asco.org/about-asco/press-center/news-releases/ca…
Natürlich muss man abwarten, wie sich offene Fragen (Sicherheit, Kosten, Nachhaltigkeit des Ansprechens...) klären lassen. Aber bei diesen Ergebnissen darf man doch optimistisch sein, dass es da Wege geben wird. Die momentane Behandlung ist ja auch nicht gerade günstig! Und ob es dann medizinisch und auch finanziell sinnvoll ist, etwas wie CAR-T ganz am Ende einzusetzen darf ja auch bezweifelt werden.
http://www.asco.org/about-asco/press-center/news-releases/ca…
Natürlich muss man abwarten, wie sich offene Fragen (Sicherheit, Kosten, Nachhaltigkeit des Ansprechens...) klären lassen. Aber bei diesen Ergebnissen darf man doch optimistisch sein, dass es da Wege geben wird. Die momentane Behandlung ist ja auch nicht gerade günstig! Und ob es dann medizinisch und auch finanziell sinnvoll ist, etwas wie CAR-T ganz am Ende einzusetzen darf ja auch bezweifelt werden.
Antwort auf Beitrag Nr.: 55.119.784 von Joschka Schröder am 11.06.17 12:17:25
--
Hi, bin nicht sicher ob es hilft, aber schau mal auf Seite 31,
"CAR T-Zellen sind ein „lebendes“ Medikament.
Sie vermehren sich im Körper des Patienten, bis
alle Krebszellen abgeräumt sind, und können
den Patienten als CAR Gedächtnis T-Zellen vor
einem möglichen Wiederauftreten der Krankheit
schützen.."https://www.badw.de/fileadmin/pub/akademieAktuell/2017/60/AA…
Zitat von Joschka Schröder: Ehrlich gesagt kann ich Dir nicht folgen.
1. Woher hast Du, dass CAR-T-Zellen lebenslang im Körper überleben sollen?
2. Und wie stellst Du Dir die Vermehrung einer reifen (modizifierten) T-Zelle vor bzw. wo wird so etwas beschrieben?
3. Und welcher Wissenschaftler behauptet, das CRS beruhe u.a. auf einer Vermehrung infundierter CAR-T-Zellen im menschlichen Körper?
Allgemein verständlich werden die Zusammenhäng (inkl. begrenzter Lebensdauer der CAR-T-Zellen) z.B. hier beschrieben -> http://www.wissensschau.de/krebs_tumor/car-t-zellen_nebenwir…
--
Hi, bin nicht sicher ob es hilft, aber schau mal auf Seite 31,
"CAR T-Zellen sind ein „lebendes“ Medikament.
Sie vermehren sich im Körper des Patienten, bis
alle Krebszellen abgeräumt sind, und können
den Patienten als CAR Gedächtnis T-Zellen vor
einem möglichen Wiederauftreten der Krankheit
schützen.."https://www.badw.de/fileadmin/pub/akademieAktuell/2017/60/AA…
Antwort auf Beitrag Nr.: 55.168.625 von RichyBerlin am 20.06.17 08:40:19ZUUUUULASSSSSSUUUUUNNNNNNNG GUUUUUUSELKUMAAAAAB!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!1
Schuppenflechte-Mittel von Morphosys-Lizenznehmer in den USA zugelassen
NEW YORK (dpa-AFX) - Ein wichtiges Mittel des Biotechunternehmens Morphosys ist in den USA zugelassen worden. Der Lizenznehmer Janssen erhielt am Donnerstag die Verkaufsgenehmigung für Tremfya (Guselkumab) zur Behandlung von mittelschwerer bis schwerer Schuppenflechte bei Erwachsenen, wie aus Unterlagen der US-Gesundheitsbehörde FDA hervorgeht. Das Mittel ist auf Basis von Morphosys' Antikörper-Bibliothek HuCAL entwickelt worden.
Morphosys hatte im Mai mitgeteilt, dass Janssen Research & Development bei der FDA ein beschleunigtes Zulassungsverfahren beantragt hatte. Ein Zulassung im dritten Quartal hatte Janssen damals für möglich gehalten./mis/la
ISIN DE0006632003
AXC0287 2017-07-13/21:29
NEW YORK (dpa-AFX) - Ein wichtiges Mittel des Biotechunternehmens Morphosys ist in den USA zugelassen worden. Der Lizenznehmer Janssen erhielt am Donnerstag die Verkaufsgenehmigung für Tremfya (Guselkumab) zur Behandlung von mittelschwerer bis schwerer Schuppenflechte bei Erwachsenen, wie aus Unterlagen der US-Gesundheitsbehörde FDA hervorgeht. Das Mittel ist auf Basis von Morphosys' Antikörper-Bibliothek HuCAL entwickelt worden.
Morphosys hatte im Mai mitgeteilt, dass Janssen Research & Development bei der FDA ein beschleunigtes Zulassungsverfahren beantragt hatte. Ein Zulassung im dritten Quartal hatte Janssen damals für möglich gehalten./mis/la
ISIN DE0006632003
AXC0287 2017-07-13/21:29
Top-Ergebnisse aus der Phase IIb-Psoriasis-Studie mit Bimekizumab (anti-IL-17A/IL-17F):
PASI90 nach 12 Wochen bis zu 79 %
PASI100 nach 12 Wochen bis zu 60 %
Das hat meines Wissens bislang noch kein Präparat geschafft, auch nicht Guselkumab.
PASI90 nach 12 Wochen bis zu 79 %
PASI100 nach 12 Wochen bis zu 60 %
Das hat meines Wissens bislang noch kein Präparat geschafft, auch nicht Guselkumab.
Antwort auf Beitrag Nr.: 55.368.077 von Joschka Schröder am 21.07.17 08:28:59Danke für die Info.
Wie ist dort das Dosierungsschema? Alle 4 Wochen?
Wie ist dort das Dosierungsschema? Alle 4 Wochen?
Antwort auf Beitrag Nr.: 55.369.133 von Ville7 am 21.07.17 10:47:19Gusel hatte in der VOYAGE-1 Studie PASI-90 von 85,1% nach 16 Wochen. Wieso sollte das schlechter sein?
Antwort auf Beitrag Nr.: 55.371.557 von deadflowers am 21.07.17 14:55:05
Da irrst Du Dich leider. Guselkumab: PASI 90 nach 16-Wochen 73,3 %. Du hast das mit dem IGA-Score verwechselt.
https://www.ncbi.nlm.nih.gov/labs/articles/28057360/
Zitat von deadflowers: Gusel hatte in der VOYAGE-1 Studie PASI-90 von 85,1% nach 16 Wochen. Wieso sollte das schlechter sein?
Da irrst Du Dich leider. Guselkumab: PASI 90 nach 16-Wochen 73,3 %. Du hast das mit dem IGA-Score verwechselt.
https://www.ncbi.nlm.nih.gov/labs/articles/28057360/
Antwort auf Beitrag Nr.: 55.372.142 von Joschka Schröder am 21.07.17 15:59:04Du hast recht. PASI-90 nach 24 Wochen dann aber auch 80,2%. Detaillierte Zahlen zu Bimekizumab finde ich nicht im Netz. Gibt's bisher nur die Pressemitteilung?
Ingesamt kein Grund zur Aufregung. Geringe Unterschiede. Über statistische Signifikanz kann man nichts sagen, die klinische Relevanz der Unterschiede dürfte nahe 0 sein. Aber natürlich ein weiterer Konkurrent, mit dem man den Kuchen teilen muss. Wobei BI noch eine ganze Weile bis zur Zulassung brauchen wird.
Ingesamt kein Grund zur Aufregung. Geringe Unterschiede. Über statistische Signifikanz kann man nichts sagen, die klinische Relevanz der Unterschiede dürfte nahe 0 sein. Aber natürlich ein weiterer Konkurrent, mit dem man den Kuchen teilen muss. Wobei BI noch eine ganze Weile bis zur Zulassung brauchen wird.
Antwort auf Beitrag Nr.: 55.372.979 von deadflowers am 21.07.17 17:56:44Ein weiterer Rückschlag 
http://www.finanznachrichten.de/nachrichten-2017-07/41264939…

http://www.finanznachrichten.de/nachrichten-2017-07/41264939…
was für ein Seuchentag!
Antwort auf Beitrag Nr.: 55.373.255 von Trendfighter am 21.07.17 18:34:43Allerdings nur in einer Indikation. Die vorhandenen Daten sollen so schlecht ja nicht sein, so dass in anderen Indikationen noch Hoffnung besteht.
Antwort auf Beitrag Nr.: 55.373.273 von milchbubi am 21.07.17 18:36:07Das mag sein mit der Hoffnung,
aber das andere ist Standard bei solchen Meldungen.
aber das andere ist Standard bei solchen Meldungen.
Antwort auf Beitrag Nr.: 55.373.303 von schnappi am 21.07.17 18:39:15ich will auch nicht aus Negativem etwas Positives machen.
Aber nachbörslich und auf Tradegate hat es jetzt nicht so ein Blutbad gegeben wie beim scheitern der vorherigen Rohrkrepierer Bima und Gante. Darum habe ich die kleine Hoffnung, das es doch etwas differenzierter gesehen wird.
Die hohe und laufend erhöhte Shortquote in den letzten Monaten macht mich auch wieder mal stutzig. Das war damals ja kurz vor dem Ausstieg von Celgene ein ähnliches Muster.......
Aber nachbörslich und auf Tradegate hat es jetzt nicht so ein Blutbad gegeben wie beim scheitern der vorherigen Rohrkrepierer Bima und Gante. Darum habe ich die kleine Hoffnung, das es doch etwas differenzierter gesehen wird.
Die hohe und laufend erhöhte Shortquote in den letzten Monaten macht mich auch wieder mal stutzig. Das war damals ja kurz vor dem Ausstieg von Celgene ein ähnliches Muster.......
Antwort auf Beitrag Nr.: 55.373.303 von schnappi am 21.07.17 18:39:15
Nach chart könnten wir Montag die 60 sehen, rechne dann wieder mit einem Anstieg auf 64/65
hm
Nach chart könnten wir Montag die 60 sehen, rechne dann wieder mit einem Anstieg auf 64/65
Guten Abend zusammen,
so eine Reaktion des Kurses aufgrund dieser Meldung ist nicht angepasst. Ich freue mich über jeden hyperaktive nervösen Pessimisten der jetzt aussteigt. Die gleiche Situation habe ich mehrfach bei Evotec erlebt. Die Zeit wird auch bei Morphosys kommen.
so eine Reaktion des Kurses aufgrund dieser Meldung ist nicht angepasst. Ich freue mich über jeden hyperaktive nervösen Pessimisten der jetzt aussteigt. Die gleiche Situation habe ich mehrfach bei Evotec erlebt. Die Zeit wird auch bei Morphosys kommen.
Antwort auf Beitrag Nr.: 55.373.582 von 1MioEuroMan am 21.07.17 19:06:29aber nicht mehr in deinem leben. diese bude ist seid nun gut25 jahren nicht in der lage irgend etwas eigenes auf die beine zu stellen.das schöngerede einiger hier ist schon seltsam.
Was ist denn hier los ? Bitte den Thread-Titel beachten..
Danke
Danke
Na Ja ich möchte darauf hinweisen das ich genau das angemahnt habe: Optimsmus für Anetumab Ravtansine war lediglich basiert auf PR aussagen aus Bayer. Es ist sehr sehr schwierig Fortschritte zu machen in der Behandlung von Fortgeschrittenen Solid Tumors. Mesothelin ist einfach ein Marker das überexprimert der Oberfläche von Krebszellen vorkommt aber nicht nur. Deswegen stellt sich die Frage für mich warum sollte das ganz grosses Erfolg haben im vergleich zu herkommlichen Chemo therapien. Die Lösung ist meine Meinung nach nicht besonders Innovativ. Es ist eine Indikation, aber ich erwarte auch für Eierstockkrebs nix. Würde mich echt überraschen. Wie ich schon früher gesagt habe, in vitro Modelle sind immernoch super schlecht, tier modellen oft nicht adequat....
Kurs wird sich erholen nach dem Quartal zahlen wann die Prognose Anpassen. Und es kommen evtl auch andere Kmeldungen (MOR103 zum beispiel).
Kurs wird sich erholen nach dem Quartal zahlen wann die Prognose Anpassen. Und es kommen evtl auch andere Kmeldungen (MOR103 zum beispiel).
Antwort auf Beitrag Nr.: 55.373.966 von Maslacak am 21.07.17 20:07:51Das ist richtig, Du hast mit Recht frühzeitig Deine Skepsis zum Ausdruck gebracht! Schon aufgrund der Phase Ib-Daten (in der gesamten Expansionskohorte bei maximal verträglicher Dosis gerade mal 19 % PR, 47 % SD, in der Mesotheliom-Kohorte 31 % PR, 44 % SD) war Vorsicht angesagt. Gleichwohl haben sich die Bayer-Verantwortlichen so weit aus dem Fenster gelehnt, dass man eigentlich mit deutlich besseren Daten in der P2-Studie hätte rechnen müssen, zumal das eine unverblindete Studie war, die Verantwortlichen also über das Fortschreiten der Studie immer gut im Bilde waren. Genau das ist der Grund, weshalb sich die Bayer-Führung zur Lachnummer gemacht hat. Wer wird Aussagen des Vorstands zu bestimmten Entwicklungsprojekten künftig noch ernst nehmen? Niemand.
Antwort auf Beitrag Nr.: 55.373.966 von Maslacak am 21.07.17 20:07:51Maslacak, was sind aus Deiner (fachlichen) Sicht die nächsten Hoffnungsträger ?
Bin am Telefon also tut mir leid für extra Tipp Fehlern. Was guselkmab Konkurrenz angeht mag sein das pasi Daten etwas besser sind aber die müssen erstmal noch Phase III machen. und bei Vermarktung Spielt auch Firmen stärke, Marketing etc eine Rolle. Dann kommt auch Geschmack von Ärzten so zu sagen was die für ein Gefühl habrn..
Ich finde es wichtiger wie sich guselkumab in weiteren Indikationen zeigen wird. In Psoriasis ist gut genug um ordentliches Umsatz zu machen denke ich. Ansonsten ist schwer zu tippen auch für mich. Ich habe gesagt ich warte auf Mor103. Da habe ich etwas Hoffnung das es positiv überraschen kann. Utomilumab bei Pfizer könnte dunkel pferd sein. Die schrauben alle wie wild bei immunomodulatoren, ist nicht ganz klar was und wie anspricht und warum sich resistenzen entwickeln....ich habe bei aacr xy Vorträge i gehört mit Erklärungen und Strategien von verschiedenen trials ... Aber utomilumab scheint verträglich zu sein ( relativ gesehen bei immunomodulatoren ist alles heftig) und da laufen mehrere Kombo trials mit pd-1 inhibitoren etc. Pfizer hält die ball flach (in Gegensatz zu Bayer) aber das heisst nix wie wir wissen
Ansonsten möchte ich gerne sagen das ich bisschen rescharchiert habe wegen bimagrumab u d Novartis entscheidung Phase II zu beginnen bei Übergewichtigen. Ja es ist ein mutiges Schritt, aber es gibt wirklich einige sehr interessante Peer reviewrmed Studien die für Verbindung sprechen. Das wäre natürlich ein richtiges "Jack Pot" wann Novartis in so eine Indikation ein medikament enwickeln würde und es ist früh (klinische Daten fehlen) aber ich ich finde die Idee garnicht schlecht.
Ich finde es wichtiger wie sich guselkumab in weiteren Indikationen zeigen wird. In Psoriasis ist gut genug um ordentliches Umsatz zu machen denke ich. Ansonsten ist schwer zu tippen auch für mich. Ich habe gesagt ich warte auf Mor103. Da habe ich etwas Hoffnung das es positiv überraschen kann. Utomilumab bei Pfizer könnte dunkel pferd sein. Die schrauben alle wie wild bei immunomodulatoren, ist nicht ganz klar was und wie anspricht und warum sich resistenzen entwickeln....ich habe bei aacr xy Vorträge i gehört mit Erklärungen und Strategien von verschiedenen trials ... Aber utomilumab scheint verträglich zu sein ( relativ gesehen bei immunomodulatoren ist alles heftig) und da laufen mehrere Kombo trials mit pd-1 inhibitoren etc. Pfizer hält die ball flach (in Gegensatz zu Bayer) aber das heisst nix wie wir wissen
Ansonsten möchte ich gerne sagen das ich bisschen rescharchiert habe wegen bimagrumab u d Novartis entscheidung Phase II zu beginnen bei Übergewichtigen. Ja es ist ein mutiges Schritt, aber es gibt wirklich einige sehr interessante Peer reviewrmed Studien die für Verbindung sprechen. Das wäre natürlich ein richtiges "Jack Pot" wann Novartis in so eine Indikation ein medikament enwickeln würde und es ist früh (klinische Daten fehlen) aber ich ich finde die Idee garnicht schlecht.
Antwort auf Beitrag Nr.: 55.376.135 von Maslacak am 22.07.17 11:21:52Danke Maslacak für deine Einschätzung. Ich bin auch auf MOR 103 gespannt. Was meinst du, wie es hier weitergeht bzw. wir die nächsten Daten sehen? Laut clinicaltrials und Morphosys müßte ich bald so weit sein. Wie groß ist bei MOR 103 das mögliche Salesvolumen? Und wie groß wäre es bei Utomilumab? Ist noch alles früh, aber so eine grobe Hausnummer würde mich interessieren.
Ich hab dir eine persönliche Nachricht geschrieben. Wenn du Zeit hast, guck doch mal bitte.
Ich hab dir eine persönliche Nachricht geschrieben. Wenn du Zeit hast, guck doch mal bitte.
Ich schreibe Anetumab ravtansine noch nicht ab. Mesotheliom ist eine sehr harte Indikation, trotzdem scheint Mesothelin ein gutes ADC-Target zu sein, da nur geringe Expression auf gesundem Gewebe. Die Dosis von 6,5 mg/kg unterstreicht dies, das ist wirklich Oberkante dessen, was die off-target Toxizität von DM4 hergibt; on-target Toxizität scheint also nicht limitierend zu sein. Ausserdem tendiert die Industrie bei ADCs (nicht nur dort) immer mehr Richtung Kombination mit Checkpoint-Inhibitoren, so kann durchaus noch etwas gehen.
Antwort auf Beitrag Nr.: 55.376.267 von yok am 22.07.17 12:08:07Aus meiner Sicht ist es realistischer, Anetumab ravtansine vollständig abzuschreiben.
Das Rennen dürften letztlich anti-mesothelin-CAR T-Tellen machen, mesothelin scheint für CAR T als Target prädestiniert. Gegenwärtig laufen dazu diverse klinische Studien verschiedener Biotech-/Pharmaunternehmen, die sich allesamt in Phase 1 oder 1/2 befinden.
BMS hat noch eine Studie mit einem eigenen anti-mesothelin-ADC laufen (darin auch ein Kombi-Arm mit Nivolumab), während Roche das eigene anti-mesothelin-Projekt (DMOT4039A) meines Wissens nicht mehr weiter fortführt.
Ich selbst bin noch immer verblüfft, dass die Bayer-Verantwortlich in unverantwortlicher Weise solch einen Mist kommuniziert haben.
Das Rennen dürften letztlich anti-mesothelin-CAR T-Tellen machen, mesothelin scheint für CAR T als Target prädestiniert. Gegenwärtig laufen dazu diverse klinische Studien verschiedener Biotech-/Pharmaunternehmen, die sich allesamt in Phase 1 oder 1/2 befinden.
BMS hat noch eine Studie mit einem eigenen anti-mesothelin-ADC laufen (darin auch ein Kombi-Arm mit Nivolumab), während Roche das eigene anti-mesothelin-Projekt (DMOT4039A) meines Wissens nicht mehr weiter fortführt.
Ich selbst bin noch immer verblüfft, dass die Bayer-Verantwortlich in unverantwortlicher Weise solch einen Mist kommuniziert haben.
Antwort auf Beitrag Nr.: 55.376.135 von Maslacak am 22.07.17 11:21:52Auch von meiner Seite einen Dank für Deine wertfreie Einschätzung. Wie meinst Du das mit dem dunklen Pferd ?
Hast Du auch eine Einschätzung zu der geplanten Novartis Phase2/3 Studie mit VAY736 (Autoimmune Hepatitis) ?
Hast Du auch eine Einschätzung zu der geplanten Novartis Phase2/3 Studie mit VAY736 (Autoimmune Hepatitis) ?
Aus meine Sich befinden sich anfängliche In Krebsbehandlung in eine neue Phase. Nachdem anfängliche Euphorie sind Erwartungen immernoch Riesig aber ist ist definitiv klar geworden das viel noch zu lernen und verstehen gibt um den richtig einzusetzten. Dazu hatten andere anti 4IBB antikörper (utolimumab) sehr unangenehme nebenwirkungene ( leber inflammation wann ich mich nicht irre). Deswgen, vermute ich ist Pfizer verhalten. Aber es ist auch klar das die daran arbeiten in verschiedenen kombinationen ( presentiert würde bei ASCO letztes jahr) gerade für fortgeschrittenen Krebsen. Die scheinen akzeptables Safety zu haben.
Ganz ehrlich ich hatte VAY 736 garnicht auf dem Schirm. Ich muss bischen nachlesen
Mesothelin is expprimieret in gesamten peritoneum, soweit ich weiss. Ja es gibt mehr davon in manchen Krebsarten. Aber warum sollte das bessser sein als cisplatinhaltige therapien? Das ist das hauptprobläm in Krebstherapie Generell: Krebszellen könnte mann auf 1000 art und weise umbringen wann mann in der Lage wäre den Spezifisch zu treffen und dabei Geesunde zellen nicht zu sehr zu beinflüssen. Und ich sehe mesothelin als Protein nicht wirklich als ideales Kandidat dieses Targeting zu erreichen.
Ich verstehe es nicht. Es kann natürlich alles sein, manchmal gibt es Überraschungen aber nachdem neuesten Daten, ich wurde mich wundern.
Ganz ehrlich ich hatte VAY 736 garnicht auf dem Schirm. Ich muss bischen nachlesen
Mesothelin is expprimieret in gesamten peritoneum, soweit ich weiss. Ja es gibt mehr davon in manchen Krebsarten. Aber warum sollte das bessser sein als cisplatinhaltige therapien? Das ist das hauptprobläm in Krebstherapie Generell: Krebszellen könnte mann auf 1000 art und weise umbringen wann mann in der Lage wäre den Spezifisch zu treffen und dabei Geesunde zellen nicht zu sehr zu beinflüssen. Und ich sehe mesothelin als Protein nicht wirklich als ideales Kandidat dieses Targeting zu erreichen.
Ich verstehe es nicht.
Es kann natürlich alles sein, manchmal gibt es Überraschungen aber nachdem neuesten Daten, ich wurde mich wundern.
In den erste Satzt fehlte Subjekt Es handelt sich natürlich um Immunotherapien. Vor in paar Jahren gab es pure Euphorie. Jetzt ist das Bild etwas differenzierter. Zwei grossen Problembereichen sind Biomarkers und Resistänzentwicklung. Die wissen es einfach nicht wann es funktionieren wird und wann nicht. Aber jetzt gibt es durch kombi projekte mit verschiedenen Präparaten versuche Immunotheraien zu verbessern. Und utomilumab ist einer davon. Ich finde es nicht schlecht.
Es handelt sich natürlich um Immunotherapien. Vor in paar Jahren gab es pure Euphorie. Jetzt ist das Bild etwas differenzierter. Zwei grossen Problembereichen sind Biomarkers und Resistänzentwicklung. Die wissen es einfach nicht wann es funktionieren wird und wann nicht. Aber jetzt gibt es durch kombi projekte mit verschiedenen Präparaten versuche Immunotheraien zu verbessern. Und utomilumab ist einer davon. Ich finde es nicht schlecht.
AUS für MOR209!
Eigentlich müsste Morphosys das noch vor Börsenbeginn melden.
https://globenewswire.com/news-release/2017/08/31/1106420/0/…
Jetzt wird auch klar, wieso man damals die Phase 1 abgebrochen und mit verändertem Dosisregime neu gestartet hat. Es war allerdings naiv zu glauben, durch das veränderte Dosisregime könne man die Bildung neutralisierender Antikörper verhindern. Mir ist rätselhaft, wie Aptevo an diesem Entwicklungsprojekt festhalten kann.
Eigentlich müsste Morphosys das noch vor Börsenbeginn melden.
https://globenewswire.com/news-release/2017/08/31/1106420/0/…
Jetzt wird auch klar, wieso man damals die Phase 1 abgebrochen und mit verändertem Dosisregime neu gestartet hat. Es war allerdings naiv zu glauben, durch das veränderte Dosisregime könne man die Bildung neutralisierender Antikörper verhindern. Mir ist rätselhaft, wie Aptevo an diesem Entwicklungsprojekt festhalten kann.
Antwort auf Beitrag Nr.: 55.643.714 von Joschka Schröder am 01.09.17 07:56:15
stimmt, habe bei Morphosys schon mal gefragt, wieso das nicht gemeldet wurde.
Auf der Präsentation bei der Commerzbank steht Mor209 ja schon nicht mehr drauf bei expected Newsflow (S. 14). Ist bei Morphosys also vielleicht schon länger quasi abgehakt?!
Zitat von Joschka Schröder: AUS für MOR209!
Eigentlich müsste Morphosys das noch vor Börsenbeginn melden.
https://globenewswire.com/news-release/2017/08/31/1106420/0/…
Jetzt wird auch klar, wieso man damals die Phase 1 abgebrochen und mit verändertem Dosisregime neu gestartet hat. Es war allerdings naiv zu glauben, durch das veränderte Dosisregime könne man die Bildung neutralisierender Antikörper verhindern. Mir ist rätselhaft, wie Aptevo an diesem Entwicklungsprojekt festhalten kann.
stimmt, habe bei Morphosys schon mal gefragt, wieso das nicht gemeldet wurde.
Auf der Präsentation bei der Commerzbank steht Mor209 ja schon nicht mehr drauf bei expected Newsflow (S. 14). Ist bei Morphosys also vielleicht schon länger quasi abgehakt?!
Am 5./6.9.2017 finden bekanntlich die Kapitalmarktkonferenzen der Morphosys AG in London und NY statt. Eigentlich führt man derartige Konferenzen nur dann durch, wenn man wirklich Neues zu berichten hat.
Ich habe zur Einstimmung einen Blick in die gängigen Patentdatenbanken geworfen, dort aber nichts Nennenswertes entdecken können. Zusammen mit Häussinger von der Uniklinik Düsseldorf hat man einen anti-BSEP-Antikörper angemeldet, der ggf. irgendwann einmal zur Therapie der progessiven familiären intrahepatischen Cholestase eingesetzt werden könnte. Ansonsten gähnende Leere.
Desweiteren ist MOR dem Vernehmen nach mit der Entwicklung bispezifischer Antikörper befasst. Dies ist aber ein Thema, das bereits von zig anderen Unternehmen abgedeckt wird.
Was den Fortgang der klinischen Studien betrifft, wird man vermutlich ein Update zu MOR208 geben. Hier schweben die Warnhinweise der Arzneimittelkommission über gehäufte Todesfälle in verschiedenen Benamustin-Kombistudien über der laufenden Phase 2/3-Studie. Hier kann nicht ausgeschlossen werden, dass für derartige Kombinationsbehandlungen künftig keine Zulassungen mehr erteilt werden.
Hmmm, das sind aber eigentlich alles keine Neuigkeiten, derentwegen man eine Kapitalmarktkonferenz durchführen würde (das Bendamustin-Problem würde man im Gegenteil vermutlich von selbst wohl eher nicht ansprechen).
Um so gespannter darf man sein, was MOR während der letzten Jahre - offenbar im Verborgenen - tatsächlich entwickelt hat und in der nächsten Woche präsentieren wird.
Ich habe zur Einstimmung einen Blick in die gängigen Patentdatenbanken geworfen, dort aber nichts Nennenswertes entdecken können. Zusammen mit Häussinger von der Uniklinik Düsseldorf hat man einen anti-BSEP-Antikörper angemeldet, der ggf. irgendwann einmal zur Therapie der progessiven familiären intrahepatischen Cholestase eingesetzt werden könnte. Ansonsten gähnende Leere.
Desweiteren ist MOR dem Vernehmen nach mit der Entwicklung bispezifischer Antikörper befasst. Dies ist aber ein Thema, das bereits von zig anderen Unternehmen abgedeckt wird.
Was den Fortgang der klinischen Studien betrifft, wird man vermutlich ein Update zu MOR208 geben. Hier schweben die Warnhinweise der Arzneimittelkommission über gehäufte Todesfälle in verschiedenen Benamustin-Kombistudien über der laufenden Phase 2/3-Studie. Hier kann nicht ausgeschlossen werden, dass für derartige Kombinationsbehandlungen künftig keine Zulassungen mehr erteilt werden.
Hmmm, das sind aber eigentlich alles keine Neuigkeiten, derentwegen man eine Kapitalmarktkonferenz durchführen würde (das Bendamustin-Problem würde man im Gegenteil vermutlich von selbst wohl eher nicht ansprechen).
Um so gespannter darf man sein, was MOR während der letzten Jahre - offenbar im Verborgenen - tatsächlich entwickelt hat und in der nächsten Woche präsentieren wird.
Antwort auf Beitrag Nr.: 55.654.124 von Joschka Schröder am 02.09.17 16:22:52
Was könnte neu sein
Die Macher von MorphoSys könnten es toll finden in tolle neue innovative Projekte einzusteigen / die selbst finanziert werden und wohl auch ein wenig dauern, wenn sowas bekannt gegeben wird, werden sich einige kurzfristige Spekulanten verabschieden und die Leerverkäufer hätten ihre Ruh - einige wü
Antwort auf Beitrag Nr.: 55.655.063 von guztaw am 02.09.17 22:16:36
Zu dicker Finger
Text vor Abschluß gesendet, eigentlich wollte ich nur noch bemerken, bei allem pro oder contra mit dem Schuppenflechtemedikament gibt es lange Zeit sichere Einnahmen, wie hoch weiß auch ich nicht
Ich sehe mir gerade die aktuelle MOR-Präsentation an und kann schon jetzt sagen, dass mir das Unternehmen immer weniger gefällt. In Teilen ist die Präsentation sogar als als unseriös zu bezeichnen.
"world-class technology platforms" ... "world-class developement organization" ... wie komplexbeladen und arm muss man sein, um sich selbst in dieser Form darzustellen?
Die präsentierte Pipeline umfasst Ladenhüter, die überhaupt nicht mehr weiterentwickelt werden.
In der Studien-Vergleichsübersicht zu MOR208 fehlen die besten Konkurrenzpräparate.
Bzgl. MOR202 wird einmal mehr auf eine bessere Infusionsverträglichkeit hingewiesen, gleichzeitig wird aber verschwiegen, dass das sonstige Nebenwirkungsprofil des MOR202 VIEL schlechter ist als das anderer anti-CD38-Mabs.
Die Aussage, MOR202 wirke so gut wie Daratumumab, steht im Widerspruch zu den bisherigen Studienergebnissen.
Werde jetzt mal weiterlesen, hoffentlich geht das nicht in dieser Form so weiter.
"world-class technology platforms" ... "world-class developement organization" ... wie komplexbeladen und arm muss man sein, um sich selbst in dieser Form darzustellen?
Die präsentierte Pipeline umfasst Ladenhüter, die überhaupt nicht mehr weiterentwickelt werden.
In der Studien-Vergleichsübersicht zu MOR208 fehlen die besten Konkurrenzpräparate.
Bzgl. MOR202 wird einmal mehr auf eine bessere Infusionsverträglichkeit hingewiesen, gleichzeitig wird aber verschwiegen, dass das sonstige Nebenwirkungsprofil des MOR202 VIEL schlechter ist als das anderer anti-CD38-Mabs.
Die Aussage, MOR202 wirke so gut wie Daratumumab, steht im Widerspruch zu den bisherigen Studienergebnissen.
Werde jetzt mal weiterlesen, hoffentlich geht das nicht in dieser Form so weiter.
So, ich hab die Präsentation jetzt in toto durchgesehen. Etwas Neues oder Innovatives kann ich leider nicht erkennen. Insbesonders die erstmals vorgestellten Entwicklungsprojekte sind alte Hüte. Einen Anti-C5AR-Antikörper hat bereits Innate Pharma in der Klinik (IPH5401) und bispezifische Mabs, die CD3 binden, gibt es bereits wie Sand am Meer. Was die Morphosys F&E seit einiger Zeit zustande bringt, ist enttäuschend. Wo bleibt eine wirklich innovative Technik, wo ein interessanter first-in-class-Antibody? Den Aufwand der Kapitalmarktkonferenzen in London und NY hätte man sich schenken können.
Antwort auf Beitrag Nr.: 55.669.386 von Joschka Schröder am 05.09.17 12:25:02Korrektur: IPH5401 steht erst unmittelbar vor Eintritt in die Klinik. In einigen Monaten wird es soweit sein.
Antwort auf Beitrag Nr.: 55.669.467 von Joschka Schröder am 05.09.17 12:33:25Die Angaben zur Immatrics- und MDA-Kooperation auf S.47 f. sind (zumindest für mich) neu.
Dies könnte mit der Entwicklung zielgerichteter Antikörper gemeint gewesen sein, was kürzlich im Parallelthread von Solideinvestierenforyou gepostet wurde.
Klingt nicht uninteressant, ist aber auch erst in der Lead Optimization (zumindest der Immatrics-Part).
Dies könnte mit der Entwicklung zielgerichteter Antikörper gemeint gewesen sein, was kürzlich im Parallelthread von Solideinvestierenforyou gepostet wurde.
Klingt nicht uninteressant, ist aber auch erst in der Lead Optimization (zumindest der Immatrics-Part).
Wer von MOR208 überzeugt ist, könnte sich ggf. überlegen, Xencor-Aktien zu kaufen. Xencor notiert gegenwärtig mit einem Enterprise Value von 632 Mio. USD. Im Fall einer Zulassung von MOR208 gibt´s 187 Mio. USD von Morphosys, was einem Drittel des derzeitigen Xencor-Unternehmenswertes entspricht.
Xencors Fc-Optimierungstechnik ist top, negativ ist, dass mit XmAb7195 ein anti-IgE-Antikörper entwickelt wird, der in klinischen Studien als Nebenwirkung Urtikaria hervorruft, mithin eine Symptomatik, die sonst in schweren Fällen mit einem anti-IgE-Antikörper (Xolair von Genentech und Novartis) therapiert wird. Insoweit sind die F&E-Kosten für XMAb7195 aus meiner Sicht herausgeschmissenes Geld. Davon abgesehen befinden sich aber mehrere durchaus interessante Präparate in der Xencor-Pipeline.
Xencors Fc-Optimierungstechnik ist top, negativ ist, dass mit XmAb7195 ein anti-IgE-Antikörper entwickelt wird, der in klinischen Studien als Nebenwirkung Urtikaria hervorruft, mithin eine Symptomatik, die sonst in schweren Fällen mit einem anti-IgE-Antikörper (Xolair von Genentech und Novartis) therapiert wird. Insoweit sind die F&E-Kosten für XMAb7195 aus meiner Sicht herausgeschmissenes Geld. Davon abgesehen befinden sich aber mehrere durchaus interessante Präparate in der Xencor-Pipeline.
Antwort auf Beitrag Nr.: 55.675.599 von Joschka Schröder am 05.09.17 23:54:10Das klingt doch hervorragend für Morphosys.
Kann zwar nicht abschätzen, welches Umsatzpotential hinter MOR208 steckt, aber bei einer Zahlung von USD 187 Mio. allein an Xencor dürfte es sich hier um sehr nennenswerte Beträge handeln.
Wann und wo wurde die Zahlung von USD 187 Mio. kommuniziert? Ich kann mich nur an eine Meldung erinnern, wonach weitere finanzielle Einzelheiten nicht bekanntgegeben werden.
Kann zwar nicht abschätzen, welches Umsatzpotential hinter MOR208 steckt, aber bei einer Zahlung von USD 187 Mio. allein an Xencor dürfte es sich hier um sehr nennenswerte Beträge handeln.
Wann und wo wurde die Zahlung von USD 187 Mio. kommuniziert? Ich kann mich nur an eine Meldung erinnern, wonach weitere finanzielle Einzelheiten nicht bekanntgegeben werden.
Antwort auf Beitrag Nr.: 55.679.085 von eltorero am 06.09.17 13:19:15Was soll daran hervorragend für Morphosys sein? Morphosys muss zahlen! Gut ist die Regelung für Xencor.
Die Daten finden sich in alten SEC-files.
Die Daten finden sich in alten SEC-files.
Antwort auf Beitrag Nr.: 55.679.334 von Joschka Schröder am 06.09.17 13:43:06
Und nicht zu knapp: http://investors.xencor.com/secfiling.cfm?filingid=1558370-1…
..."Under the terms of the MorphoSys Agreement, we received a $13 million upfront payment and $3 million for development milestone payments in 2013. If certain developmental, regulatory and sales milestones are achieved, we are also eligible to receive up to an additional $299 million in milestone payments which are comprised as follows: $62.0 million in clinical development milestones, $187 million in regulatory approval milestones and $50 million of aggregate milestone payments for the achievement of certain product sale goals. If licensed products are commercialized, we are also entitled to receive tiered royalties in the high single ‑digit to low ‑teen percent range..."
Zitat von Joschka Schröder: Was soll daran hervorragend für Morphosys sein? Morphosys muss zahlen!...
Und nicht zu knapp: http://investors.xencor.com/secfiling.cfm?filingid=1558370-1…
..."Under the terms of the MorphoSys Agreement, we received a $13 million upfront payment and $3 million for development milestone payments in 2013. If certain developmental, regulatory and sales milestones are achieved, we are also eligible to receive up to an additional $299 million in milestone payments which are comprised as follows: $62.0 million in clinical development milestones, $187 million in regulatory approval milestones and $50 million of aggregate milestone payments for the achievement of certain product sale goals. If licensed products are commercialized, we are also entitled to receive tiered royalties in the high single ‑digit to low ‑teen percent range..."
Antwort auf Beitrag Nr.: 55.679.334 von Joschka Schröder am 06.09.17 13:43:06Ich finde Deine Argumentation nicht recht nachvollziehbar... Was soll dann eigentlich für Novartis, J&J, Pfizer usw. gut an den Kooperationen mit Morphosys sein??? Die müssen auch zahlen!
Im Übrigen sollten mit Pitschnass' Post endlich die unsinnigen 187 Mio. bei Erstzulassung vom Tisch sein.
"...If certain developmental, regulatory and sales milestones are achieved, we are also eligible to receive up to an additional $299 million in milestone payments..."
Im Übrigen sollten mit Pitschnass' Post endlich die unsinnigen 187 Mio. bei Erstzulassung vom Tisch sein.
"...If certain developmental, regulatory and sales milestones are achieved, we are also eligible to receive up to an additional $299 million in milestone payments..."
Antwort auf Beitrag Nr.: 55.680.570 von Milestones am 06.09.17 15:41:45Kurz zu Deinen lustigen Anmerkungen:
1) Wieso sollen die genannten 187 Mio. USD Meilensteinzahlung bei Zulassung "unsinnig" sein? Wörtlich heißt es "$187 million in regulatory approval milestones".
2) Zu Deinem Novartis- und J&J-Vergleich: Wie hoch ist der Betrag, den Morphosys z.B. von J&J als Meilensteinzahlung für die Guselkumab-Zulassung bekommen hat? 187 Mio. USD? Oder war es ein kleiner einstelliger Millionenbetrag?
Und nun?
1) Wieso sollen die genannten 187 Mio. USD Meilensteinzahlung bei Zulassung "unsinnig" sein? Wörtlich heißt es "$187 million in regulatory approval milestones".
2) Zu Deinem Novartis- und J&J-Vergleich: Wie hoch ist der Betrag, den Morphosys z.B. von J&J als Meilensteinzahlung für die Guselkumab-Zulassung bekommen hat? 187 Mio. USD? Oder war es ein kleiner einstelliger Millionenbetrag?
Und nun?
Es kommt immer auf die Relation zwischen Meilensteinzahlung und Produktumsatz an.
Guselkumab wird wahrscheinlich Jahresumsätze in Milliardenhöhe generieren, gleichwohl ist bei Zulassung nur eine vernachlässigbare Meilensteinzahlung von J&J an Morphosys geflossen.
MOR208 wird im positiven Fall einen kleinen, dreistelligen Millionenumsatz generieren, bei Zulassung werden jedoch 187 Mio. USD von Morphosys an Xencor fließen.
Guselkumab wird wahrscheinlich Jahresumsätze in Milliardenhöhe generieren, gleichwohl ist bei Zulassung nur eine vernachlässigbare Meilensteinzahlung von J&J an Morphosys geflossen.
MOR208 wird im positiven Fall einen kleinen, dreistelligen Millionenumsatz generieren, bei Zulassung werden jedoch 187 Mio. USD von Morphosys an Xencor fließen.
Antwort auf Beitrag Nr.: 55.680.831 von Joschka Schröder am 06.09.17 16:13:03Du hast recht, Morphosys wird bei Zulassung aller geplanter Indikationen Meilensteinzahlungen in Höhe von 187 Mio. US$ leisten.
Meinst du wirklich, Moroney zahlt mehr, als er einnimmt?
Meinst du wirklich, Moroney zahlt mehr, als er einnimmt?
Antwort auf Beitrag Nr.: 55.681.404 von Milestones am 06.09.17 17:07:35Das ganze wurde letztes Jahr schon ausführlich diskutiert:
https://www.wallstreet-online.de/diskussion/1205075-3101-311…
Ville7 wollte damals bei Xencor nachfragen. Ich weiß nicht, ob er eine Antwort bekommen hat.
Wenn ich aber lese, dass im Juli 2017 $12,5 Mio für den Phase 3 Start an Xencor gegangen sind, habe ich auch nicht wirklich ein gutes Gefühl bei der Sache. Vielleicht hatte Moroney etwas zu breite Schultern als er den Deal abschloß...
In June 2017, MorphoSys AG announced the initiation of the pivotal Phase 3 portion of its Phase 2/3 clinical trial of XmAb5574/MOR208 in combination with bendamustine for the treatment of relapsed or refractory large B-cell lymphoma (B-MIND trial). MOR208 uses Xencor's XmAb Cytotoxic Fc Domain. The trial initiation triggered a milestone payment to Xencor of $12.5 million.
http://investors.xencor.com/releasedetail.cfm?releaseid=1036…
https://www.wallstreet-online.de/diskussion/1205075-3101-311…
Ville7 wollte damals bei Xencor nachfragen. Ich weiß nicht, ob er eine Antwort bekommen hat.
Wenn ich aber lese, dass im Juli 2017 $12,5 Mio für den Phase 3 Start an Xencor gegangen sind, habe ich auch nicht wirklich ein gutes Gefühl bei der Sache. Vielleicht hatte Moroney etwas zu breite Schultern als er den Deal abschloß...
In June 2017, MorphoSys AG announced the initiation of the pivotal Phase 3 portion of its Phase 2/3 clinical trial of XmAb5574/MOR208 in combination with bendamustine for the treatment of relapsed or refractory large B-cell lymphoma (B-MIND trial). MOR208 uses Xencor's XmAb Cytotoxic Fc Domain. The trial initiation triggered a milestone payment to Xencor of $12.5 million.
http://investors.xencor.com/releasedetail.cfm?releaseid=1036…
Es ist ja zumindest richtig, dass da "approval milestones" steht. Plural. Ich würde da auch der Interpretation zuneigen, dass das ein Höchstbetrag bei Zulassung in zahlreichen Indikationen ist. Der wird wohl kaum fällig sein, wenn man als Second Line Therapie in DCLBC zugelassen wird. Ich bin zwar grad auch von vielem nicht so begeistert und überlege über 65 € eine relevante Position zu verkaufen, aber gänzlich bescheuert ist Moroney ja nun auch nicht.
Wenn dass so ist, dass Morphosys USD 187 Mio. an Xencor zahlt, um einen kleinen dreistelligen Millionenumsatz zu machen und gleichzeitig nur eine relativ kleine Summe von Janssen erhält, damit die dann Jahresumsätze in Milliardenhöhe erzielen, sollte man sehen dass man die Aktie verkauft, weil man dann am Verstand des Managements zweifeln sollte.
Warum wohl sollte Morphosys die genannten Meilensteinzahlungen an Xencor vereinbart haben? Wurden sie etwa gezwungen? Wohl kaum. Es handelt sich um einen zweiseitigen Vertrag, in dem beide Seiten die wirtschaftlichen Vorteile abgewogen haben. Und wenn Morphosysy bereit ist, allein für die Zulassung USD 187 Mio. zu zahlen, dann dürften wirtschaftlichen Vorteile daraus um einiges höher sein.
Warum wohl sollte Morphosys die genannten Meilensteinzahlungen an Xencor vereinbart haben? Wurden sie etwa gezwungen? Wohl kaum. Es handelt sich um einen zweiseitigen Vertrag, in dem beide Seiten die wirtschaftlichen Vorteile abgewogen haben. Und wenn Morphosysy bereit ist, allein für die Zulassung USD 187 Mio. zu zahlen, dann dürften wirtschaftlichen Vorteile daraus um einiges höher sein.
Absurde Diskussion. Das Medikament sollte erstmal Marktreife erlangen.Coba Analyst nennt 2020 als machbar.
Als leidgeprüfter Aktionär weiss man um die Risiken.
Als leidgeprüfter Aktionär weiss man um die Risiken.
Antwort auf Beitrag Nr.: 55.681.995 von aalerich am 06.09.17 18:12:17Hoffen wir mal das es daneben geht, dann spart Morphosys die USD 187 Mio. 

Antwort auf Beitrag Nr.: 55.682.082 von eltorero am 06.09.17 18:24:13bist du besoffen oder grad auf einem ironischen Trip?
Antwort auf Beitrag Nr.: 55.683.426 von milchbubi am 06.09.17 20:56:07ok, hab erst jetzt deine vorangegangen Posts gelesen. Nehme das besoffen zurück und wir sind derselben Meinung
Antwort auf Beitrag Nr.: 55.681.599 von deadflowers am 06.09.17 17:28:17
Was hat Dich denn innerhalb der letzten acht Wochen, als MOR zuletzt über 65€ stand, so verdrossen, dass Du nun über einen Teilverkauf nachdenkst?
Zitat von deadflowers: Ich bin zwar grad auch von vielem nicht so begeistert und überlege über 65 € eine relevante Position zu verkaufen, aber gänzlich bescheuert ist Moroney ja nun auch nicht.
Was hat Dich denn innerhalb der letzten acht Wochen, als MOR zuletzt über 65€ stand, so verdrossen, dass Du nun über einen Teilverkauf nachdenkst?
Antwort auf Beitrag Nr.: 55.681.599 von deadflowers am 06.09.17 17:28:17
Eine derartige (Indikations-basierte) Vereinbarung wäre absolut ungewöhnlich. Üblich ist: Plural = Erstzulassung in verschiedenen Regionen (USA, Europa, Japan, ROW).
Zitat von deadflowers: Es ist ja zumindest richtig, dass da "approval milestones" steht. Plural. Ich würde da auch der Interpretation zuneigen, dass das ein Höchstbetrag bei Zulassung in zahlreichen Indikationen ist.
Eine derartige (Indikations-basierte) Vereinbarung wäre absolut ungewöhnlich. Üblich ist: Plural = Erstzulassung in verschiedenen Regionen (USA, Europa, Japan, ROW).
Antwort auf Beitrag Nr.: 55.681.404 von Milestones am 06.09.17 17:07:35@Milestones: Ich habe eben noch einmal meinen Beitrag 694 durchgelesen. Er gefällt mir nicht, er ist zu polemisch formuliert. Das passiert mir leider gelegentlich, wenn ich unter Zeitdruck im Forum poste. Bitte sieh es mir nach und nimm es nicht persönlich!
Antwort auf Beitrag Nr.: 55.684.149 von Joschka Schröder am 06.09.17 22:08:18
Ist ein valides Argument und durchaus möglich, dass es sich auf die Regionen bezieht. Mir kommt es dafür aber einfach sehr viel vor. Bei einer der beiden Firmen nachzufragen wäre evtl. eine Möglichkeit, das zu klären.
Zitat von Joschka Schröder:Zitat von deadflowers: Es ist ja zumindest richtig, dass da "approval milestones" steht. Plural. Ich würde da auch der Interpretation zuneigen, dass das ein Höchstbetrag bei Zulassung in zahlreichen Indikationen ist.
Eine derartige (Indikations-basierte) Vereinbarung wäre absolut ungewöhnlich. Üblich ist: Plural = Erstzulassung in verschiedenen Regionen (USA, Europa, Japan, ROW).
Ist ein valides Argument und durchaus möglich, dass es sich auf die Regionen bezieht. Mir kommt es dafür aber einfach sehr viel vor. Bei einer der beiden Firmen nachzufragen wäre evtl. eine Möglichkeit, das zu klären.
Antwort auf Beitrag Nr.: 55.683.621 von BReal am 06.09.17 21:17:58Wichtigster Grund ist das Scheitern von Anetumab Ravtansine. Da hatte sich Bayer so weit aus dem Fenster gelehnt, dass mich das echt überrascht hat. So richtig absehbar kommt eigentlich nichts nach, BPS 804 vielleicht in ner Nischenindikation.
Die Unternehmenskommunikation finde ich auch zunehmend bizarr. Und letztlich ist es einfach immer noch ein sehr großer Depotanteil, obwohl andere Sachen viel besser gelaufen sind. Denke darüber nach, etwa ein Drittel bis die Hälfte zu verkaufen.
Die Unternehmenskommunikation finde ich auch zunehmend bizarr. Und letztlich ist es einfach immer noch ein sehr großer Depotanteil, obwohl andere Sachen viel besser gelaufen sind. Denke darüber nach, etwa ein Drittel bis die Hälfte zu verkaufen.
Antwort auf Beitrag Nr.: 55.685.286 von deadflowers am 07.09.17 08:12:32Danke für Deine Antwort. Das klingt nicht unvernünftig, ich hatte meinen Bestand schon vor ein paar Jahren halbiert, der Rest verbleibt jedoch mindestens bis 2025/2030 im Depot.
Ich war in den letzten Wochen eher positiv von den Entwicklungen angetan (deshalb auch meine Frage an Dich zu Deinen Beweggründen):
- MOR103 wird nun von GSK priorisiert
- das (von Celgene letztlich voll finanzierte!) Projekt MOR202 wird für MOR kein Groschengrab werden
- wenn MOR208 wie geplant auslizensiert werden kann (USA? ROW?), werden auch die Milestones für Xencor kein Problem darstellen
- MOR107 könnte bei positiven Daten durchaus eine Überraschung werden und äußerst schnell durch die Klinik kommen
- man scheint nun endlich auch das Prinzip Phase I/II + Phase IIb/III für die eigenen Projekte adaptieren zu wollen (unsäglich, wie lange MOR202 schon herumkrebst)
- die Peptidforschung mit Immatics klingt durchaus zukunftstauglich, wenn sie funktionieren sollte
- die Patentklage lässt bislang hoffen, dass zumindest ein kleiner Betrag in einer außergerichtlichen Einigung herumkommen könnte
usw.
Ich war in den letzten Wochen eher positiv von den Entwicklungen angetan (deshalb auch meine Frage an Dich zu Deinen Beweggründen):
- MOR103 wird nun von GSK priorisiert
- das (von Celgene letztlich voll finanzierte!) Projekt MOR202 wird für MOR kein Groschengrab werden
- wenn MOR208 wie geplant auslizensiert werden kann (USA? ROW?), werden auch die Milestones für Xencor kein Problem darstellen
- MOR107 könnte bei positiven Daten durchaus eine Überraschung werden und äußerst schnell durch die Klinik kommen
- man scheint nun endlich auch das Prinzip Phase I/II + Phase IIb/III für die eigenen Projekte adaptieren zu wollen (unsäglich, wie lange MOR202 schon herumkrebst)
- die Peptidforschung mit Immatics klingt durchaus zukunftstauglich, wenn sie funktionieren sollte
- die Patentklage lässt bislang hoffen, dass zumindest ein kleiner Betrag in einer außergerichtlichen Einigung herumkommen könnte
usw.
Antwort auf Beitrag Nr.: 55.685.667 von BReal am 07.09.17 08:59:00Ich meinte eben MOR106, nicht MOR107.
Antwort auf Beitrag Nr.: 55.668.978 von Joschka Schröder am 05.09.17 11:44:32Ich teile deine Einschätzung weitgehend. Auch mir gefällt Morphosys von Jahr zu Jahr weniger. Auch und vor allem aus den von dir genannten Gründen... hohe Nase und dafür bisher außer Masse (anzahl Projekte) kaum Resultate und auch kein WOW Effekt in der Pipe oder in der Technologie und weiterhin nur Hausmannskost-Technologie Ansätze, die inzwischen jeder kann...
Der interessanteste Part der Präsentationen war, dass man das erste MOR208 Filing bei einer Zulassungsbehörde um ca. ein Jahr beschleunigen möchte und gute Chancen dafür sieht (nach aktuellem Plan dann 2019 statt 2020 auf Basis B-Mind). Nach persönlicher Interpretation aller Antworten auf Nachfragen dazu gehe ich davon aus, dass man auf Basis der aktuellen L-Mind Studie in den USA einen Zulassungsantrag einreichen möchte und befindet sich dazu mit der FDA in Gesprächen.
Insgesamt sind die abgezielten Indikationen inzwischen eine extreme crowdy space, aber dennoch: ganz so negativ sehe ich MOR208 nicht. Die neuen Medikamte wie z.B. CAR-T haben sehr starke Nebenwirkungen. Zudem ist Ritiximab oder deren Nachfolger weiter stark als SOC verbreitet und hier könnte MOR208 - wenn auch mühsam - Anteile abjagen. Zudem sind die Ergebnisse der L-Mind Studie in dieser elderly patient population eigentlich gar nicht so schlecht - die Nebenwirkungen vergleichsweise gering gegenüber neueren Ansätzen. Neben ORR scheint vor allem die DoR nicht schlecht zu sein. Also wieso nicht versuchen - die FDA sieht ja alles inzwischen viel viel lockerer. Also definitiv kein Überrendite Produkt, aber ein Verlustbringer wird das wahrscheinlich auch nicht werden...
Der interessanteste Part der Präsentationen war, dass man das erste MOR208 Filing bei einer Zulassungsbehörde um ca. ein Jahr beschleunigen möchte und gute Chancen dafür sieht (nach aktuellem Plan dann 2019 statt 2020 auf Basis B-Mind). Nach persönlicher Interpretation aller Antworten auf Nachfragen dazu gehe ich davon aus, dass man auf Basis der aktuellen L-Mind Studie in den USA einen Zulassungsantrag einreichen möchte und befindet sich dazu mit der FDA in Gesprächen.
Insgesamt sind die abgezielten Indikationen inzwischen eine extreme crowdy space, aber dennoch: ganz so negativ sehe ich MOR208 nicht. Die neuen Medikamte wie z.B. CAR-T haben sehr starke Nebenwirkungen. Zudem ist Ritiximab oder deren Nachfolger weiter stark als SOC verbreitet und hier könnte MOR208 - wenn auch mühsam - Anteile abjagen. Zudem sind die Ergebnisse der L-Mind Studie in dieser elderly patient population eigentlich gar nicht so schlecht - die Nebenwirkungen vergleichsweise gering gegenüber neueren Ansätzen. Neben ORR scheint vor allem die DoR nicht schlecht zu sein. Also wieso nicht versuchen - die FDA sieht ja alles inzwischen viel viel lockerer. Also definitiv kein Überrendite Produkt, aber ein Verlustbringer wird das wahrscheinlich auch nicht werden...
Antwort auf Beitrag Nr.: 55.684.581 von Joschka Schröder am 06.09.17 23:42:28Schwamm drüber...
Ich bin mir zwar ziemlich sicher, dass Zulassungsmeilensteine (auch) indikationsbezogen gezahlt werden, kann es aber momentan aufgrund von Zeitnot nicht verifizieren. Aber Morphosys wird garantiert für weitere Guselkumab-Zulassungen z.B. in den Indikationen RS oder MC weitere Zulassungsmeilensteinzahlungen bekommen.
Außerdem kann man Guselkumab nicht mit MOR208 vergleichen, da MOR208 viel weiter entwickelt war.
Morphosys hat bei Guselkumab ja nur die Identifizierung und Optimierung der AK-Kandidaten geleistet. Die komplette Entwicklung lag bei J&J (bzw. Janssen bzw. anfangs Centocor). Wenn du dir MOR103 ansiehst, bekommst du eher einen Eindruck der Relationen.
Morphosys kann hier abhängig von bestimmten Entwicklungsschritten und regulatorischen usw. Meilensteinen bis zu 423 Mio. € bekommen (im Vergleich zu 299 Mio. US$). Dazu noch die 22 Mio. € Upfront im Vergleich zu 13 Mio. US$ und gestaffelte Royalties im zweistelligen Mio.-bereich im Gegensatz zu (gestaffelten) hohen einstelligen bis niedrigen zweistelligen Royalties für Xencor, die wohl kaum eine 2 ganz vorne stehen haben werden... (ich rechne mit einer Range von ca. 8-15%)
Ich denke, Moroney ist ein recht guter Verhandler, der rechnen kann und ziemlich genau weiß, was er will und worauf er sich einlässt. Das hat er in der Vergangenheit ausgiebig bewiesen.
Ich bin mir zwar ziemlich sicher, dass Zulassungsmeilensteine (auch) indikationsbezogen gezahlt werden, kann es aber momentan aufgrund von Zeitnot nicht verifizieren. Aber Morphosys wird garantiert für weitere Guselkumab-Zulassungen z.B. in den Indikationen RS oder MC weitere Zulassungsmeilensteinzahlungen bekommen.
Außerdem kann man Guselkumab nicht mit MOR208 vergleichen, da MOR208 viel weiter entwickelt war.
Morphosys hat bei Guselkumab ja nur die Identifizierung und Optimierung der AK-Kandidaten geleistet. Die komplette Entwicklung lag bei J&J (bzw. Janssen bzw. anfangs Centocor). Wenn du dir MOR103 ansiehst, bekommst du eher einen Eindruck der Relationen.
Morphosys kann hier abhängig von bestimmten Entwicklungsschritten und regulatorischen usw. Meilensteinen bis zu 423 Mio. € bekommen (im Vergleich zu 299 Mio. US$). Dazu noch die 22 Mio. € Upfront im Vergleich zu 13 Mio. US$ und gestaffelte Royalties im zweistelligen Mio.-bereich im Gegensatz zu (gestaffelten) hohen einstelligen bis niedrigen zweistelligen Royalties für Xencor, die wohl kaum eine 2 ganz vorne stehen haben werden... (ich rechne mit einer Range von ca. 8-15%)
Ich denke, Moroney ist ein recht guter Verhandler, der rechnen kann und ziemlich genau weiß, was er will und worauf er sich einlässt. Das hat er in der Vergangenheit ausgiebig bewiesen.
Antwort auf Beitrag Nr.: 55.681.554 von Pitschnass am 06.09.17 17:23:20
Hatte ich damals dann doch nicht gemacht... vielleicht hat jemand anderer nachgefragt?
Zitat von Pitschnass: Ville7 wollte damals bei Xencor nachfragen. Ich weiß nicht, ob er eine Antwort bekommen hat.
Hatte ich damals dann doch nicht gemacht... vielleicht hat jemand anderer nachgefragt?
Antwort auf Beitrag Nr.: 55.687.194 von Ville7 am 07.09.17 11:18:29@Ville
MOR208 sehe ich ähnlich. Das Präparat zeigt bislang gute Studienergebnisse. Der Markt ist allerdings hart umkämpft und es gibt ausgezeichnete Konkurrenzpräparate, die aus welchen Gründen auch immer in MOR-Präsentationen nicht einmal erwähnt werden (ADCT-402, Adcetris, IMGN529, Coltuximab [falls sich ein Lizenznehmer findet], Obinutuzumab etc.). Rituximab hingegen ist ein Auslaufmodell.
@Milestones
Was das Verhandlungsgeschick Moroneys betrifft, kann ich mir kein Urteil erlauben. Bislang gibt es da wenig Greifbares. Die weit über 30 Mio. USD (bis Ende 2015 waren es 30,8 Mio. USD), die mit MOR209 versenkt worden sind, stellen schon einmal keine Glanzleistung dar. Ob sich das MOR208-Investment langfristig rechnen wird, wird man sehen.
Von MOR202 halte ich weiterhin nicht viel, die Einschätzungen der Unternehmensführung stehen im Widerspruch zur Datenlage. Für eine Überraschung könnte andererseits MOR103 sorgen.
Insgesamt scheint Morphosys derzeit fair bis ambitioniert bewertet. Einen begründeten Bewertungsspielraum nach oben sehe ich aktuell nicht.
MOR208 sehe ich ähnlich. Das Präparat zeigt bislang gute Studienergebnisse. Der Markt ist allerdings hart umkämpft und es gibt ausgezeichnete Konkurrenzpräparate, die aus welchen Gründen auch immer in MOR-Präsentationen nicht einmal erwähnt werden (ADCT-402, Adcetris, IMGN529, Coltuximab [falls sich ein Lizenznehmer findet], Obinutuzumab etc.). Rituximab hingegen ist ein Auslaufmodell.
@Milestones
Was das Verhandlungsgeschick Moroneys betrifft, kann ich mir kein Urteil erlauben. Bislang gibt es da wenig Greifbares. Die weit über 30 Mio. USD (bis Ende 2015 waren es 30,8 Mio. USD), die mit MOR209 versenkt worden sind, stellen schon einmal keine Glanzleistung dar. Ob sich das MOR208-Investment langfristig rechnen wird, wird man sehen.
Von MOR202 halte ich weiterhin nicht viel, die Einschätzungen der Unternehmensführung stehen im Widerspruch zur Datenlage. Für eine Überraschung könnte andererseits MOR103 sorgen.
Insgesamt scheint Morphosys derzeit fair bis ambitioniert bewertet. Einen begründeten Bewertungsspielraum nach oben sehe ich aktuell nicht.
Antwort auf Beitrag Nr.: 55.669.467 von Joschka Schröder am 05.09.17 12:33:25Hallo Joschka,
Wie schätzt du die Pipeline von Innate Pharma abgesehen von dem anti-C5aR , welcher im übrigen auch nicht ganz billig war, ein?
Dieser wurde zu folgenden Bedingungen eingekauft:
"The terms of the agreement provide for an upfront payment of €40m, of which €37.2m will be paid in the form of new shares in the Company and €2.8m will be paid in cash. Novo is eligible for up to €370m by way of development, regulatory and sales milestone payments and to double digit royalties on future net sales."
Welche Programme von Innate könnten noch aussichtsreich bzw. lukrativ sein?
Innate Pharma scheint ein solider European Player zu sein, welcher aktuell eher moderat bepreist wird.
Viele Grüße
Wie schätzt du die Pipeline von Innate Pharma abgesehen von dem anti-C5aR , welcher im übrigen auch nicht ganz billig war, ein?
Dieser wurde zu folgenden Bedingungen eingekauft:
"The terms of the agreement provide for an upfront payment of €40m, of which €37.2m will be paid in the form of new shares in the Company and €2.8m will be paid in cash. Novo is eligible for up to €370m by way of development, regulatory and sales milestone payments and to double digit royalties on future net sales."
Welche Programme von Innate könnten noch aussichtsreich bzw. lukrativ sein?
Innate Pharma scheint ein solider European Player zu sein, welcher aktuell eher moderat bepreist wird.
Viele Grüße
Antwort auf Beitrag Nr.: 55.707.312 von biopadawan am 10.09.17 12:11:28was hat das mit Morphosys zu tun???? Schick Joschka halt eine PN.
zum Themenkomplex MOR202, CD38, Multiples Myelom:
Ich schaue mir gerade ein spezielles Unternehmen und dessen Entwicklungspräparate an. Darunter ist auch ein BCMA-Inhibitor, der zur Behandlung des Multiplen Myeloms eingesetzt werden soll. Da BCMA verschiedentlich als besonders aussichtsreich gepriesen worden ist und beispielsweise Bluebird mit seinem anti-BCMA-CAR T Präparat herausragende klinische Studienergebnisse erzielt hat, habe ich interessehalber einmal alle derzeitigen klinischen Entwicklungspräparate mit Target BCMA zusammengestellt. Es wurde eine ziemlich lange Liste, in der alleine drei bispezifische anti-BCMAxCD3-Antikörper (Amgen, Janssen und Pfizer) und jede Menge CAR T Therapeutika waren, alle in Phase 1 (dazu noch Antikörper/ADCs). Wie soll man das alles noch bewerten?
Fakt ist jedenfalls, dass auf die anti-CD38-Mabs von Janssen/Genmab, Sanofi und Morphosys starke Konkurrenz zukommt. Unter diesen Umständen kann Morphosys froh sein, wenn sich für MOR202 überhaupt ein vorzeigbarer Partner findet, der einen nennenswerten Betrag auf den Tisch legt.
Ich schaue mir gerade ein spezielles Unternehmen und dessen Entwicklungspräparate an. Darunter ist auch ein BCMA-Inhibitor, der zur Behandlung des Multiplen Myeloms eingesetzt werden soll. Da BCMA verschiedentlich als besonders aussichtsreich gepriesen worden ist und beispielsweise Bluebird mit seinem anti-BCMA-CAR T Präparat herausragende klinische Studienergebnisse erzielt hat, habe ich interessehalber einmal alle derzeitigen klinischen Entwicklungspräparate mit Target BCMA zusammengestellt. Es wurde eine ziemlich lange Liste, in der alleine drei bispezifische anti-BCMAxCD3-Antikörper (Amgen, Janssen und Pfizer) und jede Menge CAR T Therapeutika waren, alle in Phase 1 (dazu noch Antikörper/ADCs). Wie soll man das alles noch bewerten?
Fakt ist jedenfalls, dass auf die anti-CD38-Mabs von Janssen/Genmab, Sanofi und Morphosys starke Konkurrenz zukommt. Unter diesen Umständen kann Morphosys froh sein, wenn sich für MOR202 überhaupt ein vorzeigbarer Partner findet, der einen nennenswerten Betrag auf den Tisch legt.
Antwort auf Beitrag Nr.: 55.874.479 von Joschka Schröder am 04.10.17 02:57:23Vielleicht kommt es ja bei der laufenden Klage zu einem Vergleich oder gar Sieg. In diesem Fall glaube ich, dass Mor202 dann gar nicht mehr weiterentwickelt wird.
Wer macht die Dichterlesung?
Antwort auf Beitrag Nr.: 55.881.253 von Friseuse am 04.10.17 21:03:20
Zitat von Friseuse: Wer macht die Dichterlesung?
“MOR” engineering of antibodies in ALL
http://www.bloodjournal.org/content/130/13/1487?utm_content=…
Session PO.A11 - Clinical Trials
A095 / 95 - Phase Ib study of anetumab ravtansine in combination with pemetrexed and cisplatin in patients with mesothelin-expressing epithelial mesothelioma or nonsquamous non-small cell lung cancer
Add To My Itinerary
October 28, 2017, 12:30 - 4:00 PM Hall E, Pennsylvania Convention Center
Presenter/Authors
Raffit Hassan1, Ding Wang2, John Wrangle3, Anish Thomas1, Angus Byars4, Lianne Asschert5, Rolando Atienza6, Prabhu Rajagopalan7, Annette Walter8, Jun Zhang7, Nenad Sarapa7, Hedy Kindler9. 1National Cancer Institute, Bethesda, MD; 2Henry Ford Hospital, Detriot, MI; 3Medical University of South Carolina, Charleston, SC; 4Bexon Clinical Consulting, Upper Monclair, NJ; 5Bexon Clinical Consulting, Upper Montclair, NJ; 6Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ; 7Bayer Healthcare Pharmaceuticals Inc., Whippany, NJ; 8Bayer AG, Pharmaceuticals, Berlin, Germany; 9University of Chicago Hospital, Chicago, IL
Disclosures
R. Hassan: None. D. Wang: None. J. Wrangle: None. A. Thomas: None. A. Byars: None. L. Asschert: None. R. Atienza: ; Bayer Healthcare Pharmaceuticals Inc. P. Rajagopalan: ; Bayer Healthcare Pharmaceuticals Inc. A. Walter: ; Bayer AG, Pharmaceuticals. J. Zhang: ; Bayer Healthcare Pharmaceuticals Inc. N. Sarapa: ; Bayer Healthcare Pharmaceuticals Inc.. H. Kindler: None.
Abstract
Background: Phase I study with the antibody-drug conjugate anetumab ravtansine demonstrated promising safety and efficacy as monotherapy in mesothelin-expressing tumors, and in particular for patients with advanced metastatic malignant pleural mesothelioma. Pemetrexed in combination with cisplatin is standard for first-line treatment of patients with metastatic mesothelioma and NSCLC. We therefore conducted a phase Ib study to determine the safety, tolerability, and maximum tolerated dose (MTD) of anetumab ravtansine in combination with pemetrexed/cisplatin (Pem/Cis) in subjects with mesothelin-expressing predominantly epithelial mesothelioma or nonsquamous NSCLC. Methods: Patients with histologically confirmed, unresectable, locally advanced or metastatic, mesothelin-expressing predominantly epithelial pleural or peritoneal mesothelioma or nonsquamous NSCLC, who previously failed ≤3 prior lines of chemotherapy, were eligible. Patients were prescreened based on obligatory tumor staining for mesothelin as determined by the Ventana MSLN (SP74) immunohistochemistry assay. Anetumab ravtansine was administered by 1-hour IV infusion on day 1 of every 21-day treatment cycle (Q3W). Pem (500 mg/m2) and Cis (75 mg/m2) were administered as IV infusions Q3W. Serial plasma samples were collected and analyzed for anetumab ravtansine and its components, pemetrexed, total and free platinum. Response to therapy was assessed by RECIST 1.1 and modified RECIST criteria for mesothelioma. Results: As of June 12, 2017, 17 patients had been treated and had received ≥2 cycles of treatment, including 7 patients with pleural mesothelioma, 9 with peritoneal mesothelioma, and 1 with NSCLC. Three patients were treated with anetumab ravtansine 5.5 mg/kg in combination with Pem/Cis with no dose-limiting toxicity (DLT). Six patients were then treated with anetumab ravtansine 6.5 mg/kg in combination with Pem/Cis, again without DLTs. This dose in combination was deemed as the MTD. An additional 8 patients were enrolled in an MTD expansion cohort. Anetumab ravtansine-related adverse events were mainly grade (G)1 and G2. G1/G2 eye toxicities were possibly more frequent with the combination than in historical data. There was one G3 corneal microcystic event and one G3 hypertension. There was a 50% overall response rate (all partial responses) from 16 evaluable patients. Of 13 patients treated at the 6.5 mg/kg MTD dose of anetumab ravtansine in combination with Pem/Cis, the overall response rate was 46%. Based on comparisons to historical data and within-subject comparisons in small number of subjects, clinically relevant PK interaction was not observed. Conclusions: Anetumab ravtansine at 6.5 mg/kg dosed Q3W in combination with standard dosing of Pem/Cis had a manageable toxicity profile without evidence of PK interaction and with early signs of clinical efficacy. Anetumab ravtansine at 6.5 mg/kg in combination with Pem 500 mg/m2 and Cis 75 mg/m2, all administered in a Q3W regimen, is thus feasible and the recommended dose for future study.
A095 / 95 - Phase Ib study of anetumab ravtansine in combination with pemetrexed and cisplatin in patients with mesothelin-expressing epithelial mesothelioma or nonsquamous non-small cell lung cancer
Add To My Itinerary
October 28, 2017, 12:30 - 4:00 PM Hall E, Pennsylvania Convention Center
Presenter/Authors
Raffit Hassan1, Ding Wang2, John Wrangle3, Anish Thomas1, Angus Byars4, Lianne Asschert5, Rolando Atienza6, Prabhu Rajagopalan7, Annette Walter8, Jun Zhang7, Nenad Sarapa7, Hedy Kindler9. 1National Cancer Institute, Bethesda, MD; 2Henry Ford Hospital, Detriot, MI; 3Medical University of South Carolina, Charleston, SC; 4Bexon Clinical Consulting, Upper Monclair, NJ; 5Bexon Clinical Consulting, Upper Montclair, NJ; 6Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ; 7Bayer Healthcare Pharmaceuticals Inc., Whippany, NJ; 8Bayer AG, Pharmaceuticals, Berlin, Germany; 9University of Chicago Hospital, Chicago, IL
Disclosures
R. Hassan: None. D. Wang: None. J. Wrangle: None. A. Thomas: None. A. Byars: None. L. Asschert: None. R. Atienza: ; Bayer Healthcare Pharmaceuticals Inc. P. Rajagopalan: ; Bayer Healthcare Pharmaceuticals Inc. A. Walter: ; Bayer AG, Pharmaceuticals. J. Zhang: ; Bayer Healthcare Pharmaceuticals Inc. N. Sarapa: ; Bayer Healthcare Pharmaceuticals Inc.. H. Kindler: None.
Abstract
Background: Phase I study with the antibody-drug conjugate anetumab ravtansine demonstrated promising safety and efficacy as monotherapy in mesothelin-expressing tumors, and in particular for patients with advanced metastatic malignant pleural mesothelioma. Pemetrexed in combination with cisplatin is standard for first-line treatment of patients with metastatic mesothelioma and NSCLC. We therefore conducted a phase Ib study to determine the safety, tolerability, and maximum tolerated dose (MTD) of anetumab ravtansine in combination with pemetrexed/cisplatin (Pem/Cis) in subjects with mesothelin-expressing predominantly epithelial mesothelioma or nonsquamous NSCLC. Methods: Patients with histologically confirmed, unresectable, locally advanced or metastatic, mesothelin-expressing predominantly epithelial pleural or peritoneal mesothelioma or nonsquamous NSCLC, who previously failed ≤3 prior lines of chemotherapy, were eligible. Patients were prescreened based on obligatory tumor staining for mesothelin as determined by the Ventana MSLN (SP74) immunohistochemistry assay. Anetumab ravtansine was administered by 1-hour IV infusion on day 1 of every 21-day treatment cycle (Q3W). Pem (500 mg/m2) and Cis (75 mg/m2) were administered as IV infusions Q3W. Serial plasma samples were collected and analyzed for anetumab ravtansine and its components, pemetrexed, total and free platinum. Response to therapy was assessed by RECIST 1.1 and modified RECIST criteria for mesothelioma. Results: As of June 12, 2017, 17 patients had been treated and had received ≥2 cycles of treatment, including 7 patients with pleural mesothelioma, 9 with peritoneal mesothelioma, and 1 with NSCLC. Three patients were treated with anetumab ravtansine 5.5 mg/kg in combination with Pem/Cis with no dose-limiting toxicity (DLT). Six patients were then treated with anetumab ravtansine 6.5 mg/kg in combination with Pem/Cis, again without DLTs. This dose in combination was deemed as the MTD. An additional 8 patients were enrolled in an MTD expansion cohort. Anetumab ravtansine-related adverse events were mainly grade (G)1 and G2. G1/G2 eye toxicities were possibly more frequent with the combination than in historical data. There was one G3 corneal microcystic event and one G3 hypertension. There was a 50% overall response rate (all partial responses) from 16 evaluable patients. Of 13 patients treated at the 6.5 mg/kg MTD dose of anetumab ravtansine in combination with Pem/Cis, the overall response rate was 46%. Based on comparisons to historical data and within-subject comparisons in small number of subjects, clinically relevant PK interaction was not observed. Conclusions: Anetumab ravtansine at 6.5 mg/kg dosed Q3W in combination with standard dosing of Pem/Cis had a manageable toxicity profile without evidence of PK interaction and with early signs of clinical efficacy. Anetumab ravtansine at 6.5 mg/kg in combination with Pem 500 mg/m2 and Cis 75 mg/m2, all administered in a Q3W regimen, is thus feasible and the recommended dose for future study.
Antwort auf Beitrag Nr.: 55.961.269 von Ville7 am 16.10.17 19:53:33Macht Bayer also doch wie angekündigt weiter. Danke für den Link Ville.
Ob die Gesamtansprechrate von 46% bzw 50% gut ist kann ich nicht beurteilen. Für Bayer aber anscheinend gut genug. Sehr schön
Ob die Gesamtansprechrate von 46% bzw 50% gut ist kann ich nicht beurteilen. Für Bayer aber anscheinend gut genug. Sehr schön
Recht interessant: J&J hat offensichtlich den bislang in Phase 3 befindlichen anti-CD123-Mab talacotuzumab aus dem Programm genommen. talacotuzumab wurde mit demselben Verfahren von Xencor FC-optimiert wie MOR208. Eigentlich müßte der Xencor-Kurs stärker nachgeben, möglicherweise haben sich die Neuigkeiten aber auch nur noch nicht herumgesprochen (wenigstens wird es in den Endpoints News erwähnt).
Antwort auf Beitrag Nr.: 55.966.665 von Joschka Schröder am 17.10.17 16:01:12
Oh, das sind schlechte Nachrichten für Morphosys 😔
Zitat von Joschka Schröder: Recht interessant: J&J hat offensichtlich den bislang in Phase 3 befindlichen anti-CD123-Mab talacotuzumab aus dem Programm genommen. talacotuzumab wurde mit demselben Verfahren von Xencor FC-optimiert wie MOR208. Eigentlich müßte der Xencor-Kurs stärker nachgeben, möglicherweise haben sich die Neuigkeiten aber auch nur noch nicht herumgesprochen (wenigstens wird es in den Endpoints News erwähnt).
Oh, das sind schlechte Nachrichten für Morphosys 😔
Antwort auf Beitrag Nr.: 55.966.917 von hinz12 am 17.10.17 16:34:04Wieso sind das schlechte News für Morphosys?
Antwort auf Beitrag Nr.: 55.966.998 von Ville7 am 17.10.17 16:44:32Ich vermute auch, dass es am Target lag, an CD123 sind schon mehrere Präparate gescheitert, zuletzt JNJ-63709178 (anti CD123 x CD3). Erwähnenswert ist es aber allemal, weil es einmal mehr zeigt, dass noch in Phase 3 überraschend Studien scheitern können.
Offtopic:
Da Xencor hier schonmal erwähnt wurde und es (noch) keinen eigenen Diskussionszweig gibt erlaube ich mir nachfolgende Meldung mal einzustellen:
http://acrabstracts.org/abstract/final-results-of-an-open-la…
Gruß
Da Xencor hier schonmal erwähnt wurde und es (noch) keinen eigenen Diskussionszweig gibt erlaube ich mir nachfolgende Meldung mal einzustellen:
http://acrabstracts.org/abstract/final-results-of-an-open-la…
Gruß
Der Grund für die gegenwärtige Kursschwäche der Morphosys-Aktie liegt in aktuellen Phase-3-Daten des guselkumab-Konkurrenzen risankizumab (ant-IL-23p19) begründet! Ich schaue mir gerade die Daten an und werde später noch etwas etwas schreiben.
Die Konkurrenz in der Indikation Psoriasis ist hammerhart. risankizmab mit Top-Daten aus 3 Phase-3-Studien. Dabei wurde der Antikörper nur alle 12 Wochen appliziert (jeweils 150 mg)!
Ergebnisse:
PASI 90 nach 16 Wochen: zwischen 72 % und 75 % (guselkumab zwischen 70 % und 73,3 %)
PASI 100 nach 16 Wochen: zwischen 36 % und 51 % (guselkumab 34,1 % und 37,4 %)
PASI 90 nach 1 Jahr: 82 % und 81 % (guselkumab 76,3 %)
PASI 100 nach 1 Jahr: 56 % und 60 % (guselkumab 47,4 %)
Stelara und Humira wurden in den Vergleichsstudien um Längen geschlagen.
risankizumab wurde von Boehringer Ingelheim entwicklelt und an AbbVie auslizensiert. risankizumab adressiert dasselbe Target wie guselkumab.
Ergebnisse:
PASI 90 nach 16 Wochen: zwischen 72 % und 75 % (guselkumab zwischen 70 % und 73,3 %)
PASI 100 nach 16 Wochen: zwischen 36 % und 51 % (guselkumab 34,1 % und 37,4 %)
PASI 90 nach 1 Jahr: 82 % und 81 % (guselkumab 76,3 %)
PASI 100 nach 1 Jahr: 56 % und 60 % (guselkumab 47,4 %)
Stelara und Humira wurden in den Vergleichsstudien um Längen geschlagen.
risankizumab wurde von Boehringer Ingelheim entwicklelt und an AbbVie auslizensiert. risankizumab adressiert dasselbe Target wie guselkumab.
ASH 2017 abstracts - Veröffentlichung
"Abstracts will be available online beginning November 1, 2017, at 9:00 a.m. Eastern Time."Bei Morphosys Fokus auf MOR208 L-Mind Daten.
Wenn Celgene weiter so bei Lenalidomide zulangt, dann wird eine L-MIND Behandlung sehr sehr teuer. Hier die Preisentwicklung. Daran sieht man auch wie Celgene es schaffte so massiv zu "wachsen" (persönliche Meinung: unverschämt!):
Antwort auf Beitrag Nr.: 56.038.365 von Ville7 am 27.10.17 07:22:41
Meine Fragen hierzu:
- Warum wohl ist die letzte Veröffentlichung zu diesem Wirkstoff von 2012?
- Kann es sein, dass gsk die Vereinbarung gar nicht zum Zwecke der Erreichung der Produktreife eingegangen ist, sondern vielmehr, um zu vermeiden, dass ein aussichtsreiches Konkurrenzprodukt Marktreife erlangt (ich kenne das Wirkstoffportfolio von gsk nicht)?
Immerhin war gemäß Jahresbericht 2013 MOR 103 in der Pipeline bereits Phase 2 der klinischen Entwicklung erreicht.
MOR 103
MOR 103 wurde 2013 an gsk auslizensiert. Seitdem liegt die gesamte Verantwortung für die Weiterentwicklung bei gsk.Meine Fragen hierzu:
- Warum wohl ist die letzte Veröffentlichung zu diesem Wirkstoff von 2012?
- Kann es sein, dass gsk die Vereinbarung gar nicht zum Zwecke der Erreichung der Produktreife eingegangen ist, sondern vielmehr, um zu vermeiden, dass ein aussichtsreiches Konkurrenzprodukt Marktreife erlangt (ich kenne das Wirkstoffportfolio von gsk nicht)?
Immerhin war gemäß Jahresbericht 2013 MOR 103 in der Pipeline bereits Phase 2 der klinischen Entwicklung erreicht.
Das stimmt nicht ganz. GSK hat zwei grossen trials angefangen in zwei indikation und Ergebnisse von ersten ( osteoarthritis) werden noch in diesem Jahr erwartet. Ja es hat gedauert bis die weiter entwicklung fortgesetzt haben, aber trial dauert auch seine Zeit. Bald musste es soweit sein.
Antwort auf Beitrag Nr.: 56.038.365 von Ville7 am 27.10.17 07:22:41Passend dazu:
https://www.statnews.com/2017/10/30/celgene-hugin-trump/
https://www.statnews.com/2017/10/30/celgene-hugin-trump/
Das war mir bislang nicht bewußt ... Bayer testet anetumab ravtansine in der Indikation AML -> https://ash.confex.com/ash/2017/webprogram/Paper105740.html
Wieso man einen Behandlungserfolg mit MOR208 möglicherweise durch die nachfolgende Gabe eines anti-CD22-Antikörpers prolongieren könnte -> https://www.ncbi.nlm.nih.gov/pubmed/29155426
und off topic zu fatalen Nebenwirkungen von CAR-T-Zell-Therapien - lesenswert!
-> http://www.sciencemag.org/news/2017/11/cancer-immunotherapy-…
und off topic zu fatalen Nebenwirkungen von CAR-T-Zell-Therapien - lesenswert!
-> http://www.sciencemag.org/news/2017/11/cancer-immunotherapy-…
Antwort auf Beitrag Nr.: 56.244.680 von Joschka Schröder am 22.11.17 12:09:42bzgl. der fatalen wirkungen habe ich mich mit meinen nem laienwissen, welches ich hier allerdings aus angst vor blamage besser für mich behalte, schon öfter gewundert, dass diese therapie so hochgelobt wird.
nach dem bescheidenen wissen das ich habe bzw. das was ich bisher darüber weiß würde ich selbst als patient schon ins grübeln kommen u mir überlegen ob ich diesen schritt gehen würde. außer ich wäre eh beretis dem tod geweiht u das mein letzter strohhalm.
scheiß krebs.....
nach dem bescheidenen wissen das ich habe bzw. das was ich bisher darüber weiß würde ich selbst als patient schon ins grübeln kommen u mir überlegen ob ich diesen schritt gehen würde. außer ich wäre eh beretis dem tod geweiht u das mein letzter strohhalm.
scheiß krebs.....
Abermals sehr starke Daten einer weiteren Phase 3-Studie mit dem anti-IL-23p19-Antikörper risankizumab der Firmen Abbvie/Boehringer Ingelheim in der Indikation "moderate bis schwere Plaque-Psoriasis", u.a. mit
PASI 75 nach 16 Wochen = 79 % (Placebo 8 %)
PASI 90 nach 16 Wochen = 73 % (Placebo 2 %)
PASI 100 nach 16 Wochen = 47 % (Placebo 1 %).
risankizumab adressiert dasselbe Target wie guselkumab (J&J/Morphosys).
PASI 75 nach 16 Wochen = 79 % (Placebo 8 %)
PASI 90 nach 16 Wochen = 73 % (Placebo 2 %)
PASI 100 nach 16 Wochen = 47 % (Placebo 1 %).
risankizumab adressiert dasselbe Target wie guselkumab (J&J/Morphosys).
Antwort auf Beitrag Nr.: 56.364.177 von Joschka Schröder am 04.12.17 22:54:34
Im cross trial - Vergleich sieht Guselkumab dieses mal insgesamt aber nicht durchweg unterlegen aus:

Zitat von Joschka Schröder: Abermals sehr starke Daten einer weiteren Phase 3-Studie mit dem anti-IL-23p19-Antikörper risankizumab der Firmen Abbvie/Boehringer Ingelheim in der Indikation "moderate bis schwere Plaque-Psoriasis", u.a. mit
PASI 75 nach 16 Wochen = 79 % (Placebo 8 %)
PASI 90 nach 16 Wochen = 73 % (Placebo 2 %)
PASI 100 nach 16 Wochen = 47 % (Placebo 1 %).
risankizumab adressiert dasselbe Target wie guselkumab (J&J/Morphosys).
Im cross trial - Vergleich sieht Guselkumab dieses mal insgesamt aber nicht durchweg unterlegen aus:
Antwort auf Beitrag Nr.: 56.364.177 von Joschka Schröder am 04.12.17 22:54:34
Sorry, Tippfehler, PASI75 = 89 %!
@Ville7: Nach den Ergebnissen sämtlicher Phase III-Studien scheinen risankizumab und guselkumab gleichwertig.
Zitat von Joschka Schröder: Abermals sehr starke Daten einer weiteren Phase 3-Studie mit dem anti-IL-23p19-Antikörper risankizumab der Firmen Abbvie/Boehringer Ingelheim in der Indikation "moderate bis schwere Plaque-Psoriasis", u.a. mit
PASI 75 nach 16 Wochen = 79 % (Placebo 8 %)
PASI 90 nach 16 Wochen = 73 % (Placebo 2 %)
PASI 100 nach 16 Wochen = 47 % (Placebo 1 %).
risankizumab adressiert dasselbe Target wie guselkumab (J&J/Morphosys).
Sorry, Tippfehler, PASI75 = 89 %!
@Ville7: Nach den Ergebnissen sämtlicher Phase III-Studien scheinen risankizumab und guselkumab gleichwertig.
Hier noch die Links zu den Ergebnissen aller vier risankizumab-P3-Studien
Nr. 1 bis 3 -> https://news.abbvie.com/news/press-releases/risankizumab-mee…" target="_blank" rel="nofollow ugc noopener">https://news.abbvie.com/news/press-releases/risankizumab-mee…
Nr. 4 -> https://news.abbvie.com/news/press-releases/risankizumab-mee…" target="_blank" rel="nofollow ugc noopener">https://news.abbvie.com/news/press-releases/risankizumab-mee…
Nr. 1 bis 3 -> https://news.abbvie.com/news/press-releases/risankizumab-mee…" target="_blank" rel="nofollow ugc noopener">https://news.abbvie.com/news/press-releases/risankizumab-mee…
Nr. 4 -> https://news.abbvie.com/news/press-releases/risankizumab-mee…" target="_blank" rel="nofollow ugc noopener">https://news.abbvie.com/news/press-releases/risankizumab-mee…
Antwort auf Beitrag Nr.: 56.365.383 von Joschka Schröder am 05.12.17 08:17:46Ich denke, dass wenn beide Präparate auf dem Markt sein sollten, es nicht mehr um ein paar Prozentpunkte hin oder her gehen wird. Was m.E. dann viel wichtiger ist, ist wie gut die beiden Medikamente vermarktet werden.
Es gab im August nach der Zulassung von Tremfya die Meldung, dass das Medikament 2 Wochen nach der Zulassung nur 50% der Dermatologen bekannt sei. Das fand ich schon etwas ernüchternd und zeigt, dass auch sog. potentielle Blockbuster keine Selbstläufer sind sondern die Unternehmen sehr viel investieren müssen, um es bekannt zu machen und erfolgreich auf dem Markt zu etablieren.
Hier würde mich eine Einschätzung interessieren wie aus eurer Sicht Abbvie/Boehringer Ingelheim bzw. Jannsen (und auch MOR) aufgestellt sind .
Es gab im August nach der Zulassung von Tremfya die Meldung, dass das Medikament 2 Wochen nach der Zulassung nur 50% der Dermatologen bekannt sei. Das fand ich schon etwas ernüchternd und zeigt, dass auch sog. potentielle Blockbuster keine Selbstläufer sind sondern die Unternehmen sehr viel investieren müssen, um es bekannt zu machen und erfolgreich auf dem Markt zu etablieren.
Hier würde mich eine Einschätzung interessieren wie aus eurer Sicht Abbvie/Boehringer Ingelheim bzw. Jannsen (und auch MOR) aufgestellt sind .
Antwort auf Beitrag Nr.: 56.365.722 von Pitschnass am 05.12.17 08:48:56Sowohl AbbVie als auch Johnson & Johnson besitzen Top-Vertriebsmannschaften.
Mit Amgens/Xencors AMG 424 kommt nun ein weiterer - in diesem Fall bispezifischer - Antikörper in die Klinik, der CD38 adressiert. Konkurrenz für MOR208, daratumumab und isatuximab.
Antwort auf Beitrag Nr.: 56.368.293 von Joschka Schröder am 05.12.17 15:05:46
Sorry, MOR202 natürlich.
Zitat von Joschka Schröder: Konkurrenz für MOR208
Sorry, MOR202 natürlich.
Antwort auf Beitrag Nr.: 56.365.356 von Joschka Schröder am 05.12.17 08:14:34Wenn guselkumab und risankizumab gleichwertig sind und JNJ einen mindestens 18-monatigen Vorsprung hat, weil AbbVie 2019 als Ziel für den Marktstart angibt, dann ist doch guselkumab klar im Vorteil, oder ?
Antwort auf Beitrag Nr.: 56.376.424 von -weitblick- am 06.12.17 14:03:05Man könnte es auch so sehen: guselkumab wird sich erst einmal an secukinumab/Cosentyx abarbeiten. Bis guselkumab in der Lage ist, größere Marktanteile zu gewinnen, wird risankizumab auf dem Markt sein. Ich bin leider kein Helleseher, gehe bei meinen Überlegungen aber davon aus, dass sich guselkumab und risankizumab den IL-23p19-Markt teilen werden, in kleinerem Umfang könnte noch tildrakizumab hinzukommen.
Antwort auf Beitrag Nr.: 56.382.829 von Joschka Schröder am 06.12.17 21:47:53Hellseher sind wir natürlich alle nicht, dann bräuchten wir hier nicht diskutieren ;-).
Wenn es so kommt wie Du erwartest und sich AbbVie und JNJ den Markt teilen fände ich das nicht schlecht.
AbbVie erwartet bis 2025 nur bei Psoriasis durch Risankizumab einem Umsatz von 3mrd$ plus weitere Indikationen (+2mrd$). Siehe Strategic Update Folie 24 vom 3Q17 Call:
http://investors.abbvie.com/phoenix.zhtml?c=251551&p=irol-Ev…
Wenn es so kommt wie Du erwartest und sich AbbVie und JNJ den Markt teilen fände ich das nicht schlecht.
AbbVie erwartet bis 2025 nur bei Psoriasis durch Risankizumab einem Umsatz von 3mrd$ plus weitere Indikationen (+2mrd$). Siehe Strategic Update Folie 24 vom 3Q17 Call:
http://investors.abbvie.com/phoenix.zhtml?c=251551&p=irol-Ev…
zu MOR208:
Eindrucksvolle Daten aus der Kombistudie Obinutuzumab + LEN + CHOP mit 98 % overall response rate und 90 % complete response in der Indikation DLBCL. Über 90 % PFS nach 21 Monaten (vom Präsentationsfoto abgelesen). Präsentation durch Jason Westin beim ASH-Meeting.
Eindrucksvolle Daten aus der Kombistudie Obinutuzumab + LEN + CHOP mit 98 % overall response rate und 90 % complete response in der Indikation DLBCL. Über 90 % PFS nach 21 Monaten (vom Präsentationsfoto abgelesen). Präsentation durch Jason Westin beim ASH-Meeting.
Antwort auf Beitrag Nr.: 56.414.851 von Joschka Schröder am 10.12.17 08:21:46Ich denke eine derart gelagerte Frontline Studie wird auch Morphosys fahren (müssen): MOR208+LEN+CHOP.
German biotechs would love to be American
https://global.handelsblatt.com/companies-markets/german-bio…
weiter zur MOR208:
Die DLBCL-Indikation ist wirklich stark überlaufen, man kommt kaum noch nach, die ganzen Studien zu verfolgen. Eine gute Nachricht für die betroffenen Patienten!
Roche hat heute Ergebnisse aus einer Dreier-Kombinationsstudie mit dem anti-CD79b-ADC polatuzumab vedotin (stammt aus einer Kooperation mit Deattle Genetics) plus bendamustin plus rituximab veröffentlicht (s.u.).
Weitere Studien hat Roche mit obinutuzumab, rituximab, venetoclax und atezolizumab in unterschiedlichen Querkombinationen am laufen.
Bislang zeichnet sich MOR208 im Vergleich zu vielen anderen Therapien in einem nennenswerten Punkt aus: Die Dauer der klinischen Antwort! Beispiel: DoR (duration of response) bei Monotherapie mit MOR208 zuletzt 20,1 Monate (s. Präsentation zum letztjährigen ASH-Meeting). In unten zitierter Roche-Dreifachkombi hingegen "nur" 8,8 Monate (zu berücksichtigen wäre noch, dass sich die jeweiligen Patientenkollektive zwangsläufig nicht identisch zusammensetzen). Die bisherigen Daten zur Dauer der klinischen Antwort und des progressionsfreien Überlebens lassen vermuten, dass es MOR208 gelingen wird, ein gewisses Stück vom DLBCL-Markt zu okkupieren. Positiv ist dies natürlich insbesondere auch für Xencor, das bei Zulassung eine Meilensteinzahlung in Höhe von 187 Mio. USD kassieren wird und Royalties im hohen einstelligen bis niedrigen zweistelligen Prozentbereich kassieren wird..
Phase II data showed Roche’s investigational polatuzumab vedotin plus bendamustine and MabThera/Rituxan (BR) increased complete response rates compared to BR alone in previously treated aggressive lymphoma
Polatuzumab vedotin combination demonstrated improvements across multiple efficacy measures compared to BR alone in relapsed or refractory diffuse large B-cell lymphoma
These data led to FDA Breakthrough Therapy and EMA PRIME (PRIority MEdicines) designations
Results will be presented at the 59th American Society of Hematology Annual Meeting
Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced positive results from the randomised phase II GO29365 study that compared polatuzumab vedotin in combination with bendamustine plus MabThera®/Rituxan® (rituximab) (BR) against BR alone in people with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who are not candidates for haematopoietic stem cell transplant. The study met its primary endpoint, demonstrating that the addition of polatuzumab vedotin to BR increased complete response (CR) rates from 15% to 40% (p=0.012) at the end of treatment, as measured by positron emission tomography (PET) and assessed by an independent review committee (IRC). No unexpected safety signals were observed with the addition of polatuzumab vedotin to BR.
“As many as forty percent of people with diffuse large B-cell lymphoma do not respond to initial therapy or experience the return of their disease, at which point their treatment options are limited and the prognosis is poor,” said Sandra Horning, MD, Roche’s Chief Medical Officer and Head of Global Product Development. “The promising efficacy observed for polatuzumab vedotin in this study supports its potential as a new treatment option for people previously treated for this aggressive blood cancer, and we look forward to discussing the results with health authorities.”
The data will be presented in a poster session on Sunday, 10 December at the 59th American Society of Hematology (ASH) Annual Meeting by Laurie Sehn, MD, British Columbia Cancer Agency/University of British Columbia.
The results showed:
Polatuzumab vedotin plus BR significantly improved CR rates from 15% with BR alone to 40% (p=0.012), as measured by PET and assessed by IRC. A CR means no cancer could be detected at that time.
The benefit observed was consistent across secondary endpoints, including improvements in investigator-assessed best objective response (OR; CR and partial response, PR) and CR with polatuzumab vedotin plus BR (70.0% OR, 57.5% CR) compared to BR alone (32.5% OR, CR 20.0%).
Exploratory endpoints also improved with the addition of polatuzumab vedotin to BR:
Patients treated with polatuzumab vedotin plus BR lived longer than those receiving BR alone (median overall survival; 11.8 months vs. 4.7 months; HR 0.35; 95% CI 0.19-0.67; p=0.0008).
The addition of polatuzumab vedotin also increased the time until disease worsening or death (median progression-free survival: 6.7 months vs. 2.0 months; HR 0.31; 95% CI 0.18-0.55; p<0. 0001), and the time between first response to treatment and disease worsening (duration of response: 8.8 months vs. 3.7 months).
No unexpected safety signals were observed with the addition of polatuzumab vedotin to BR. The most common Grade 3-4 adverse events with polatuzumab vedotin plus BR compared to BR alone, respectively, were low white blood cell count (46.2% vs. 35.9%), low white blood cell count with fever (10.3% vs. 5.1%), low platelet count (33.3% vs. 20.5%), anaemia (25.6% vs. 12.8%) and infections (17.9% vs. 17.9%).
Based on results from this study, polatuzumab vedotin was recently granted Breakthrough Therapy Designation by the US Food and Drug Administration and PRIME (PRIority MEdicines) designation by the European Medicines Agency for the treatment of people with relapsed or refractory DLBCL. There are a number of ongoing studies evaluating the efficacy and safety of polatuzumab vedotin for several types of non-Hodgkin lymphoma, including combinations with Gazyva® /Gazyvaro® (obinutuzumab), MabThera/Rituxan, Venclexta™/Venclyxto™ (venetoclax) and Tecentriq® (atezolizumab).
About the GO29365 study
GO29365 is a global, phase Ib/II randomised study evaluating the safety, tolerability and activity of polatuzumab vedotin in combination with MabThera /Rituxan (rituximab) or Gazyva /Gazyvaro (obinutuzumab) plus bendamustine in relapsed or refractory (R/R) follicular lymphoma or diffuse large B-cell lymphoma (DLBCL). The phase II stage randomised 80 patients with heavily pre-treated R/R DLBCL to receive either bendamustine plus MabThera/Rituxan (BR), or BR in combination with polatuzumab vedotin. Patients enrolled had received a median of two prior therapies (a range of 1-7 prior therapies in the polatuzumab vedotin arm and range of 1-5 prior therapies in the BR alone arm). The primary endpoint was complete response (CR) at the end of treatment, as measured by positron emission tomography (PET) and assessed by an independent review committee (IRC). Secondary endpoints included objective response (OR; CR and partial response, PR) by investigator assessment and best objective response at the end of treatment by investigator and IRC assessment. Exploratory endpoints included duration of response (DOR), progression-free survival (PFS), event-free survival (EFS) and overall survival (OS).
About polatuzumab vedotin
Polatuzumab vedotin is a first-in-class anti-CD79b antibody drug conjugate (ADC) currently being investigated for the treatment of several types of non-Hodgkin lymphoma (NHL). The CD79b protein is highly specific and expressed in the majority of types of B-cell NHL, making it a promising target for the development of new therapies.1 Polatuzumab vedotin binds to CD79b and destroys these B-cells via a targeted approach, which is thought to minimise the effects on normal cells while maximising tumour cell death. Polatuzumab vedotin is being developed by Roche utilising Seattle Genetics ADC technology.
About diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL.2 DLBCL is an aggressive (fast-growing) type of NHL, which is generally responsive to treatment in the frontline.3 However, as many as 40% of patients will relapse, at which time salvage therapy options are limited and survival is short.3 Approximately 123,000 people worldwide are estimated to be diagnosed with DLBCL each year.4
Die DLBCL-Indikation ist wirklich stark überlaufen, man kommt kaum noch nach, die ganzen Studien zu verfolgen. Eine gute Nachricht für die betroffenen Patienten!
Roche hat heute Ergebnisse aus einer Dreier-Kombinationsstudie mit dem anti-CD79b-ADC polatuzumab vedotin (stammt aus einer Kooperation mit Deattle Genetics) plus bendamustin plus rituximab veröffentlicht (s.u.).
Weitere Studien hat Roche mit obinutuzumab, rituximab, venetoclax und atezolizumab in unterschiedlichen Querkombinationen am laufen.
Bislang zeichnet sich MOR208 im Vergleich zu vielen anderen Therapien in einem nennenswerten Punkt aus: Die Dauer der klinischen Antwort! Beispiel: DoR (duration of response) bei Monotherapie mit MOR208 zuletzt 20,1 Monate (s. Präsentation zum letztjährigen ASH-Meeting). In unten zitierter Roche-Dreifachkombi hingegen "nur" 8,8 Monate (zu berücksichtigen wäre noch, dass sich die jeweiligen Patientenkollektive zwangsläufig nicht identisch zusammensetzen). Die bisherigen Daten zur Dauer der klinischen Antwort und des progressionsfreien Überlebens lassen vermuten, dass es MOR208 gelingen wird, ein gewisses Stück vom DLBCL-Markt zu okkupieren. Positiv ist dies natürlich insbesondere auch für Xencor, das bei Zulassung eine Meilensteinzahlung in Höhe von 187 Mio. USD kassieren wird und Royalties im hohen einstelligen bis niedrigen zweistelligen Prozentbereich kassieren wird..
Phase II data showed Roche’s investigational polatuzumab vedotin plus bendamustine and MabThera/Rituxan (BR) increased complete response rates compared to BR alone in previously treated aggressive lymphoma
Polatuzumab vedotin combination demonstrated improvements across multiple efficacy measures compared to BR alone in relapsed or refractory diffuse large B-cell lymphoma
These data led to FDA Breakthrough Therapy and EMA PRIME (PRIority MEdicines) designations
Results will be presented at the 59th American Society of Hematology Annual Meeting
Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced positive results from the randomised phase II GO29365 study that compared polatuzumab vedotin in combination with bendamustine plus MabThera®/Rituxan® (rituximab) (BR) against BR alone in people with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who are not candidates for haematopoietic stem cell transplant. The study met its primary endpoint, demonstrating that the addition of polatuzumab vedotin to BR increased complete response (CR) rates from 15% to 40% (p=0.012) at the end of treatment, as measured by positron emission tomography (PET) and assessed by an independent review committee (IRC). No unexpected safety signals were observed with the addition of polatuzumab vedotin to BR.
“As many as forty percent of people with diffuse large B-cell lymphoma do not respond to initial therapy or experience the return of their disease, at which point their treatment options are limited and the prognosis is poor,” said Sandra Horning, MD, Roche’s Chief Medical Officer and Head of Global Product Development. “The promising efficacy observed for polatuzumab vedotin in this study supports its potential as a new treatment option for people previously treated for this aggressive blood cancer, and we look forward to discussing the results with health authorities.”
The data will be presented in a poster session on Sunday, 10 December at the 59th American Society of Hematology (ASH) Annual Meeting by Laurie Sehn, MD, British Columbia Cancer Agency/University of British Columbia.
The results showed:
Polatuzumab vedotin plus BR significantly improved CR rates from 15% with BR alone to 40% (p=0.012), as measured by PET and assessed by IRC. A CR means no cancer could be detected at that time.
The benefit observed was consistent across secondary endpoints, including improvements in investigator-assessed best objective response (OR; CR and partial response, PR) and CR with polatuzumab vedotin plus BR (70.0% OR, 57.5% CR) compared to BR alone (32.5% OR, CR 20.0%).
Exploratory endpoints also improved with the addition of polatuzumab vedotin to BR:
Patients treated with polatuzumab vedotin plus BR lived longer than those receiving BR alone (median overall survival; 11.8 months vs. 4.7 months; HR 0.35; 95% CI 0.19-0.67; p=0.0008).
The addition of polatuzumab vedotin also increased the time until disease worsening or death (median progression-free survival: 6.7 months vs. 2.0 months; HR 0.31; 95% CI 0.18-0.55; p<0. 0001), and the time between first response to treatment and disease worsening (duration of response: 8.8 months vs. 3.7 months).
No unexpected safety signals were observed with the addition of polatuzumab vedotin to BR. The most common Grade 3-4 adverse events with polatuzumab vedotin plus BR compared to BR alone, respectively, were low white blood cell count (46.2% vs. 35.9%), low white blood cell count with fever (10.3% vs. 5.1%), low platelet count (33.3% vs. 20.5%), anaemia (25.6% vs. 12.8%) and infections (17.9% vs. 17.9%).
Based on results from this study, polatuzumab vedotin was recently granted Breakthrough Therapy Designation by the US Food and Drug Administration and PRIME (PRIority MEdicines) designation by the European Medicines Agency for the treatment of people with relapsed or refractory DLBCL. There are a number of ongoing studies evaluating the efficacy and safety of polatuzumab vedotin for several types of non-Hodgkin lymphoma, including combinations with Gazyva® /Gazyvaro® (obinutuzumab), MabThera/Rituxan, Venclexta™/Venclyxto™ (venetoclax) and Tecentriq® (atezolizumab).
About the GO29365 study
GO29365 is a global, phase Ib/II randomised study evaluating the safety, tolerability and activity of polatuzumab vedotin in combination with MabThera /Rituxan (rituximab) or Gazyva /Gazyvaro (obinutuzumab) plus bendamustine in relapsed or refractory (R/R) follicular lymphoma or diffuse large B-cell lymphoma (DLBCL). The phase II stage randomised 80 patients with heavily pre-treated R/R DLBCL to receive either bendamustine plus MabThera/Rituxan (BR), or BR in combination with polatuzumab vedotin. Patients enrolled had received a median of two prior therapies (a range of 1-7 prior therapies in the polatuzumab vedotin arm and range of 1-5 prior therapies in the BR alone arm). The primary endpoint was complete response (CR) at the end of treatment, as measured by positron emission tomography (PET) and assessed by an independent review committee (IRC). Secondary endpoints included objective response (OR; CR and partial response, PR) by investigator assessment and best objective response at the end of treatment by investigator and IRC assessment. Exploratory endpoints included duration of response (DOR), progression-free survival (PFS), event-free survival (EFS) and overall survival (OS).
About polatuzumab vedotin
Polatuzumab vedotin is a first-in-class anti-CD79b antibody drug conjugate (ADC) currently being investigated for the treatment of several types of non-Hodgkin lymphoma (NHL). The CD79b protein is highly specific and expressed in the majority of types of B-cell NHL, making it a promising target for the development of new therapies.1 Polatuzumab vedotin binds to CD79b and destroys these B-cells via a targeted approach, which is thought to minimise the effects on normal cells while maximising tumour cell death. Polatuzumab vedotin is being developed by Roche utilising Seattle Genetics ADC technology.
About diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL.2 DLBCL is an aggressive (fast-growing) type of NHL, which is generally responsive to treatment in the frontline.3 However, as many as 40% of patients will relapse, at which time salvage therapy options are limited and survival is short.3 Approximately 123,000 people worldwide are estimated to be diagnosed with DLBCL each year.4
Antwort auf Beitrag Nr.: 56.417.553 von Joschka Schröder am 10.12.17 18:06:19
Sehe ich auch so. Die Dauer der Antwort ist ein wirklich sehr wichtiges Feature, das nicht nur dem Patienten hilft, sondern auch eine wichtige ökonomische Komponente hat, denn somit kann ich das Produkt sehr lange verabreichen und deutlich höhere Umsätze generieren.
Dieser DLBCL-Markt ist in seiner Dynamik aber nicht wirklich prognostizierbar und birgt weiterhin ziemliche Risiken.
Neue Player und Produkte werden auf den Markt kommen, z.B. bispezifische anti-CD20 oder anti-CD19 mabs, die genau die Funktion von MOR208 wahrnehmen sollen - mit vielleicht noch besserer und längerer Wirksamkeit.
Zudem wird die weitere Entwicklung, z.B. in Frontline als MOR208-CHOP oder MOR208-LEN-CHOP recht teuer werden.
Zitat von Joschka Schröder: Die bisherigen Daten zur Dauer der klinischen Antwort und des progressionsfreien Überlebens lassen vermuten, dass es MOR208 gelingen wird, ein gewisses Stück vom DLBCL-Markt zu okkupieren.
Sehe ich auch so. Die Dauer der Antwort ist ein wirklich sehr wichtiges Feature, das nicht nur dem Patienten hilft, sondern auch eine wichtige ökonomische Komponente hat, denn somit kann ich das Produkt sehr lange verabreichen und deutlich höhere Umsätze generieren.
Dieser DLBCL-Markt ist in seiner Dynamik aber nicht wirklich prognostizierbar und birgt weiterhin ziemliche Risiken.
Neue Player und Produkte werden auf den Markt kommen, z.B. bispezifische anti-CD20 oder anti-CD19 mabs, die genau die Funktion von MOR208 wahrnehmen sollen - mit vielleicht noch besserer und längerer Wirksamkeit.
Zudem wird die weitere Entwicklung, z.B. in Frontline als MOR208-CHOP oder MOR208-LEN-CHOP recht teuer werden.
Sanofi wird während der nächsten Monate einen Zulassungsantrag für ihren anti-CD38-Antikörper isatuximab stellen.
-> https://globenewswire.com/news-release/2017/12/13/1261024/0/…
Bin gespannt, ob Morphosys auch hier im Fall einer Zulassung Patentverletzungen reklamieren und Schadensersatz geltend machen wird.
-> https://globenewswire.com/news-release/2017/12/13/1261024/0/…
Bin gespannt, ob Morphosys auch hier im Fall einer Zulassung Patentverletzungen reklamieren und Schadensersatz geltend machen wird.
Antwort auf Beitrag Nr.: 56.417.553 von Joschka Schröder am 10.12.17 18:06:19
Sorry Joschka, aber ich verstehe nicht, warum Du immer wieder auf dieser falschen Interpretation der Zulassungsmeilensteine herumreitest. Es ist absurd, zu glauben, dass die 187 Mio US$ in einem Stück bei Erstzulassung in einer einzigen Indikaton in einem einzigen Markt fällig werden. Das widerspricht sämtlichen Erfahrungen aus der Vertragsgestaltung vergleichbarer Deals anderer Unternehmen. Es widerspricht auch den spärlichen bekannten Details aus MOR-Verträgen...
Es wäre auch kein Vorteil, sondern ein sehr großer Nachteil für Xencor (wenn wir mal davon ausgehen, dass die Lenker von Morphosys nicht vollkommen bescheuert sind...), denn ein Medikament für das jemand bereit wäre, einen solch gigantischen Zulassungsmeilenstein zu zahlen, sollte doch ein entsprechendes Potential besitzen. Xencor wäre damit aber von allen zukünftigen Erfolgen, bis auf gestaffelte Meilensteinzahlungen in den niedrigen 2-stelligen Bereich hinein, ausgeschlossen. Meinst du, das ist vernünftig?
Kurz und gut - ich hab's schon das letzte Mal gesagt, als du diese Behauptung aufgestellt hast: Die 187 Mio. US$ sind die Summe aller möglichen zulassungsrelevanten Meilensteinzahlungen für MOR 208.
Zitat von Joschka Schröder: ...Positiv ist dies natürlich insbesondere auch für Xencor, das bei Zulassung eine Meilensteinzahlung in Höhe von 187 Mio. USD kassieren wird und Royalties im hohen einstelligen bis niedrigen zweistelligen Prozentbereich kassieren wird...
Sorry Joschka, aber ich verstehe nicht, warum Du immer wieder auf dieser falschen Interpretation der Zulassungsmeilensteine herumreitest. Es ist absurd, zu glauben, dass die 187 Mio US$ in einem Stück bei Erstzulassung in einer einzigen Indikaton in einem einzigen Markt fällig werden. Das widerspricht sämtlichen Erfahrungen aus der Vertragsgestaltung vergleichbarer Deals anderer Unternehmen. Es widerspricht auch den spärlichen bekannten Details aus MOR-Verträgen...
Es wäre auch kein Vorteil, sondern ein sehr großer Nachteil für Xencor (wenn wir mal davon ausgehen, dass die Lenker von Morphosys nicht vollkommen bescheuert sind...), denn ein Medikament für das jemand bereit wäre, einen solch gigantischen Zulassungsmeilenstein zu zahlen, sollte doch ein entsprechendes Potential besitzen. Xencor wäre damit aber von allen zukünftigen Erfolgen, bis auf gestaffelte Meilensteinzahlungen in den niedrigen 2-stelligen Bereich hinein, ausgeschlossen. Meinst du, das ist vernünftig?
Kurz und gut - ich hab's schon das letzte Mal gesagt, als du diese Behauptung aufgestellt hast: Die 187 Mio. US$ sind die Summe aller möglichen zulassungsrelevanten Meilensteinzahlungen für MOR 208.
Positive top line results from the UCB Phase 2b BE ACTIVE study underscore the potential of bimekizumab to significantly improve joint and skin symptoms in PsA patients
http://www.pipelinereview.com/index.php/2017122066895/Antibo…
http://www.pipelinereview.com/index.php/2017122066895/Antibo…
Antwort auf Beitrag Nr.: 56.500.547 von Milestones am 20.12.17 10:20:04@Milestones
Über das Thema Meilensteinzahlung(en)/Zulassung wurde schon so oft und ausführlich in verschiedenen Threads diskutiert, dass es nun wirklich nicht mehr eines Hinweises bedarfs, dass sich die Gesamtsumme i.H.v. 187 Mio. USD aus verschiedenen Teilzahlungen zusammensetzt. Branchenüblich wäre, aber das wurde auch schon bis zum geht nicht mehr wiederholt, eine Differenzierung nach Regionen (z.B. USA, Europa, RoW), deutlich seltener, aber auch vorstellbar, wäre noch eine zusätzliche Indikationsklausel. Details wird man erfahren, wenn es soweit ist.
Aber wie gesagt: Das wurde alles in schon so oft durchdekliniert, dass es vergeudete Energie ist, sich daran zu reiben, wenn vereinfachend von einer Meilensteinzahlung bei Zulassung die Rede ist. Jeder weiß, was gemeint ist.
Deine anderen Erläuterungen ("Es wäre auch kein Vorteil, sondern ein sehr großer Nachteil für Xencor (wenn wir mal davon ausgehen, dass die Lenker von Morphosys nicht vollkommen bescheuert sind...), denn ein Medikament für das jemand bereit wäre, einen solch gigantischen Zulassungsmeilenstein zu zahlen, sollte doch ein entsprechendes Potential besitzen. Xencor wäre damit aber von allen zukünftigen Erfolgen, bis auf gestaffelte Meilensteinzahlungen in den niedrigen 2-stelligen Bereich hinein, ausgeschlossen") kann ich nicht recht nachvollziehen. Deiner etwas schrägen Argumentation zufolge, muss einer der beiden Vertragspartner (aus Deiner Sicht: Xencor) "bescheuert" sein.
Fakt ist doch, dass die Verträge vor etlichen Jahren abgeschlossen worden sind, als die künftige Marktentwicklung noch nicht absehbar war. Jeder Vertragspartner hatte bei Vertragsabschluss ein bestimmtes Szenario vor Augen. Für welchen Vertragspartner sich das Vertragswerk letztlich als attraktiver erweisen wird, wird sich erst später - retrospektiv - beurteilen lassen. Stand Dezember 2017 scheinen die Regelungen unter rein finanziellen Aspekten eher für Xencor vorteilhaft. Dies ist aber nur eine Momentaufnahme unter der Bedingung, dass es keine nennenswerte indikationsbezogene Aufsplittung der Zulassungsmeilensteine gibt.
Über das Thema Meilensteinzahlung(en)/Zulassung wurde schon so oft und ausführlich in verschiedenen Threads diskutiert, dass es nun wirklich nicht mehr eines Hinweises bedarfs, dass sich die Gesamtsumme i.H.v. 187 Mio. USD aus verschiedenen Teilzahlungen zusammensetzt. Branchenüblich wäre, aber das wurde auch schon bis zum geht nicht mehr wiederholt, eine Differenzierung nach Regionen (z.B. USA, Europa, RoW), deutlich seltener, aber auch vorstellbar, wäre noch eine zusätzliche Indikationsklausel. Details wird man erfahren, wenn es soweit ist.
Aber wie gesagt: Das wurde alles in schon so oft durchdekliniert, dass es vergeudete Energie ist, sich daran zu reiben, wenn vereinfachend von einer Meilensteinzahlung bei Zulassung die Rede ist. Jeder weiß, was gemeint ist.
Deine anderen Erläuterungen ("Es wäre auch kein Vorteil, sondern ein sehr großer Nachteil für Xencor (wenn wir mal davon ausgehen, dass die Lenker von Morphosys nicht vollkommen bescheuert sind...), denn ein Medikament für das jemand bereit wäre, einen solch gigantischen Zulassungsmeilenstein zu zahlen, sollte doch ein entsprechendes Potential besitzen. Xencor wäre damit aber von allen zukünftigen Erfolgen, bis auf gestaffelte Meilensteinzahlungen in den niedrigen 2-stelligen Bereich hinein, ausgeschlossen") kann ich nicht recht nachvollziehen. Deiner etwas schrägen Argumentation zufolge, muss einer der beiden Vertragspartner (aus Deiner Sicht: Xencor) "bescheuert" sein.
Fakt ist doch, dass die Verträge vor etlichen Jahren abgeschlossen worden sind, als die künftige Marktentwicklung noch nicht absehbar war. Jeder Vertragspartner hatte bei Vertragsabschluss ein bestimmtes Szenario vor Augen. Für welchen Vertragspartner sich das Vertragswerk letztlich als attraktiver erweisen wird, wird sich erst später - retrospektiv - beurteilen lassen. Stand Dezember 2017 scheinen die Regelungen unter rein finanziellen Aspekten eher für Xencor vorteilhaft. Dies ist aber nur eine Momentaufnahme unter der Bedingung, dass es keine nennenswerte indikationsbezogene Aufsplittung der Zulassungsmeilensteine gibt.
Studie unterstützt Hoffnung auf Möglichkeit kognitiver Wirksamkeit von Gantenerumab.
"No Amyloid, No Memory Problem—Even with ApoE4?"https://www.alzforum.org/news/research-news/no-amyloid-no-me…
Aktuelles Paper zu bimagrumab:
Effects of bimagrumab, an activin receptor type II inhibitor, on pituitary neurohormonal axes.
https://www.ncbi.nlm.nih.gov/pubmed/29566437[url]" target="_blank" rel="nofollow ugc noopener">
https://www.ncbi.nlm.nih.gov/pubmed/29566437[url]
Effects of bimagrumab, an activin receptor type II inhibitor, on pituitary neurohormonal axes.
https://www.ncbi.nlm.nih.gov/pubmed/29566437[url]" target="_blank" rel="nofollow ugc noopener">
https://www.ncbi.nlm.nih.gov/pubmed/29566437[url]
Antwort auf Beitrag Nr.: 57.374.294 von Joschka Schröder am 23.03.18 22:03:06Jetzt müsste ich nur noch verstehen, wie die Werte einzuschätzen sind und warum es bei Männern (soweit ich es verstanden habe) keinen Effekt gegeben hat.
Kannst mir das jemand so erklären, dass ich das auch verstehe?
Kannst mir das jemand so erklären, dass ich das auch verstehe?
Ach ja, die alte Amyloid-Hypothese ....
https://endpts.com/merck-study-may-signal-doom-for-a-broad-g…
Merck study may signal doom for a broad group of pivotal Alzheimer’s studies
by john carroll — on May 3, 2018 08:53 AM EDT
Updated: 09:17 AM
The BACE theory in Alzheimer’s R&D is simple. Cut off the flow of amyloid beta to the brain and you can eliminate what is widely believed — though not proven — to be a cause of the disease. Do that, and you could bend the course of this devastating illness in millions of people with mild to moderate forms of the disease.
And Merck $MRK just spent a fortune to demonstrate that it may well be completely wrong.
To be sure, Merck ran a clean study for verubecestat, the leading BACE drug in the clinic, and displayed the data on 1,958 patients for all to see today in the New England Journal of Medicine. Investigators carefully tracked amyloid beta flows in cerebrospinal cords and found that the drug did what it was intended to do, with a dramatic reduction of the toxic protein.
It had no effect, with patients in the two dosage groups tracking in parallel decline on both cognition and function, the two classic measures for Alzheimer’s.
The conclusion they reached is that the damage already present in the brains of patients with Alzheimer’s may be too extensive to treat with any BACE drug. And they also concede that the amyloid theory itself may be just flat wrong.
This suggests that once dementia is present, disease progression may be independent of Aβ production or, alternatively, that the amyloid hypothesis of Alzheimer’s disease may not be correct. Because Aβ deposition takes place years before clinical symptoms become apparent, it has been proposed that treatments targeting amyloid should be implemented early in the disease process, before the onset of clinical symptoms.
Soon after this study failed, Merck also threw in the towel on their second pivotal trial, noting it too was a flop. Those data are still being evaluated, but it underscores the belief that all of the BACE studies — including those at Eli Lilly $LLY, partnered with AstraZeneca$AZN, or Biogen $BIIB, allied with Eisai — are headed straight to failure.
Biogen is also rolling the dice on aducanumab, which the company has touted as a leading amyloid beta therapy. But with investigators in the field openly wondering whether the amyloid theory has lured a long lineup into a clinical disaster zone, it’s likely to face growing skepticism that it can develop a safe, effective therapy with just one drug.
This doesn’t by any means eliminate work in the area. True, Pfizer recently pulled out after spending hundreds of millions of dollars on their programs. But startups like Denali believe that new and better technology can give them better odds at success, while Celgene is jumping in with its own new pipeline. Others want to see if combination approaches using tau and amyloid beta together could work.
Merck’s suggestion about going even earlier in the disease process has also prompted a range of studies in pre-symptomatic patients, while the FDA has signaled its interest in coming up with biomarkers to help speed new studies.
After more than 200 R&D projects ended in disaster, though, Alzheimer’s is looking like an increasingly daunting challenge, with no clear path forward that would inspire confidence among patients with the disease.
https://endpts.com/merck-study-may-signal-doom-for-a-broad-g…
Merck study may signal doom for a broad group of pivotal Alzheimer’s studies
by john carroll — on May 3, 2018 08:53 AM EDT
Updated: 09:17 AM
The BACE theory in Alzheimer’s R&D is simple. Cut off the flow of amyloid beta to the brain and you can eliminate what is widely believed — though not proven — to be a cause of the disease. Do that, and you could bend the course of this devastating illness in millions of people with mild to moderate forms of the disease.
And Merck $MRK just spent a fortune to demonstrate that it may well be completely wrong.
To be sure, Merck ran a clean study for verubecestat, the leading BACE drug in the clinic, and displayed the data on 1,958 patients for all to see today in the New England Journal of Medicine. Investigators carefully tracked amyloid beta flows in cerebrospinal cords and found that the drug did what it was intended to do, with a dramatic reduction of the toxic protein.
It had no effect, with patients in the two dosage groups tracking in parallel decline on both cognition and function, the two classic measures for Alzheimer’s.
The conclusion they reached is that the damage already present in the brains of patients with Alzheimer’s may be too extensive to treat with any BACE drug. And they also concede that the amyloid theory itself may be just flat wrong.
This suggests that once dementia is present, disease progression may be independent of Aβ production or, alternatively, that the amyloid hypothesis of Alzheimer’s disease may not be correct. Because Aβ deposition takes place years before clinical symptoms become apparent, it has been proposed that treatments targeting amyloid should be implemented early in the disease process, before the onset of clinical symptoms.
Soon after this study failed, Merck also threw in the towel on their second pivotal trial, noting it too was a flop. Those data are still being evaluated, but it underscores the belief that all of the BACE studies — including those at Eli Lilly $LLY, partnered with AstraZeneca$AZN, or Biogen $BIIB, allied with Eisai — are headed straight to failure.
Biogen is also rolling the dice on aducanumab, which the company has touted as a leading amyloid beta therapy. But with investigators in the field openly wondering whether the amyloid theory has lured a long lineup into a clinical disaster zone, it’s likely to face growing skepticism that it can develop a safe, effective therapy with just one drug.
This doesn’t by any means eliminate work in the area. True, Pfizer recently pulled out after spending hundreds of millions of dollars on their programs. But startups like Denali believe that new and better technology can give them better odds at success, while Celgene is jumping in with its own new pipeline. Others want to see if combination approaches using tau and amyloid beta together could work.
Merck’s suggestion about going even earlier in the disease process has also prompted a range of studies in pre-symptomatic patients, while the FDA has signaled its interest in coming up with biomarkers to help speed new studies.
After more than 200 R&D projects ended in disaster, though, Alzheimer’s is looking like an increasingly daunting challenge, with no clear path forward that would inspire confidence among patients with the disease.
Betrifft Guselkumab:
Ich habe mir heute diverse Studiendaten von UCBs Bimekizumab (anti-IL-17A/F) angesehen. In der Indikation "Psoriatische Arthritis" toppen die Phase 2b-Daten die Daten der Konkurrenz deutlich. Nach dreimaliger Gabe in Woche 2, 3 und 6 wurden in Woche 8 bereits 89 % PASI90 (Placebo 1%) und in Woche 20 sogar 93 % PASI90 (Placebo abermals 1 %) erzielt Die ACR50-Werte in Woche 8 bzw. 20 wurden von 40 % bzw. 57 % der Patienten erreicht.
Man darf gespannt sein, zu welchen Ergebnissen der multispezifische Antikörper UCB0159 führen wird, der außer IL-17A/F noch TNF adressiert und zusätzlich (zwecks Verlängerung der Halbwertszeit) an Albumin bindet.
Ich habe mir heute diverse Studiendaten von UCBs Bimekizumab (anti-IL-17A/F) angesehen. In der Indikation "Psoriatische Arthritis" toppen die Phase 2b-Daten die Daten der Konkurrenz deutlich. Nach dreimaliger Gabe in Woche 2, 3 und 6 wurden in Woche 8 bereits 89 % PASI90 (Placebo 1%) und in Woche 20 sogar 93 % PASI90 (Placebo abermals 1 %) erzielt Die ACR50-Werte in Woche 8 bzw. 20 wurden von 40 % bzw. 57 % der Patienten erreicht.
Man darf gespannt sein, zu welchen Ergebnissen der multispezifische Antikörper UCB0159 führen wird, der außer IL-17A/F noch TNF adressiert und zusätzlich (zwecks Verlängerung der Halbwertszeit) an Albumin bindet.
Bei diesjährigen EULAR Meeting wird GSK wohl keine Ergebnisse zu MOR103 berichten, konnte in der Abstracts-Übersicht jedenfalls nichts dergleichen finden.
Antwort auf Beitrag Nr.: 57.705.709 von Joschka Schröder am 06.05.18 23:27:52
Ab wann würde Janssen denn hier Konkurrenz bekommen? Wie viel Vorsprung haben sie?
Zitat von Joschka Schröder: Betrifft Guselkumab:
Ich habe mir heute diverse Studiendaten von UCBs Bimekizumab (anti-IL-17A/F) angesehen. In der Indikation "Psoriatische Arthritis" toppen die Phase 2b-Daten die Daten der Konkurrenz deutlich. Nach dreimaliger Gabe in Woche 2, 3 und 6 wurden in Woche 8 bereits 89 % PASI90 (Placebo 1%) und in Woche 20 sogar 93 % PASI90 (Placebo abermals 1 %) erzielt Die ACR50-Werte in Woche 8 bzw. 20 wurden von 40 % bzw. 57 % der Patienten erreicht.
Man darf gespannt sein, zu welchen Ergebnissen der multispezifische Antikörper UCB0159 führen wird, der außer IL-17A/F noch TNF adressiert und zusätzlich (zwecks Verlängerung der Halbwertszeit) an Albumin bindet.
Ab wann würde Janssen denn hier Konkurrenz bekommen? Wie viel Vorsprung haben sie?
Novartis ARROW trial to assess mechanistic superiority of direct IL-17A inhibition (Cosentyx®) over IL-23 inhibition (Tremfya®*)
ARROW study to assess the mechanistic superiority of direct IL-17A inhibition (Cosentyx) over IL-23 inhibition (Tremfya®*) in clearing of Stelara®*-resistant psoriatic plaques[1]
This is the 100th trial with Cosentyx in the last 10 years, adding to the wealth of data[2]. Cosentyx has been prescribed to more than 150,000 patients to date[3]
Cosentyx has a broad head-to-head study program that includes FIXTURE, CLEAR, CLARITY, SURPASS and EXCEED clinical superiority trials[4]-[8]
Basel, May 15, 2018 - Novartis announced today the plan to initiate ARROW, a head-to-head proof of concept study to assess the mechanistic superiority of the direct inhibition of IL-17A with Cosentyx® (secukinumab) over the inhibition of IL-23 with Tremfya®* (guselkumab) in patients with psoriatic plaques resistant to treatment with Stelara®*[1]. Study results are expected in 2019.
"We know there are different immune mechanisms driving the clinical manifestations of psoriasis, including the involvement of joints, scalp, nails, palms and soles psoriasis. Results from the ARROW trial could help us learn more about the differences between the direct targeting of IL-17A and IL-23 in psoriasis," said Kristian Reich, M.D., Ph.D., Georg-August-University Göttingen and Dermatologikum Hamburg, Germany. "It's great to see Novartis leading the science in psoriasis, psoriatic arthritis and ankylosing spondylitis."
"Treating all manifestations of psoriasis in the most effective way is crucial for patients and physicians. We are proud to be driving scientific activities to elucidate the biologic pathways in these diseases with the goal of transforming treatment options for patients," said Shreeram Aradhye, Chief Medical Officer and Global Head, Medical Affairs, Novartis Pharmaceuticals.
The ARROW study objective is to assess the mechanistic superiority of Cosentyx® over Tremfya®* in controlling clinical activity in psoriatic plaques resistant to treatment with Stelara®*[1]. IL-23 independent pathways of IL-17A release are potentially involved in inflammation of other persistent localizations, including joints, scalp, nails, palms and soles psoriasis[9]. Up to 90% of people with psoriasis may develop nail or palmoplantar psoriasis[10],[11]. Nail psoriasis is an important predictor of psoriatic arthritis (PsA) which affects up to 40% of patients with psoriasis[12].
Cosentyx is the first fully-human biologic that specifically inhibitsinterleukin-17A (IL-17A), a cornerstone cytokine involved in the inflammation and development of psoriasis, ankylosing spondylitis (AS), and psoriatic arthritis (PsA)[13]-[16]. IL-17A is produced by both IL-23 dependent and IL-23 independent pathways, by various cells from both the innate immune system (which can be triggered by mechanical stress) and the adaptive immune system[9]. By acting directly on IL-17A, Cosentyx inhibits this cornerstone cytokine irrespective of where the IL-17A comes from[13].
This study is now the 100th trial with Cosentyx in the last 10 years, adding to the wealth of data[2]. Cosentyx has a broad head-to-head study program that includes FIXTURE, CLEAR, CLARITY, SURPASS and EXCEED clinical superiority trials[1],[4]-[8]. To date, Cosentyx has been prescribed to more than 150,000 patients with psoriasis, PsA and AS worldwide[3].
About ARROW[1]
ARROW (CAIN457A2403) is a global multicenter open label randomized proof of concept Phase 2a study designed to assess the mechanistic superiority of secukinumab 300 mg over guselkumab 100 mg in achieving the clearing of psoriatic skin plaques resistant to treatment with ustekinumab after 16 weeks. 40 patients will be randomized 1:1 to secukinumab or guselkumab and treated for 16 weeks. The primary endpoint of the study is the percentage of patients achieving clear or almost clear status of the ustekinumab-resistant plaques as assessed through the Total Clinical Score (TCS =<2). The mechanistic exploratory endpoints of this study will explore the hypothesis that the direct targeting of IL-17A is able to overcome IL-23 independent mechanisms of resistance, providing a more complete approach to the control of alternative pathways of psoriasis inflammation.
ARROW study to assess the mechanistic superiority of direct IL-17A inhibition (Cosentyx) over IL-23 inhibition (Tremfya®*) in clearing of Stelara®*-resistant psoriatic plaques[1]
This is the 100th trial with Cosentyx in the last 10 years, adding to the wealth of data[2]. Cosentyx has been prescribed to more than 150,000 patients to date[3]
Cosentyx has a broad head-to-head study program that includes FIXTURE, CLEAR, CLARITY, SURPASS and EXCEED clinical superiority trials[4]-[8]
Basel, May 15, 2018 - Novartis announced today the plan to initiate ARROW, a head-to-head proof of concept study to assess the mechanistic superiority of the direct inhibition of IL-17A with Cosentyx® (secukinumab) over the inhibition of IL-23 with Tremfya®* (guselkumab) in patients with psoriatic plaques resistant to treatment with Stelara®*[1]. Study results are expected in 2019.
"We know there are different immune mechanisms driving the clinical manifestations of psoriasis, including the involvement of joints, scalp, nails, palms and soles psoriasis. Results from the ARROW trial could help us learn more about the differences between the direct targeting of IL-17A and IL-23 in psoriasis," said Kristian Reich, M.D., Ph.D., Georg-August-University Göttingen and Dermatologikum Hamburg, Germany. "It's great to see Novartis leading the science in psoriasis, psoriatic arthritis and ankylosing spondylitis."
"Treating all manifestations of psoriasis in the most effective way is crucial for patients and physicians. We are proud to be driving scientific activities to elucidate the biologic pathways in these diseases with the goal of transforming treatment options for patients," said Shreeram Aradhye, Chief Medical Officer and Global Head, Medical Affairs, Novartis Pharmaceuticals.
The ARROW study objective is to assess the mechanistic superiority of Cosentyx® over Tremfya®* in controlling clinical activity in psoriatic plaques resistant to treatment with Stelara®*[1]. IL-23 independent pathways of IL-17A release are potentially involved in inflammation of other persistent localizations, including joints, scalp, nails, palms and soles psoriasis[9]. Up to 90% of people with psoriasis may develop nail or palmoplantar psoriasis[10],[11]. Nail psoriasis is an important predictor of psoriatic arthritis (PsA) which affects up to 40% of patients with psoriasis[12].
Cosentyx is the first fully-human biologic that specifically inhibitsinterleukin-17A (IL-17A), a cornerstone cytokine involved in the inflammation and development of psoriasis, ankylosing spondylitis (AS), and psoriatic arthritis (PsA)[13]-[16]. IL-17A is produced by both IL-23 dependent and IL-23 independent pathways, by various cells from both the innate immune system (which can be triggered by mechanical stress) and the adaptive immune system[9]. By acting directly on IL-17A, Cosentyx inhibits this cornerstone cytokine irrespective of where the IL-17A comes from[13].
This study is now the 100th trial with Cosentyx in the last 10 years, adding to the wealth of data[2]. Cosentyx has a broad head-to-head study program that includes FIXTURE, CLEAR, CLARITY, SURPASS and EXCEED clinical superiority trials[1],[4]-[8]. To date, Cosentyx has been prescribed to more than 150,000 patients with psoriasis, PsA and AS worldwide[3].
About ARROW[1]
ARROW (CAIN457A2403) is a global multicenter open label randomized proof of concept Phase 2a study designed to assess the mechanistic superiority of secukinumab 300 mg over guselkumab 100 mg in achieving the clearing of psoriatic skin plaques resistant to treatment with ustekinumab after 16 weeks. 40 patients will be randomized 1:1 to secukinumab or guselkumab and treated for 16 weeks. The primary endpoint of the study is the percentage of patients achieving clear or almost clear status of the ustekinumab-resistant plaques as assessed through the Total Clinical Score (TCS =<2). The mechanistic exploratory endpoints of this study will explore the hypothesis that the direct targeting of IL-17A is able to overcome IL-23 independent mechanisms of resistance, providing a more complete approach to the control of alternative pathways of psoriasis inflammation.
Antwort auf Beitrag Nr.: 55.149.869 von deadflowers am 16.06.17 09:14:36
Kritischer Artikel im NEJM zu CAR-T-Therapien und deren Kosten und Nutzen für Patienten und Public Health:
https://www.nejm.org/doi/full/10.1056/NEJMp1807382
…
The two approved CAR-T therapies have boxed warnings regarding serious side effects, and each costs about $400,000. Ancillary costs include initial leukapheresis and inpatient stays that may be necessitated by frequent treatment complications, which may result in administration of tocilizumab (up to four doses at $2,500 per dose). Hernandez and colleagues estimate that these ancillary costs would average around $33,000 per patient; media reports suggest a figure 10 times as high.2
…
…
CAR-T therapies have broken new ground on many fronts — they have shown efficacy in patients who previously had few options, but they cost multiple times what any previously approved cancer therapy costs. Their rapid approval based on small, uncontrolled studies reflects their promise. But they are no panacea. Ms. Kearney died a few weeks after receiving her dose of the CAR-T therapy that cost nearly $400,000, and she endured an extended hospital and ICU stay along the way. She told WBUR reporter Richard Knox that she knew death was a possible outcome, and she was grateful for the chance to receive a possibly effective treatment — a reminder of what’s at stake for Medicare patients when CMS considers CAR-T therapy coverage. If Medicare chooses to cover this therapy, it should think carefully about how to do it.
…
Zitat von deadflowers: Eine der großen offenen Fragen zu CAR-T ist allerdings, wie das alles finanzierbar sein soll. Das betrifft die Therapie selbst und die sehr teure lebenlange IVIG Gabe. Wenn man das in der Breite einsetzt, läuft das ganzes System aus dem Ruder. M.E. unbezahlbar, und eigentlich nicht vorstellbar, dass es bei Patienten zum Einsatz kommt, wo es noch andere Therapiemöglichkeiten gibt.
Kritischer Artikel im NEJM zu CAR-T-Therapien und deren Kosten und Nutzen für Patienten und Public Health:
https://www.nejm.org/doi/full/10.1056/NEJMp1807382
…
The two approved CAR-T therapies have boxed warnings regarding serious side effects, and each costs about $400,000. Ancillary costs include initial leukapheresis and inpatient stays that may be necessitated by frequent treatment complications, which may result in administration of tocilizumab (up to four doses at $2,500 per dose). Hernandez and colleagues estimate that these ancillary costs would average around $33,000 per patient; media reports suggest a figure 10 times as high.2
…
…
CAR-T therapies have broken new ground on many fronts — they have shown efficacy in patients who previously had few options, but they cost multiple times what any previously approved cancer therapy costs. Their rapid approval based on small, uncontrolled studies reflects their promise. But they are no panacea. Ms. Kearney died a few weeks after receiving her dose of the CAR-T therapy that cost nearly $400,000, and she endured an extended hospital and ICU stay along the way. She told WBUR reporter Richard Knox that she knew death was a possible outcome, and she was grateful for the chance to receive a possibly effective treatment — a reminder of what’s at stake for Medicare patients when CMS considers CAR-T therapy coverage. If Medicare chooses to cover this therapy, it should think carefully about how to do it.
…
Antwort auf Beitrag Nr.: 58.461.202 von rollingovermilestones am 16.08.18 10:51:11News zu Gantenerumab:
https://www.alzforum.org/news/conference-coverage/four-immun…
…At least four drugs have now demonstrated the ability to clear plaques from the brain: aducanumab, gantenerumab, Lilly’s LY3002813, and BAN2401…
…It clinched the case that these antibodies can mop up brain amyloid, bringing many people with early symptomatic Alzheimer’s disease below the threshold for amyloid positivity. At one to two years, this clearance took a long time. But still: Gregory Klein at Roche claimed that two years of treatment with high-dose gantenerumab essentially resets a person’s trajectory of amyloid accumulation. “We are setting back the clock by 15 years,” Klein told the audience…
https://www.alzforum.org/news/conference-coverage/four-immun…
…At least four drugs have now demonstrated the ability to clear plaques from the brain: aducanumab, gantenerumab, Lilly’s LY3002813, and BAN2401…
…It clinched the case that these antibodies can mop up brain amyloid, bringing many people with early symptomatic Alzheimer’s disease below the threshold for amyloid positivity. At one to two years, this clearance took a long time. But still: Gregory Klein at Roche claimed that two years of treatment with high-dose gantenerumab essentially resets a person’s trajectory of amyloid accumulation. “We are setting back the clock by 15 years,” Klein told the audience…
Antwort auf Beitrag Nr.: 55.004.922 von Joschka Schröder am 24.05.17 01:00:56
https://orf.at/stories/3118329/
Evenity mit dem Workstoff Romosozumab ist in den USA seit Jänner zugelassen.
Zitat von Joschka Schröder: Mir ist immer noch rätselhaft, wieso Novartis die Entwicklung des von Morphosys konstruierten anti-sclorostin-Antikörpers BPS804 eingestellt (bzw. für eine Nischenindikation auslizensiert) hat, wo doch die präklinischen Ergebnisse so hervorragend waren und auch in den ersten klinischen Untersuchungen (Einmalgabe) bestätigt worden sind.
Zitat von Joschka Schröder: Ich bin auf das anti-sclerostin-Thema am vergangenen Wochenende gestoßen, als bekannt wurde, dass während der P3-Evenity-Studie unter dem von Amgen und UCB entwickelten Konkurrenzantikörper romosozumab gehäuft kardiovaskuläre Probleme aufgetreten sind, die (zumindest) zu einer Verzögerung der Zulassung führen werden. Novartis´Datenanalyse entnehme ich, dass unter BPS804 keine derartigen Nebenwirkungen beobachtet worden sind. Um so bedauerlicher ist es, dass die klinische Entwicklung des BPS804 von Novartis nicht weiter fortgeführt worden ist.Ich krame diese alte Posting hervor, weil ich auf folgenden Artikel gestoßen bin:
https://orf.at/stories/3118329/
Evenity mit dem Workstoff Romosozumab ist in den USA seit Jänner zugelassen.
Trotz potenzieller Nebenwirkungen (Nicht geeignet ist es somit für Patientinnen, die im Vorjahr einen Herzinfarkt oder Schlaganfall hatten.) hofft die Universitätsprofessorin auf eine baldige Zulassung auch in Europa, denn – „derzeit sind keine anderen Medikamente in der Pipeline“
https://www.reuters.com/article/brief-vivoryon-says-morphosy…
Schade, MOR kombiniert jetzt wohl doch nicht mit dem niedermolekularen CD47-SIRPalpha Molekül von Vivoryon.
Die Zurückhaltung damit hatte sich bereits abgezeichnet.
Ich halte den Ansatz für SEHR vielversprechend.
Deshalb bleibt zu hoffen, dass der Ansatz weiterverfolgt wird. Es gibt ja diesbezüglich noch andere Kandidaten!
Schade, MOR kombiniert jetzt wohl doch nicht mit dem niedermolekularen CD47-SIRPalpha Molekül von Vivoryon.
Die Zurückhaltung damit hatte sich bereits abgezeichnet.
Ich halte den Ansatz für SEHR vielversprechend.
Deshalb bleibt zu hoffen, dass der Ansatz weiterverfolgt wird. Es gibt ja diesbezüglich noch andere Kandidaten!
vivoryon war von anfang an money down the drain für morphosys.
Antwort auf Beitrag Nr.: 63.493.208 von rollingovermilestones am 29.04.20 09:28:58Vielleicht musste Enzelsberger deshalb gehen:???
....* Im April 2020 beschloss MorphoSys, die im Rahmen der Vereinbarung vom Juli 2019 mit Vivoryon Therapeutics gewährte exklusive Lizenzoption auf Vivoryons niedermolekulare QPCTL-Inhibitoren im Bereich der Onkologie nicht auszuüben. Diese Entscheidung beruhte auf der umfassenden Analyse der Daten aus präklinischen Validierungsstudien. MorphoSys besitzt eine Minderheitsbeteiligung an Vivoryon Therapeutics, die auf einer Kapitalbeteiligung im Jahr 2019 basiert....
Dennoch hoffe ich persönlich, dass der Ansatz dahinter von MOR weiterverfolgt wird!
FTSV ging für 5 Riesen weg, IMAB sucht jemanden für 2 und TRIL wäre für 0,4 zu haben.
OSE kostet nur 0,1 ist aber bereits mit BI liiert...
Es gibt noch ein paar mehr...
....* Im April 2020 beschloss MorphoSys, die im Rahmen der Vereinbarung vom Juli 2019 mit Vivoryon Therapeutics gewährte exklusive Lizenzoption auf Vivoryons niedermolekulare QPCTL-Inhibitoren im Bereich der Onkologie nicht auszuüben. Diese Entscheidung beruhte auf der umfassenden Analyse der Daten aus präklinischen Validierungsstudien. MorphoSys besitzt eine Minderheitsbeteiligung an Vivoryon Therapeutics, die auf einer Kapitalbeteiligung im Jahr 2019 basiert....
Dennoch hoffe ich persönlich, dass der Ansatz dahinter von MOR weiterverfolgt wird!
FTSV ging für 5 Riesen weg, IMAB sucht jemanden für 2 und TRIL wäre für 0,4 zu haben.
OSE kostet nur 0,1 ist aber bereits mit BI liiert...
Es gibt noch ein paar mehr...
Beitrag zu dieser Diskussion schreiben
Zu dieser Diskussion können keine Beiträge mehr verfasst werden, da der letzte Beitrag vor mehr als zwei Jahren verfasst wurde und die Diskussion daraufhin archiviert wurde.
Bitte wenden Sie sich an feedback@wallstreet-online.de und erfragen Sie die Reaktivierung der Diskussion oder starten Sie eine neue Diskussion.
Investoren beobachten auch:
| Wertpapier | Perf. % |
|---|---|
| +0,62 | |
| +0,05 | |
| -6,34 | |
| +3,29 | |
| -0,79 | |
| +0,84 | |
| +0,82 | |
| +0,06 | |
| -1,47 | |
| +1,10 |
Meistdiskutiert
| Wertpapier | Beiträge | |
|---|---|---|
| 222 | ||
| 167 | ||
| 75 | ||
| 57 | ||
| 49 | ||
| 46 | ||
| 46 | ||
| 45 | ||
| 34 | ||
| 31 |




















08:00 Uhr · wO Chartvergleich · Bayer |
23.04.24 · wO Newsflash · American Express |
23.04.24 · dpa-AFX · Morphosys |
23.04.24 · wallstreetONLINE Redaktion · Bristol-Myers Squibb |
21.04.24 · wO Chartvergleich · ATOSS Software |
20.04.24 · wO Chartvergleich · ABB |
17.04.24 · Der Finanzinvestor · Bristol-Myers Squibb |
16.04.24 · kapitalerhoehungen.de · Morphosys |
11.04.24 · dpa-AFX · Morphosys |
| Zeit | Titel |
|---|---|
| 06:57 Uhr | |
| 23.04.24 | |
| 07.02.24 | |
| 06.02.24 | |
| 16.01.24 | |
| 05.01.24 | |
| 11.12.23 |