



Biotech Depot 2009 - 500 Beiträge pro Seite
eröffnet am 11.01.09 15:53:11 von
neuester Beitrag 03.01.10 16:55:57 von
neuester Beitrag 03.01.10 16:55:57 von
Beiträge: 85
ID: 1.147.459
ID: 1.147.459
Aufrufe heute: 0
Gesamt: 11.891
Gesamt: 11.891
Aktive User: 0
Top-Diskussionen
| Titel | letzter Beitrag | Aufrufe |
|---|---|---|
| vor 8 Minuten | 1449 | |
| vor 10 Minuten | 1221 | |
| vor 49 Minuten | 1086 | |
| vor 9 Minuten | 855 | |
| heute 06:46 | 735 | |
| vor 33 Minuten | 598 | |
| 20.04.24, 12:11 | 489 | |
| vor 20 Minuten | 486 |
Meistdiskutierte Wertpapiere
| Platz | vorher | Wertpapier | Kurs | Perf. % | Anzahl | ||
|---|---|---|---|---|---|---|---|
| 1. | 1. | 18.026,14 | +0,54 | 243 | |||
| 2. | 2. | 1,2700 | -7,97 | 98 | |||
| 3. | 3. | 0,1905 | +0,79 | 89 | |||
| 4. | 4. | 161,50 | +1,78 | 80 | |||
| 5. | 5. | 9,3600 | +1,24 | 75 | |||
| 6. | 6. | 7,0220 | +0,34 | 47 | |||
| 7. | 8. | 0,0160 | -24,17 | 38 | |||
| 8. | 7. | 22,220 | +0,77 | 37 |
Hallo!
Infolge der Finanzkrise war die Entwicklung meines Biotech-Depots im letzten Jahr (Thread: Biotech Depot 2008)) ebenfalls recht mau. Naja, mal schauen, was dieses Jahr bringt... zwei schwere Krisen in nichtmal 10 Jahren - ich hoffe ja nicht, dass man jetzt damit in regelmäßigen Abständen zu rechnen hat. Nun heißt es wieder... neues Jahr, neues Glück!
Das Depot hat sich zum letzten Jahr nicht wesentlich geändert... nur eine Position Seattle Genetics (SGEN) ist dazu gekommen. Wichtigste Position ist nachwievor Genmab, gefolgt von Onyx, Cubist und Regeneron. Damit sieht es aktuell so aus:
19,0% Genmab http://finance.yahoo.com/q?s=GEN.CO
11,0% Onyx http://finance.yahoo.com/q?s=ONXX
7,9% Cubist http://finance.yahoo.com/q?s=CBST
7,7% Regeneron http://finance.yahoo.com/q?s=REGN
5,5% Evotec http://finance.yahoo.com/q?s=EVT.DE
5,2% Isis http://finance.yahoo.com/q?s=ISIS
5,2% Exelixis http://finance.yahoo.com/q?s=EXEL
4,9% Medigene http://finance.yahoo.com/q?s=MDG.DE
4,3% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
4,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,9% Micromet http://finance.yahoo.com/q?s=MITI
3,7% Addex http://finance.yahoo.com/q?s=ADXN.SW
3,4% Progenics http://finance.yahoo.com/q?s=PGNX
3,0% Arena http://finance.yahoo.com/q?s=ARNA
2,9% Array http://finance.yahoo.com/q?s=ARRY
2,8% NicOx http://finance.yahoo.com/q?s=COX.PA
2,5% Rigel http://finance.yahoo.com/q?s=RIGL
1,6% Sucampo http://finance.yahoo.com/q?s=SCMP
1,3% Incyte http://finance.yahoo.com/q?s=INCY
Onyx könnte etwas reduziert werden, NicOx und Sucampo werde ich wohl nicht länger halten, ebenso ist Evotec im Moment eigentlich zu hoch gewichtet. Rigel würde ich zusammen mit Incyte etwas aufstocken. Interessant finde ich Neurosearch, vielleicht noch Basilea.
Ich orientiere mich ein wenig am Nasdaq Biotech Index (NBI)... ein Überblick seiner Werte findet sich hier: Thread: Nasdaq Biotech Index [NBI] - Charts aller enthaltenen Werte

Ein interessantes Biotech-Depot mit gutes Infos ist meiner Meinung nach http://www.hammerstockblog.com/
Nun also allen ein gutes und erfolgreiches Jahr 2009!
mfg ipollit
Infolge der Finanzkrise war die Entwicklung meines Biotech-Depots im letzten Jahr (Thread: Biotech Depot 2008)) ebenfalls recht mau. Naja, mal schauen, was dieses Jahr bringt... zwei schwere Krisen in nichtmal 10 Jahren - ich hoffe ja nicht, dass man jetzt damit in regelmäßigen Abständen zu rechnen hat. Nun heißt es wieder... neues Jahr, neues Glück!
Das Depot hat sich zum letzten Jahr nicht wesentlich geändert... nur eine Position Seattle Genetics (SGEN) ist dazu gekommen. Wichtigste Position ist nachwievor Genmab, gefolgt von Onyx, Cubist und Regeneron. Damit sieht es aktuell so aus:
19,0% Genmab http://finance.yahoo.com/q?s=GEN.CO
11,0% Onyx http://finance.yahoo.com/q?s=ONXX
7,9% Cubist http://finance.yahoo.com/q?s=CBST
7,7% Regeneron http://finance.yahoo.com/q?s=REGN
5,5% Evotec http://finance.yahoo.com/q?s=EVT.DE
5,2% Isis http://finance.yahoo.com/q?s=ISIS
5,2% Exelixis http://finance.yahoo.com/q?s=EXEL
4,9% Medigene http://finance.yahoo.com/q?s=MDG.DE
4,3% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
4,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,9% Micromet http://finance.yahoo.com/q?s=MITI
3,7% Addex http://finance.yahoo.com/q?s=ADXN.SW
3,4% Progenics http://finance.yahoo.com/q?s=PGNX
3,0% Arena http://finance.yahoo.com/q?s=ARNA
2,9% Array http://finance.yahoo.com/q?s=ARRY
2,8% NicOx http://finance.yahoo.com/q?s=COX.PA
2,5% Rigel http://finance.yahoo.com/q?s=RIGL
1,6% Sucampo http://finance.yahoo.com/q?s=SCMP
1,3% Incyte http://finance.yahoo.com/q?s=INCY
Onyx könnte etwas reduziert werden, NicOx und Sucampo werde ich wohl nicht länger halten, ebenso ist Evotec im Moment eigentlich zu hoch gewichtet. Rigel würde ich zusammen mit Incyte etwas aufstocken. Interessant finde ich Neurosearch, vielleicht noch Basilea.
Ich orientiere mich ein wenig am Nasdaq Biotech Index (NBI)... ein Überblick seiner Werte findet sich hier: Thread: Nasdaq Biotech Index [NBI] - Charts aller enthaltenen Werte
Ein interessantes Biotech-Depot mit gutes Infos ist meiner Meinung nach http://www.hammerstockblog.com/
Nun also allen ein gutes und erfolgreiches Jahr 2009!

mfg ipollit
die einzelnen Charts...
Genmab http://www.genmab.com/

Onyx http://www.onyx-pharm.com/

Cubist http://www.cubist.com/

Regeneron http://www.regeneron.com/

Evotec http://www.evotec.com/

Isis
Exelixis http://www.exelixis.com/

Medigene http://www.medigene.de/

Seattle Genetics http://www.seagen.com/

ViroPharma http://www.viropharma.com/

Micromet http://www.micromet.de/

Addex http://www.addexpharma.com/

Progenics http://www.progenics.com/

Arena http://www.arenapharm.com/

Array http://www.arraybiopharma.com/

NicOx http://www.nicox.com/

Rigel http://www.rigel.com/

Sucampo http://www.sucampo.com/

Incyte http://www.incyte.com/

mfg ipollit" target="_blank" rel="nofollow ugc noopener">http://www.isispharm.com/
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit" target="_blank" rel="nofollow ugc noopener">
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit" target="_blank" rel="nofollow ugc noopener">http://www.isispharm.com/
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit
Genmab http://www.genmab.com/
Onyx http://www.onyx-pharm.com/
Cubist http://www.cubist.com/
Regeneron http://www.regeneron.com/
Evotec http://www.evotec.com/
Isis
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit" target="_blank" rel="nofollow ugc noopener">http://www.isispharm.com/
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit" target="_blank" rel="nofollow ugc noopener">
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit" target="_blank" rel="nofollow ugc noopener">http://www.isispharm.com/
Exelixis http://www.exelixis.com/
Medigene http://www.medigene.de/
Seattle Genetics http://www.seagen.com/
ViroPharma http://www.viropharma.com/
Micromet http://www.micromet.de/
Addex http://www.addexpharma.com/
Progenics http://www.progenics.com/
Arena http://www.arenapharm.com/
Array http://www.arraybiopharma.com/
NicOx http://www.nicox.com/
Rigel http://www.rigel.com/
Sucampo http://www.sucampo.com/
Incyte http://www.incyte.com/
mfg ipollit
Antwort auf Beitrag Nr.: 36.350.858 von ipollit am 11.01.09 15:53:11Kauf doch mal Xoma + Paion 

Nimm noch MorphoSys mit rein und das Depot ist absolut top! Du brauchst jedenfalls keinen Biotech-Fonds, denn Du hast ja fast schon deinen eigenen. Auch über Celgene und Genzyme würde ich noch nachdenken...
Genmab wurde zuletzt etwas belastet durch die Zwischenergebnisse der PIII-Zulassungstudie von Zalutumumab gegen Krebs im Bereich von Kopf und Hals. Die Studie wird nun bis zum Ende weitergeführt und nicht vorzeitig abgebrochen, weil bereits vor Ende der Studie eine klare Wirksamkeit von Zalutumumab belegbar wäre. Dies heißt also nicht, dass Zalutumumab, das ein direkter Konkurrent zu Imclone's Erbitux sein soll, nicht wirksam ist, sondern nur, dass es nicht zu einer vorzeitigen Zulassung kommt. Die Endgültigen Ergebnisse könnten nun Ende des Jahres kommen und ein Partnerschaftsdeal+Zulassung in 2010.
Dafür sieht es mit Arzerra (Konkurrent von Rituxan) immer noch sehr gut aus... in Kürze sollte zusammen mit GSK der erste Zulassungantrag (CLL) gestellt werden und dann entsprechend die Zulassung im Sommer erfolgen. Genmab wird etwa 25% Royalties für Arzerra von GSK erhalten. In 2010 könnte es eine weitere Zulassung gegen NHL geben... angeblich kommen PIII-Ergebnisse bereits in diesem Quartal.
Goldman Sachs erwartet für Genmab 2008 einen Umsatz von ca. 125 Mio USD, für 2009 250 Mio USD und für 2010 500 Mio USD... entsprechend soll der Cash bis 2010 auf 500 Mio USD steigen (aktuelle MK ca. 1,75 Mrd USD bei ca. 300 Mio USD Cash). Das Ergebnis soll 2009 bei 4 DKK und 2010 bei 35 DKK pro Aktie liegen (Kurs aktuell bei 220 DKK).
**********
Genmab Shares Slide on Interim Zalutumumab Data
By Cormac Sheridan
BioWorld International Correspondent
Shares in Genmab A/S were off by more than 7 percent during trading Tuesday following an interim update on a Phase III pivotal study of zalutumumab (HuMax-EGFr) in head and neck cancer.
The Copenhagen, Denmark-based company said the data it received at the halfway point did not fulfill criteria for an early halt on grounds of exceptional efficacy. That was sufficient to prompt a slide, as the share price had been boosted "by a growing expectation during the last three to six months that the study would be stopped at the interim stage," analyst Mark Clark of ING Wholesale in London, told BioWorld International.
"Had it met the stop criteria at the interim stage, there would have been the possibility of doing a partnership deal this year," he said. "We're now back to the original timeline, which is late 2009."
The stock (COPENHAGEN:GEN) was changing hands at DKK213.25 (US$38.38) early afternoon Tuesday, down DKK17.50 on the previous day's close of DKK230.75. It hit a low of DKK211.50 earlier in the day.
"Obviously the criteria for stopping it were pretty tough," Clark said. However, analysts interpreted the company's "obvious enthusiasm" for the molecule during its third quarter analyst call last year as an indication that the drug appeared to be performing well.
Patients in the trial - who were randomized to the treatment and control arms on a 2:1 basis - were surviving beyond six months, when best supported care historically delivered about 4.5 months of survival. "You can argue whether or not the company allowed those expectations to run," he said.
The company reiterated its enthusiasm for the program Monday. "Anecdotally, we hear from some of the sites that they are very pleased with how the trial is going," Genmab CEO Lisa Drakeman told analysts on a conference call Monday.
"We've got very good feedback," Chief Scientific Officer Jan van de Winkel said on the call. "I'm fairly optimistic about the outcome. I think it will be fine."
An independent data monitoring committee did conclude that the benefit-risk profile of zalutumumab, which inhibits epidermal growth factor receptor, is acceptable. However, it is not releasing any efficacy data at this point, Drakeman said, in order to avoid the danger of introducing bias. Enrollment of up to 273 patients with squamous cell carcinoma of the head and neck, who are refractory to or intolerant of standard platinum-based chemotherapy, will continue.
The trial, which has received fast-track designation from the FDA, has enrolled 212 subjects so far. Participants are being randomized into one of two treatment groups: zalutumumab in combination with best supportive care or best supportive care alone. The primary endpoint is survival from randomization until death. A final analysis will be performed once 231 deaths have occurred.
Genmab expects to report the data by the year-end, and to file for a BLA in early 2010. The company does not plan to finalize a partnering deal in advance. "I think we need to stick with our plan, which is to wait for data," Drakeman said.
A Phase III study of zalutumumab in combination with radiotherapy in 600 previously untreated head and neck cancer patients, which is being conducted in cooperation with the Danish Head and Neck Cancer Study Group, is ongoing.
Published January 7, 2009

mfg ipollit
Dafür sieht es mit Arzerra (Konkurrent von Rituxan) immer noch sehr gut aus... in Kürze sollte zusammen mit GSK der erste Zulassungantrag (CLL) gestellt werden und dann entsprechend die Zulassung im Sommer erfolgen. Genmab wird etwa 25% Royalties für Arzerra von GSK erhalten. In 2010 könnte es eine weitere Zulassung gegen NHL geben... angeblich kommen PIII-Ergebnisse bereits in diesem Quartal.
Goldman Sachs erwartet für Genmab 2008 einen Umsatz von ca. 125 Mio USD, für 2009 250 Mio USD und für 2010 500 Mio USD... entsprechend soll der Cash bis 2010 auf 500 Mio USD steigen (aktuelle MK ca. 1,75 Mrd USD bei ca. 300 Mio USD Cash). Das Ergebnis soll 2009 bei 4 DKK und 2010 bei 35 DKK pro Aktie liegen (Kurs aktuell bei 220 DKK).
**********
Genmab Shares Slide on Interim Zalutumumab Data
By Cormac Sheridan
BioWorld International Correspondent
Shares in Genmab A/S were off by more than 7 percent during trading Tuesday following an interim update on a Phase III pivotal study of zalutumumab (HuMax-EGFr) in head and neck cancer.
The Copenhagen, Denmark-based company said the data it received at the halfway point did not fulfill criteria for an early halt on grounds of exceptional efficacy. That was sufficient to prompt a slide, as the share price had been boosted "by a growing expectation during the last three to six months that the study would be stopped at the interim stage," analyst Mark Clark of ING Wholesale in London, told BioWorld International.
"Had it met the stop criteria at the interim stage, there would have been the possibility of doing a partnership deal this year," he said. "We're now back to the original timeline, which is late 2009."
The stock (COPENHAGEN:GEN) was changing hands at DKK213.25 (US$38.38) early afternoon Tuesday, down DKK17.50 on the previous day's close of DKK230.75. It hit a low of DKK211.50 earlier in the day.
"Obviously the criteria for stopping it were pretty tough," Clark said. However, analysts interpreted the company's "obvious enthusiasm" for the molecule during its third quarter analyst call last year as an indication that the drug appeared to be performing well.
Patients in the trial - who were randomized to the treatment and control arms on a 2:1 basis - were surviving beyond six months, when best supported care historically delivered about 4.5 months of survival. "You can argue whether or not the company allowed those expectations to run," he said.
The company reiterated its enthusiasm for the program Monday. "Anecdotally, we hear from some of the sites that they are very pleased with how the trial is going," Genmab CEO Lisa Drakeman told analysts on a conference call Monday.
"We've got very good feedback," Chief Scientific Officer Jan van de Winkel said on the call. "I'm fairly optimistic about the outcome. I think it will be fine."
An independent data monitoring committee did conclude that the benefit-risk profile of zalutumumab, which inhibits epidermal growth factor receptor, is acceptable. However, it is not releasing any efficacy data at this point, Drakeman said, in order to avoid the danger of introducing bias. Enrollment of up to 273 patients with squamous cell carcinoma of the head and neck, who are refractory to or intolerant of standard platinum-based chemotherapy, will continue.
The trial, which has received fast-track designation from the FDA, has enrolled 212 subjects so far. Participants are being randomized into one of two treatment groups: zalutumumab in combination with best supportive care or best supportive care alone. The primary endpoint is survival from randomization until death. A final analysis will be performed once 231 deaths have occurred.
Genmab expects to report the data by the year-end, and to file for a BLA in early 2010. The company does not plan to finalize a partnering deal in advance. "I think we need to stick with our plan, which is to wait for data," Drakeman said.
A Phase III study of zalutumumab in combination with radiotherapy in 600 previously untreated head and neck cancer patients, which is being conducted in cooperation with the Danish Head and Neck Cancer Study Group, is ongoing.
Published January 7, 2009
mfg ipollit
Trading Spotlight
Cubist (CBST) entwickelt sich meiner Meinung nach auch recht gut. Cubicine ist sehr erfolgreich. Die aktuellen Schätzungen sind laut yahoo ein Umsatz von 434 Mio USD für 2008 und 574 Mio USD für 2009... und ein Ergebnis von 1,28 bzw 1,74 USD pro Aktie. Bei einem Kurs von 24,56 USD entspricht dies einem KGV08e von 19 und KGV09e von 14. Cubist hat für Cubicine mittelfristig einen Umsatz von 750 Mio USD angepeilt.
Letztes Jahr wurde Ecallantide (DX-88) gegen Blutverlust in PII von Dyax einlizensiert... meiner Meinung nach eine Indikation mit Potential (Dyax könnte bald für DX-88 bereits eine FDA-Zulassung gegen HAE erhalten als Konkurrent von Jerini's Icatibant).

Nun hat Cubist einen weiteren Kandidaten einlizensiert... der am weitesten fortgeschrittene RNAi-Kandidat von Alnylam, ALN-RSV01 in PII...
http://www.tmcnet.com/usubmit/2009/01/09/3902909.htm
[January 09, 2009]
Cubist, Alnylam Partner in $102.5M RNAi Drugs Deal
(BioWorld Today Via Acquire Media NewsEdge) Massachusetts firms Cubist Pharmaceuticals Inc. and Alnylam Pharmaceuticals Inc. have formed a 50-50 North American partnership to develop and commercialize Alnylam's respiratory syncytial virus (RSV)-specific RNAi therapeutics.
But Wall Street apparently was not so keen on the deal, sending both firms' shares down slightly Friday. Shares of Lexington, Mass.-based Cubist (NASDAQ:CBST) closed at $24.56, a loss of 63 cents, while shares of Cambridge, Mass.-based Alnylam (NASDAQ:ALNY) fell 88 cents, to close at $23.12.
Under the agreement, Alnylam will receive $20 million up front and could bank an additional $82.5 million based on certain development and sales milestones.
The company also could gain double-digit royalties on net sales outside of North America and Asia.
The partnership gives Cubist sole commercialization rights of the ALN-RSV program worldwide outside of Asia, where the rights are held by Japanese firm Kyowa Hakko Kirin Co. Ltd. under a $93 million deal signed with Alnylam last June. (See BioWorld Today, June 20, 2008.)
Cubist and Alnylam will share equally development responsibilities and profits in North America. After achieving certain development milestones, Alnylam could opt to convert the North American co-development and profit share to a royalty-bearing license with development and sales milestones.
The RSV-specific RNAi therapeutic program includes Alnylam's ALN-RSV01, which currently is being studied in a double-blind, randomized, placebo-controlled Phase II trial to assess the drug's safety and tolerability in adult lung transplant patients naturally infected with RSV, said Jason Rhodes, Alnylam's vice president of business development.
The trial also will evaluate the antiviral activity of ALN-RSV01 in patients with a naturally acquired RSV lower respiratory tract infection.
The firm expects to have data from the trial by the second half of this year, Rhodes told BioWorld Today. Following the completion of that trial, Alnylam expects to begin a trial of ALN-RSV01 in pediatric patients with RSV, he noted.
Results reported by Alnylam of a double-blind, randomized, placebo-controlled Phase II trial, known as GEMINI, showed that intranasally administered ALN-RSV01 demonstrated statistically significant antiviral efficacy with about a 40 percent relative reduction in RSV infection rate and a 95 percent increase in the number of infection-free patients compared with placebo.
The company's RSV-specific RNAi program also includes other potent and specific second-generation RNAi-based RSV inhibitors in preclinical development.
RSV is a common, highly contagious virus that causes infections in both the upper- and lower respiratory tract in children and adults, Rhodes explained.
There are about 18 million infections reported annually in most developed countries, resulting in about 1 million hospitalizations, he said.
In the U.S. alone, there are about 125,000 pediatric and 175,000 adult hospitalizations annually attributed to RSV, Rhodes said.
While RSV infection typically results in cold-like symptoms, it can lead to more serious respiratory illnesses such as croup, pneumonia and bronchiolitis, and in some cases, be deadly.
RSV infection in infants also has been linked to the development of childhood asthma.
There currently are no treatments that are broadly used to successfully treat RSV, Rhodes noted.
Pediatric patients especially, said Steven Gilman, Cubists' chief scientific officer, are "in dire need" of an effective treatment for the infection.
"RSV is an important disease for children, and there is a very large unmet medical need," he told BioWorld Today.
Given the RSV program's potential to be used in a fairly broad outpatient population, Rhodes said, it was always Alnylam's strategy to partner it.
"At this stage, we don't plan to build a broad outpatient commercial organization at this point in the company's development," he said.
Alnylam had a "broad range" of discussions with several pharmaceutical companies ranging from large multinational pharma companies to local biopharmaceutical firms.
Rhodes said his firm chose Cubist to partner on the RSV RNAi program because of its expertise in acute care and infectious disease products.
He noted Cubist's success in commercializing Cubicin (daptomycin for injection), the first antibiotic in a new class of anti-infectives called lipopeptides.
Cubist also has an agreement with London-based AstraZeneca plc to handle U.S. hospital sales for its intravenous antibiotic Merrem (meropenem for injection).
Cubist's product pipeline also includes ecallantide, a recombinant human protein in Phase II clinical trials for the prevention of blood loss during cardiothoracic surgery, and two development programs for drugs to treat Gram-negative infections and Clostridium difficile-associated diarrhea.
In addition, Rhodes said, Cubist is located just "15 minutes west" of Alnylam.
"So if you are going to have a partner where they are in a true 50-50 deal where you have to be working together to co-develop the product, it's obviously helpful to have them be local," he said.
Beyond the location, Rhodes said, the firms have a "culture similarity" that makes them "very compatible."
"Cubist is a superb partner," he added.
The deal "sits well" with Cubist's strategy to build its pipeline, Gilman said.
"We are very excited about the progress of RNAi therapeutics. This is a new area of science and new technology. The science together with the medical need for RSV will be a very dramatic, potentially exciting and rewarding program for us," he noted.
Analyst Thomas Russo, of R.W. Baird & Co., called Alnylam's partnership with Cubist "another disciplined deal" for the RSV RNAi program.
"We like it," he said.
But Piper Jaffray analyst Edward A. Tenthoff called the deal's terms "disappointing."
"In comparison, Kyowa paid $15 million up front plus $78 million in potential milestones last June for Asian rights, indicating that the value of the RSV program has declined," he said in a research note.
**************
http://seekingalpha.com/article/114245-cubist-and-alnylam-an…
Cubist and Alnylam, An Odd Partnership
by: Eben Tessari January 11, 2009 | about stocks: ALNY / CBST
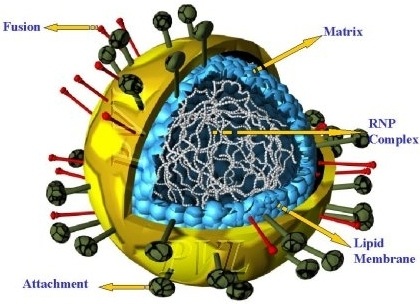
Cubist (CBST) and Alnylam (ALNY) have formed a strategic collaboration to develop and commercialize Alnylam’s ALN-RSV program. The RSV-specific RNAi therapeutic program includes ALN-RSV01, which is currently in Phase II clinical development for the treatment of respiratory syncytial virus (RSV) infection in adult lung transplant patients, as well as several other potent and specific second-generation RNAi-based RSV inhibitors in pre-clinical studies.
Deal Terms
————————-
- $20M upfront
- $82.5M in potential sales and milestone payments
- 50/50 Co-development and profit sharing agreement in US
- Cubist has full commercialization rights EX-US and Asia
- Alnylam receives double digit royalties EX-US and Asia
Cubist? Really? It’s not that I don’t have respect for Cubist, I do. It just strikes me as an odd partnership choice. Why couldn’t Alnylam land another one of those megadeals it’s known for; one of those that In Vivo appoints its deal of the year? Might it be because all of the big pharma players said no? The GEMINI study (small phase IIa with ALN-RSV01) published about a year ago was supposedly the proof that RNAi could work in humans. Well, if so, where is the obscene upfront?
The trial in question was small and mostly artificial. The patients were infected and treated with RSV in their nose… not their lungs like it would be in actual patients. When Alnylam can get the drug to the lower lung and have it reduce viral load and improved clinical symptoms, I’ll be impressed. In addition, to be approved this drug must show some benefit regarding a reduction of days in the hospital or reduction in symptoms, two topics GEMINI didn’t even begin to address.
I could be wrong here, but my guess is that these issues are why Alnylam partnered this drug with Cubist and not with big pharma. Overall, the deal is probably fairly valued, though not by recent Alnylam standards; ALNY is trading down 6% on the day as I write this so it seems that the market was a bit disappointed.
The street better get used to it; I’ve said it before and I’ll say it again: I don’t believe in RNAi as a human therapy for a wide range of diseases (if any). Now, that doesn’t mean I don’t want to see it succeed or want the companies to fizzle out and die; cutting edge research is essential to both medicine and the economy… just don’t ask me to invest in Alnylam.
*************
http://triangle.bizjournals.com/triangle/othercities/boston/…
Monday, January 5, 2009, 3:52pm EST | Modified: Wednesday, January 7, 2009, 1:32pm
Cubist receives $90M credit
Cubist Pharmaceuticals, the Lexington, Mass.-based biotechnology company, has opened a $90 million revolving line of credit with the Royal Bank of Scotland. The money is to be used for general corporate expenses, according to a document filed with the Securities and Exchange Commission.
Cubist (Nasdaq: CBST) has also filed two Investigational New Drug applications with the Food and Drug Administration, indicating its plans to begin clinical trials on the drug targets. The company has one drug, an intravenous antibiotic called Cubicin, already on the market. The company focuses on research, development and commercialization of acute care pharmaceutical products.
The company’s 2007 revenue was $294.6 million. Cubist’s shares were trading at $25.08 in late Monday afternoon trading. The previous close was $25.05. The company’s stock has traded between $16.25 and $28.74 over the past year.
***********
http://www.canadianbusiness.com/markets/market_news/article.…
From The Associated Press, December 8, 2008 - 3:40 PM
Cubist slides following second downgrade in 2 days; analyst cites Cubicin patent risk
NEW YORK (AP) - Shares of Cubist Pharmaceuticals Inc. sank Monday as concerns swirled about the patent protection for its antibiotic Cubicin, which provides almost all of the company's revenue.
Leerink Swann analyst Howard Liang lowered his rating to "Underperform" from "Market Perform" because of his view that investors are overlooking the risk that a generic drug maker will challenge the patents protecting Cubicin. He said Cubist shares might be worth $10 if the patents were successfully challenged.
Shares of the Lexington, Mass., company fell $2.34, or 8.7 percent, to $24.64 in afternoon trading.
Cubicin is vulnerable to generic competition because the Food and Drug Administration approved it more than five years ago, in September 2003. The patents on the drug are set to expire in 2016 and 2019, but generic drug companies could challenge those patents earlier by saying they are not valid or don't apply to their version of the drug.
If such a challenge were successful, Liang estimated Cubist shares would be worth $10.
Cubist said it has not been notified of any patent challenges for Cubicin.
With competing stocks slumping and Cubist shares trading at their highest levels in almost seven years, he said, investors are no longer accounting for that risk, which is particularly important to Cubist because Cubicin is its only marketed drug.
Cubist reported $112.4 million in revenue in the third quarter. Sales of Cubicin accounted for $110.6 million of that total.
The analyst described Cubicin as the most "successful intravenous antibiotic U.S. launch in history," and said demand for Cubicin to treat antibiotic-resistant infections has grown significantly. But Cubist's other drugs are in preclinical testing, and he said there is little excitement about its blood loss drug DX-88.
Liang was the second analyst in two days to cut his rating on the stock, following an Oppenheimer downgrade Friday. Kevin DeGeeter cited the weak patent protection for Cubicin as one of the reasons for his downgrade.
mfg ipollit
Letztes Jahr wurde Ecallantide (DX-88) gegen Blutverlust in PII von Dyax einlizensiert... meiner Meinung nach eine Indikation mit Potential (Dyax könnte bald für DX-88 bereits eine FDA-Zulassung gegen HAE erhalten als Konkurrent von Jerini's Icatibant).
Nun hat Cubist einen weiteren Kandidaten einlizensiert... der am weitesten fortgeschrittene RNAi-Kandidat von Alnylam, ALN-RSV01 in PII...
http://www.tmcnet.com/usubmit/2009/01/09/3902909.htm
[January 09, 2009]
Cubist, Alnylam Partner in $102.5M RNAi Drugs Deal
(BioWorld Today Via Acquire Media NewsEdge) Massachusetts firms Cubist Pharmaceuticals Inc. and Alnylam Pharmaceuticals Inc. have formed a 50-50 North American partnership to develop and commercialize Alnylam's respiratory syncytial virus (RSV)-specific RNAi therapeutics.
But Wall Street apparently was not so keen on the deal, sending both firms' shares down slightly Friday. Shares of Lexington, Mass.-based Cubist (NASDAQ:CBST) closed at $24.56, a loss of 63 cents, while shares of Cambridge, Mass.-based Alnylam (NASDAQ:ALNY) fell 88 cents, to close at $23.12.
Under the agreement, Alnylam will receive $20 million up front and could bank an additional $82.5 million based on certain development and sales milestones.
The company also could gain double-digit royalties on net sales outside of North America and Asia.
The partnership gives Cubist sole commercialization rights of the ALN-RSV program worldwide outside of Asia, where the rights are held by Japanese firm Kyowa Hakko Kirin Co. Ltd. under a $93 million deal signed with Alnylam last June. (See BioWorld Today, June 20, 2008.)
Cubist and Alnylam will share equally development responsibilities and profits in North America. After achieving certain development milestones, Alnylam could opt to convert the North American co-development and profit share to a royalty-bearing license with development and sales milestones.
The RSV-specific RNAi therapeutic program includes Alnylam's ALN-RSV01, which currently is being studied in a double-blind, randomized, placebo-controlled Phase II trial to assess the drug's safety and tolerability in adult lung transplant patients naturally infected with RSV, said Jason Rhodes, Alnylam's vice president of business development.
The trial also will evaluate the antiviral activity of ALN-RSV01 in patients with a naturally acquired RSV lower respiratory tract infection.
The firm expects to have data from the trial by the second half of this year, Rhodes told BioWorld Today. Following the completion of that trial, Alnylam expects to begin a trial of ALN-RSV01 in pediatric patients with RSV, he noted.
Results reported by Alnylam of a double-blind, randomized, placebo-controlled Phase II trial, known as GEMINI, showed that intranasally administered ALN-RSV01 demonstrated statistically significant antiviral efficacy with about a 40 percent relative reduction in RSV infection rate and a 95 percent increase in the number of infection-free patients compared with placebo.
The company's RSV-specific RNAi program also includes other potent and specific second-generation RNAi-based RSV inhibitors in preclinical development.
RSV is a common, highly contagious virus that causes infections in both the upper- and lower respiratory tract in children and adults, Rhodes explained.
There are about 18 million infections reported annually in most developed countries, resulting in about 1 million hospitalizations, he said.
In the U.S. alone, there are about 125,000 pediatric and 175,000 adult hospitalizations annually attributed to RSV, Rhodes said.
While RSV infection typically results in cold-like symptoms, it can lead to more serious respiratory illnesses such as croup, pneumonia and bronchiolitis, and in some cases, be deadly.
RSV infection in infants also has been linked to the development of childhood asthma.
There currently are no treatments that are broadly used to successfully treat RSV, Rhodes noted.
Pediatric patients especially, said Steven Gilman, Cubists' chief scientific officer, are "in dire need" of an effective treatment for the infection.
"RSV is an important disease for children, and there is a very large unmet medical need," he told BioWorld Today.
Given the RSV program's potential to be used in a fairly broad outpatient population, Rhodes said, it was always Alnylam's strategy to partner it.
"At this stage, we don't plan to build a broad outpatient commercial organization at this point in the company's development," he said.
Alnylam had a "broad range" of discussions with several pharmaceutical companies ranging from large multinational pharma companies to local biopharmaceutical firms.
Rhodes said his firm chose Cubist to partner on the RSV RNAi program because of its expertise in acute care and infectious disease products.
He noted Cubist's success in commercializing Cubicin (daptomycin for injection), the first antibiotic in a new class of anti-infectives called lipopeptides.
Cubist also has an agreement with London-based AstraZeneca plc to handle U.S. hospital sales for its intravenous antibiotic Merrem (meropenem for injection).
Cubist's product pipeline also includes ecallantide, a recombinant human protein in Phase II clinical trials for the prevention of blood loss during cardiothoracic surgery, and two development programs for drugs to treat Gram-negative infections and Clostridium difficile-associated diarrhea.
In addition, Rhodes said, Cubist is located just "15 minutes west" of Alnylam.
"So if you are going to have a partner where they are in a true 50-50 deal where you have to be working together to co-develop the product, it's obviously helpful to have them be local," he said.
Beyond the location, Rhodes said, the firms have a "culture similarity" that makes them "very compatible."
"Cubist is a superb partner," he added.
The deal "sits well" with Cubist's strategy to build its pipeline, Gilman said.
"We are very excited about the progress of RNAi therapeutics. This is a new area of science and new technology. The science together with the medical need for RSV will be a very dramatic, potentially exciting and rewarding program for us," he noted.
Analyst Thomas Russo, of R.W. Baird & Co., called Alnylam's partnership with Cubist "another disciplined deal" for the RSV RNAi program.
"We like it," he said.
But Piper Jaffray analyst Edward A. Tenthoff called the deal's terms "disappointing."
"In comparison, Kyowa paid $15 million up front plus $78 million in potential milestones last June for Asian rights, indicating that the value of the RSV program has declined," he said in a research note.
**************
http://seekingalpha.com/article/114245-cubist-and-alnylam-an…
Cubist and Alnylam, An Odd Partnership
by: Eben Tessari January 11, 2009 | about stocks: ALNY / CBST
Cubist (CBST) and Alnylam (ALNY) have formed a strategic collaboration to develop and commercialize Alnylam’s ALN-RSV program. The RSV-specific RNAi therapeutic program includes ALN-RSV01, which is currently in Phase II clinical development for the treatment of respiratory syncytial virus (RSV) infection in adult lung transplant patients, as well as several other potent and specific second-generation RNAi-based RSV inhibitors in pre-clinical studies.
Deal Terms
————————-
- $20M upfront
- $82.5M in potential sales and milestone payments
- 50/50 Co-development and profit sharing agreement in US
- Cubist has full commercialization rights EX-US and Asia
- Alnylam receives double digit royalties EX-US and Asia
Cubist? Really? It’s not that I don’t have respect for Cubist, I do. It just strikes me as an odd partnership choice. Why couldn’t Alnylam land another one of those megadeals it’s known for; one of those that In Vivo appoints its deal of the year? Might it be because all of the big pharma players said no? The GEMINI study (small phase IIa with ALN-RSV01) published about a year ago was supposedly the proof that RNAi could work in humans. Well, if so, where is the obscene upfront?
The trial in question was small and mostly artificial. The patients were infected and treated with RSV in their nose… not their lungs like it would be in actual patients. When Alnylam can get the drug to the lower lung and have it reduce viral load and improved clinical symptoms, I’ll be impressed. In addition, to be approved this drug must show some benefit regarding a reduction of days in the hospital or reduction in symptoms, two topics GEMINI didn’t even begin to address.
I could be wrong here, but my guess is that these issues are why Alnylam partnered this drug with Cubist and not with big pharma. Overall, the deal is probably fairly valued, though not by recent Alnylam standards; ALNY is trading down 6% on the day as I write this so it seems that the market was a bit disappointed.
The street better get used to it; I’ve said it before and I’ll say it again: I don’t believe in RNAi as a human therapy for a wide range of diseases (if any). Now, that doesn’t mean I don’t want to see it succeed or want the companies to fizzle out and die; cutting edge research is essential to both medicine and the economy… just don’t ask me to invest in Alnylam.
*************
http://triangle.bizjournals.com/triangle/othercities/boston/…
Monday, January 5, 2009, 3:52pm EST | Modified: Wednesday, January 7, 2009, 1:32pm
Cubist receives $90M credit
Cubist Pharmaceuticals, the Lexington, Mass.-based biotechnology company, has opened a $90 million revolving line of credit with the Royal Bank of Scotland. The money is to be used for general corporate expenses, according to a document filed with the Securities and Exchange Commission.
Cubist (Nasdaq: CBST) has also filed two Investigational New Drug applications with the Food and Drug Administration, indicating its plans to begin clinical trials on the drug targets. The company has one drug, an intravenous antibiotic called Cubicin, already on the market. The company focuses on research, development and commercialization of acute care pharmaceutical products.
The company’s 2007 revenue was $294.6 million. Cubist’s shares were trading at $25.08 in late Monday afternoon trading. The previous close was $25.05. The company’s stock has traded between $16.25 and $28.74 over the past year.
***********
http://www.canadianbusiness.com/markets/market_news/article.…
From The Associated Press, December 8, 2008 - 3:40 PM
Cubist slides following second downgrade in 2 days; analyst cites Cubicin patent risk
NEW YORK (AP) - Shares of Cubist Pharmaceuticals Inc. sank Monday as concerns swirled about the patent protection for its antibiotic Cubicin, which provides almost all of the company's revenue.
Leerink Swann analyst Howard Liang lowered his rating to "Underperform" from "Market Perform" because of his view that investors are overlooking the risk that a generic drug maker will challenge the patents protecting Cubicin. He said Cubist shares might be worth $10 if the patents were successfully challenged.
Shares of the Lexington, Mass., company fell $2.34, or 8.7 percent, to $24.64 in afternoon trading.
Cubicin is vulnerable to generic competition because the Food and Drug Administration approved it more than five years ago, in September 2003. The patents on the drug are set to expire in 2016 and 2019, but generic drug companies could challenge those patents earlier by saying they are not valid or don't apply to their version of the drug.
If such a challenge were successful, Liang estimated Cubist shares would be worth $10.
Cubist said it has not been notified of any patent challenges for Cubicin.
With competing stocks slumping and Cubist shares trading at their highest levels in almost seven years, he said, investors are no longer accounting for that risk, which is particularly important to Cubist because Cubicin is its only marketed drug.
Cubist reported $112.4 million in revenue in the third quarter. Sales of Cubicin accounted for $110.6 million of that total.
The analyst described Cubicin as the most "successful intravenous antibiotic U.S. launch in history," and said demand for Cubicin to treat antibiotic-resistant infections has grown significantly. But Cubist's other drugs are in preclinical testing, and he said there is little excitement about its blood loss drug DX-88.
Liang was the second analyst in two days to cut his rating on the stock, following an Oppenheimer downgrade Friday. Kevin DeGeeter cited the weak patent protection for Cubicin as one of the reasons for his downgrade.
mfg ipollit
Zu Rigel und R788, dem oralen Syk Kinase Hemmer gegen RA in PII... hier ist die Frage, wie am Ende das Nebenwirkungsprofil aussieht. Jedenfalls scheint das Management in den nächsten Monaten einen Partnerschafts-Deal für R788 erreichen zu wollen.
http://www.hammerstockblog.com/biotech-portfolio-updates-rig…
Assuming this trend continues in 2009, it is crucial to identify small and medium companies with candidates whose activity has already been proven in clinical trials. One of the most interesting companies that fall into this category is Rigel Pharmaceutical (RIGL). The company is currently developing a validated drug with blockbuster potential, and is expected to announce major collaboration deal during the first quarter.
Despite the imminent nature of the deal, investors should be aware of the risk factors that naturally come along with negotiations of this type. In addition, there are specific issues with Rigel that might affect the exact terms of the deals, let alone the ability to finalize it. Nevertheless, when all aspects are taken into account, it seems that at current price levels, Rigel represents an attractive investment opportunity.
In contrast to other companies that try to be as vague as possible about the timing of future deals, Rigel is going out of its way to reassure investors that it will have a deal in place by the end of the first quarter. Rigel’s lead drug, R788, is currently being evaluated in two comparative trials in patients with rheumatoid arthritis (RA), a $14 billion indication. During most of 2008, R788 was considered to be one the most promising drugs in the biotech industry, but an update at last year’s ACR meeting raised doubts regarding the safety profile of R788, as reviewed in my recent article on Rigel. According to the company, the safety data from the ACR meeting did not affect its negotiation leverage, as the potential partners had access to the data before it was published, so nothing came as a surprise to them. Trying to be as explicit as possible, Rigel managers state that none of the companies with whom it was in advanced discussions dropped out of the race as a result of the safety issues.
The safety issue most investors are worried about is the increase in blood pressure R788 seems to induce. With current regulatory climate, and considering that RA patients are more prone to develop high blood pressure, this safety signal will probably be closely watched in future trials. Nevertheless, for the same reason, future trials will probably demonstrate that there is no material risk because in the vast majority of cases, the slight blood pressure increase will be manageable by hypertension drugs. Even in cases where the elevation in blood pressure is not manageable, the patient can be taken off the drug and the blood pressure returns to its normal levels. Evidently, if R788 gets approved, it is plausible that some patients will be more sensitive to this side effect and show a too steep of an increase in blood pressure. These patients will have to be taken of the drug but they still represent a small portion of the overall market.
In order to become a successful drug, R788 does not have to outperform the currently approved drugs for RA, instead, it should just offer patients a more convenient treatment with comparable efficacy. The mainstay treatment in RA is TNF-inhibitors, which are injectable biologics. As an oral agent, R788 could be preferred by patients who will have to pick between a monthly injection and a twice-a-day pill. Having an oral drug in the market will certainly help Rigel and its partner to squeeze R788 before the injectable agents, and become a standard first line treatment. This is in contrast to recently approved agents for RA such as Orencia and Rituxan, which are usually given to patients who do not derive sufficient benefit from TNF inhibitors.
According to Rigel, the deal will include a profit sharing arrangement in the U.S, royalties on sales outside of the U.S and a generous upfront payment. Therefore, Rigel will have to fund some of the development costs as well as establish a dedicated sales force in the U.S in return to the co-promotion rights. Building a sales infrastructure in some parts of the U.S typically requires substantial investment, but from Rigel’s perspective, this move makes a lot of sense given the unique nature of the RA market. In terms of incidence, RA occurs in a relatively small number of patients every year, however, because RA is an incurable chronic disease, patients receive treatment for years and decades. Therefore, this multi-billion market can be served by a relatively small sales force, one that can be built by a small company like Rigel.
Judging by the depressed share price, the market is skeptic with respect to the amount of money Rigel can get for R788. Ironically, the company’s current market cap is close to its market cap during 2007, before R788 demonstrated such a potent activity in RA. In my opinion, there are several factors including the impressive efficacy in a randomized trial, the blockbuster potential of R788, and the fact that it is an oral agent, which turn it into a very attractive drug for large pharmas, especially those who already have a RA franchise.
mfg ipollit
http://www.hammerstockblog.com/biotech-portfolio-updates-rig…
Assuming this trend continues in 2009, it is crucial to identify small and medium companies with candidates whose activity has already been proven in clinical trials. One of the most interesting companies that fall into this category is Rigel Pharmaceutical (RIGL). The company is currently developing a validated drug with blockbuster potential, and is expected to announce major collaboration deal during the first quarter.
Despite the imminent nature of the deal, investors should be aware of the risk factors that naturally come along with negotiations of this type. In addition, there are specific issues with Rigel that might affect the exact terms of the deals, let alone the ability to finalize it. Nevertheless, when all aspects are taken into account, it seems that at current price levels, Rigel represents an attractive investment opportunity.
In contrast to other companies that try to be as vague as possible about the timing of future deals, Rigel is going out of its way to reassure investors that it will have a deal in place by the end of the first quarter. Rigel’s lead drug, R788, is currently being evaluated in two comparative trials in patients with rheumatoid arthritis (RA), a $14 billion indication. During most of 2008, R788 was considered to be one the most promising drugs in the biotech industry, but an update at last year’s ACR meeting raised doubts regarding the safety profile of R788, as reviewed in my recent article on Rigel. According to the company, the safety data from the ACR meeting did not affect its negotiation leverage, as the potential partners had access to the data before it was published, so nothing came as a surprise to them. Trying to be as explicit as possible, Rigel managers state that none of the companies with whom it was in advanced discussions dropped out of the race as a result of the safety issues.
The safety issue most investors are worried about is the increase in blood pressure R788 seems to induce. With current regulatory climate, and considering that RA patients are more prone to develop high blood pressure, this safety signal will probably be closely watched in future trials. Nevertheless, for the same reason, future trials will probably demonstrate that there is no material risk because in the vast majority of cases, the slight blood pressure increase will be manageable by hypertension drugs. Even in cases where the elevation in blood pressure is not manageable, the patient can be taken off the drug and the blood pressure returns to its normal levels. Evidently, if R788 gets approved, it is plausible that some patients will be more sensitive to this side effect and show a too steep of an increase in blood pressure. These patients will have to be taken of the drug but they still represent a small portion of the overall market.
In order to become a successful drug, R788 does not have to outperform the currently approved drugs for RA, instead, it should just offer patients a more convenient treatment with comparable efficacy. The mainstay treatment in RA is TNF-inhibitors, which are injectable biologics. As an oral agent, R788 could be preferred by patients who will have to pick between a monthly injection and a twice-a-day pill. Having an oral drug in the market will certainly help Rigel and its partner to squeeze R788 before the injectable agents, and become a standard first line treatment. This is in contrast to recently approved agents for RA such as Orencia and Rituxan, which are usually given to patients who do not derive sufficient benefit from TNF inhibitors.
According to Rigel, the deal will include a profit sharing arrangement in the U.S, royalties on sales outside of the U.S and a generous upfront payment. Therefore, Rigel will have to fund some of the development costs as well as establish a dedicated sales force in the U.S in return to the co-promotion rights. Building a sales infrastructure in some parts of the U.S typically requires substantial investment, but from Rigel’s perspective, this move makes a lot of sense given the unique nature of the RA market. In terms of incidence, RA occurs in a relatively small number of patients every year, however, because RA is an incurable chronic disease, patients receive treatment for years and decades. Therefore, this multi-billion market can be served by a relatively small sales force, one that can be built by a small company like Rigel.
Judging by the depressed share price, the market is skeptic with respect to the amount of money Rigel can get for R788. Ironically, the company’s current market cap is close to its market cap during 2007, before R788 demonstrated such a potent activity in RA. In my opinion, there are several factors including the impressive efficacy in a randomized trial, the blockbuster potential of R788, and the fact that it is an oral agent, which turn it into a very attractive drug for large pharmas, especially those who already have a RA franchise.
mfg ipollit
Viropharma...
http://www.rttnews.com/ArticleView.aspx?Id=811877&SMap=1
ViroPharma - Nothing To Sneeze At
12/29/2008 5:59 AM ET
(RTTNews) - Shares of biotech company ViroPharma Inc. (VPHM: News ) appear to be bucking the market downturn, and have gained nearly 55% over the past year. In contrast, the Amex Biotechnology Index (^BTK), the benchmark index for the biotechnology sector, has tumbled 20% during the same time period.
ViroPharma's stock traded as high as $111.62 on March 6, 2000. The disappointing initial trial results of the company's investigational drug Picovir (pleconaril) against viral meningitis and cold, released in April 2000, sent the stock into a tailspin. Since then, it has been a rollercoaster ride for the stock.
In the last twelve months, ViroPharma stock has traded in the range of $7.94 - $15.16. As on December 26, the company had a market cap of $1.02 billion based on the stock's closing price of $13.
Company Overview
ViroPharma, based in the State of Delaware was founded in 1994 by Claude Nash and Mark McKinlay. The company focuses its drug development activities on viral diseases including cytomegalovirus and hepatitis C.
ViroPharma went public on November 19, 1996 offering 2.25 million shares at $7 each. Up till now, the company has been deriving all its sales from its only marketed drug -- Vancocin, which is used to treat deadly bacterial infections. The recent acquisition of Lev Pharmaceuticals Inc. offers an opportunity for ViroPharma to diversify its sales base. With the FDA approving Lev Pharma's Cinryze, a drug to treat hereditary angioedema as recently as October, ViroPharma will no longer be a one-trick pony
Vancocin - A Financial Lifesaver
ViroPharma, which was counting on its experimental common-cold drug Picovir as a source of revenue, met with disappointment following the FDA's rejection of the drug over safety concerns in June 2002.
In November 2004, ViroPharma acquired the American rights to antibiotic Vancocin from Eli Lilly & Co. (LLY), which had developed the drug, for $116 million. Vancocin is the oral capsule formulation of antibiotic Vancomycin hydrochloride. (Vancomycin is available as a powder for injection and as capsules or solution for oral use).
ViroPharma began marketing its flagship product, Vancocin in 2004. Eli Lilly sells the drug outside of the United States. ViroPharma also pays 35% of the net sales of Vancocin to Eli Lilly.
As on date, Vancocin is the only FDA-approved product to treat severe Clostridium difficile infection, also referred to as CDI. Clostridium difficile infection is one of the most common and devastating hospital-acquired infections. The bacterial infection occurs when patients are treated with antibiotics for other diseases. A prolonged treatment with antibiotics has a negative impact on beneficial bacteria that live in gastrointestinal tract, making the patients prone to bacterial infection. Closteridium difficile can cause diarrhea, ranging from a mild disturbance to a very severe illness with ulceration and bleeding from the colon. In severe conditions, the bacterium can cause perforation of the intestine leading to peritonitis, which can be fatal.
The Clostridium difficile infection is one of the most serious problems facing the U.S. healthcare system. The most commonly used drug to treat Clostridium difficile infection is the generic antibiotic Metronidazole. However, if the infection is severe, Vancocin is the only approved treatment.
Since its commercialization, sales of Vancocin have had an impressive growth. The patent covering Vancocin expired long back in 1996.
So .....Why doesn't the drug have an approved generic yet?
Vancocin works by targeting the Clostridium difficile bacterium in the gastrointestinal tract. Because of the complexities in the way the drug works, in order for a generic copy of Vancocin to be approved by the FDA, it has to prove itself to be truly bioequivalent to Vancocin in comparative clinical trials.
Usually generic drugs do not have to go through clinical trials and they are approved based on the bioequivalence data from the laboratory. No generic drug company has come forward to conduct clinical trials for generic Vancocin to establish the bioequivalence because of the huge expenses involved in conducting clinical trials.
According to experts, "an ineffective generic version of Vancocin will prove fatal because patients treated with ineffective generic Vancocin will have no second chances if it fails".
FDA's Changing Stance
In March 2006, the FDA's Office of Generic Drugs, or OGD revised the standard for the approval of generic copies of Vancocin. The OGD claimed that Vancocin is eligible for a BCS-based (Biopharmaceutics Classification System) biowaiver and that a clinical trial for generic Vancomycin is not necessary if it exhibits rapid dissolution in 'in vitro' test (lab setting). Rapid dissolution in 'in vitro' testing is one of the criteria for granting BCS-based biowaiver.
Since then, speculation has been rife that the FDA might approve a generic Vancocin based on the in vitro dissolution testing. On March 17, 2006 ViroPharma filed its citizen petition, asking the FDA to stay approval of generic Vancocin based solely on in vitro bioequivalence testing.
The BCS, a tool for classifying drugs based upon their aqueous solubility and intestinal permeability, is used by regulatory agencies to grant waivers from in vivo bioavailability and/or bioequivalence studies.
ViroPharma came to know of the new bioequivalence standard for generic Vancocin through a report issued by a trading firm Infinium Capital. Though ViroPharma eventually received verbal confirmation from the OGD about the change in standards, the FDA refused to supply ViroPharma with copies of FDA's correspondence with Infinium or anyone else related to the revised standards.
In its SEC filing dated May 31, 2006, ViroPharma stated that a press agency conducted an email interview with the FDA, in which the FDA sent an email to the press agency on March 17, 2006 stating that "[OGD] has recently revised the bioequivalence recommendations for oral Vancomycin from a clinical trial with bioequivalence endpoints to an in vitro method involving dissolution testing"
After ViroPharma's request to the FDA for records relating to the regulatory agency's decision on the bioequivalence standards for generic copies of Vancocin went unanswered, the company appealed to the Department of Health and Human Services, or HHS in November 2007. This time too, the HHS acknowledged receipt of ViroPharma's administrative appeal, but never responded.
As recently as December 15, 2008 the FDA revealed its draft guidance for establishing bioequivalence to Vancocin. Under the proposed draft guidance, apart from dissolution testing in a lab setting, a generic version of Vancocin should significantly contain the same inactive ingredients (Qualitative sameness - Q1) in substantially the same quantities (Quantitative sameness - Q2 ) as branded Vancocin.
According to the FDA's new guidance, if the generic versions do not have 'qualitative sameness' and 'quantitative sameness' as that of Vancocin, they need to prove their 'bioequivalence to Vancocin' in comparative clinical trials. The proposed draft guidance is open for public comment.
Meanwhile, on December 18, ViroPharma filed a suit against the Department of Health and Human Services and the FDA under the Freedom of Information Act, seeking administrative records related to the FDA's decision in March 2006 revised bioequivalence standard for generic Vancocin. The company has also requested the FDA for the administrative record relating to the regulatory agency's Vancocin bioequivalence guidance of December 15, 2008.
Thomas Doyle, ViroPharma's vice president, strategic initiatives is of the view that the FDA's Vancocin bioequivalence guidance of December 15, 2008, represents a deviation from the proposed OGD's (Office of Generic Drugs) March 2006 guidance.
.....How? All along, the OGD has asserted that Vancocin capsules are highly soluble and rapidly dissolving, and hence eligible for a BCS-based biowaiver. However, according to ViroPharma, Vancocin is not a BCS drug. The information released by the FDA as recently as December 15, also confirms that Vancocin is not rapidly dissolving. The confirmation was based on a Vancocin dissolution study completed by the FDA in February 2008.
With the new study not supporting OGD's March 2006 bioequivalence approach, the FDA has now proposed that a generic copy of Vancocin does not need to demonstrate rapid dissolution and that it requires "comparable" dissolution and Q1/Q2 sameness with respect to inactive ingredients.
The draft guidance has a 60 day public comment period to review. So a clear picture about generic competition for Vancocin will emerge once the draft guidance is finalized.
Last year, Vancocin's sales totaled $203.7 million, up 22% over 2006. For the first nine months of 2008, the drug raked in sales of $182.3 million, reflecting an increase of 17% from the comparable year-ago period. For full year of 2008, ViroPharma expects Vancocin sales to range between $235 million and $245 million. Analysts estimate sales of $233.3 million.
Shedding The One-Trick Pony Image
Seeking to diversify its sales base, in July 2008, ViroPharma agreed to acquire Lev Pharmaceuticals for $442.9 million. Under the terms of the agreement, ViroPharma paid $2.25 in cash plus $0.50 in stock for each Lev share. The purchase price represented a premium of 49% over Lev's share price prior to the deal announcement. On achievement of certain regulatory and commercial milestones, Lev shareholders will also receive an additional $1.00 per share. Including the additional payments, the deal has a potential value of $617.5 million.
Lev Pharmaceuticals is a little-known drug company, which was trading on the Over the Counter Bulletin Board Exchange when the deal was announced. Following the announcement of the deal on July 15, ViroPharma stock fell 15% to $10.62 as investors felt the acquisition was pricey and bid down the stock.
However, the acquisition seems to be paying off for ViroPharma. In October, the FDA approved Lev Pharma's Cinryze as a prophylactic (preventive) treatment for hereditary angioedema, or HAE, a rare and potentially life-threatening genetic disease. Lev Pharma had sought FDA approval of Cinryze, both as prophylactic and acute treatments of HAE attack. But the FDA approved Cinryze only as a prophylactic treatment and sought additional data to approve the drug in acute treatment of HAE.
Cinryze is the first product to be approved in the U.S. for HAE, which affects about 6,000 to 10,000 Americans. HAE is characterized by rapid swelling of the hands, feet, limbs, face, intestinal tract or airway.
Being an orphan drug, Cinryze will get seven years of marketing exclusivity. Cowen and Co. analyst Rachel McMinn estimates Cinryze to cost $3,400 per dose, or about $350,000 per year. Susquehanna analysts project annual sales of Cinryze to peak at about $127 million. According to analysts, an HAE drug approved as a prophylactic treatment brings in more revenue per patient, compared to an acute treatment setting.
ViroPharma, which has acquired Lev Pharma, has sought an expanded label indication for Cinryze for acute treatment of HAE. However, in the market for acute treatment of HAE, Cinryze has a number of possible contenders like CSL Behring's Berinert, Dyax Corp's DX-88 and Jerini AG's Firazyr.
While CSL Behring's Berinert is already under review by the FDA, Dyax Corp. completed the submission of Biologics License Application for DX-88 on September 24. DX-88 is expected to be approved by the FDA for acute HAE around mid-2009. The German company Jerini was issued a not approvable letter for Firazyr in April by the FDA. (A not approvable letter lists the deficiencies in the application and explains why the application cannot be approved).
However, for the prevention of HAE, ViroPharma's Cinryze remains the only FDA-approved drug till date.
Product Pipeline
The most promising candidate in ViroPharma's pipeline is Camvia (maribavir), a Phase 3 antiviral compound for the prevention of CMV (cytomegalovirus) disease in stem cell and solid organ transplant patients. ViroPharma expects to file a New Drug Application for Camvia with the FDA next year.
ViroPharma acquired worldwide rights (excluding Japan) to Camvia from GlaxoSmithKline in August 2003 for an up-front cash payment of $3.5 million, and will make additional milestone payments upon regulatory clearance.
The approved drugs for CMV disease, which are currently in the market carry black box warnings and have associated toxicities. Roche's Valganciclovir, the market leader for prevention of CMV disease, is associated with serious adverse effects and the drug's labeling carries a black box warning. In phase III trials, ViroPharma's Camvia has been found to be safe and well tolerated.
Camvia was granted orphan drug status in February 2007. According to ViroPharma, peak global sales of Camvia could be between $400 million and $500 million annually. The patents covering Camvia expire in 2015.
The Drugs That Never Made It To Market
Picovir
After data from clinical trials that evaluated Picovir (pleconaril) against meningitis proved unimpressive, ViroPharma began testing the drug for respiratory infections. But those results also turned out negative. It was only then that ViroPharma started trying Picovir against common cold. The compound used in Picovir was licensed by ViroPharma from Sanofi-Synthelabo, known as Sanofi-Aventis since 2004.
According to ViroPharma, Picovir in clinical trials, shortened the duration of cold to 6.3 days from 7.3 days. The company submitted its new drug application for Picovir in July 2001. However, in June 2002, an FDA panel unanimously rejected the drug, saying the potential risks far outweighed the small beneficial effect.
After Picovir was rejected by the FDA, ViroPharma re-licensed the drug to Schering Plough in 2003. Schering Plough is developing pleconaril in an intranasal formulation for common cold and asthma exacerbations and the drug has completed phase II trials.
HCV-796
HCV-796, a drug co-developed by Wyeth and ViroPharma was in a phase II trial when the companies abandoned the development of the drug in April 2008 due to safety concerns. In August 2007, the companies halted the trial as some patients treated with HCV-796 developed elevated liver enzymes, indicating liver damage.
Financials
The company has been profitable since 2005. Prior to the 2004 acquisition of Vancocin, the company incurred losses.
Net income for the third-quarter ended September 30, 2008 increased to $27.07 million or $0.33 per share from $21.28 million or $0.26 per share in the year-ago quarter. Quarterly revenue climbed to $65.9 million, up from $50.9 million in the comparable quarter a year before. Thus far, the company has posted 15 consecutive quarters of positive cash flow and profitability.
Free cash flow at the end of the recent third quarter was $75.53 million. Net debt at the end of the quarter was $250 million.
Upward Revision Of Estimates
Analysts are bullish on the company's earnings growth prospects and over the last 90 days, they have revised their estimates for 2008 by 10 cents to $0.99 per share.
Stock Performance
After trading in a range for almost a year, the stock broke out of the range and gained almost 50% in middle of 2008, and has managed to remain above its 200-day MA. The stock has some resistance around the $15 level.
Closing Thoughts
The threat of generic competition has continued to remain an overhang on ViroPharma, ever since the Office of Generic Drugs revealed the revised bioequivalence standard for generic Vancocin in 2006. The FDA's recent proposed draft guidance for establishing bioequivalence to Vancocin only strengthens the generic risk.
In addition to generic competition, Vancocin also faces a threat from Optimer Pharmaceuticals' (OPTR) OPT-80, which is currently under late-stage testing. In a phase III study, OPT-80 was found to have higher cure and lower relapse rates than Vancocin in the treatment of Closteridium difficile infection.
But that said, unlike in the past, ViroPharma has new sources of revenue to offset decline in Vancocin sales. The long-term growth potential of the company now rests on the success of Cinryze, which was recently approved by the FDA, and Camvia, which is expected to reach the market around 2010 or early 2011.
by RTT Staff Writer[/i]
mfg ipollit
http://www.rttnews.com/ArticleView.aspx?Id=811877&SMap=1
ViroPharma - Nothing To Sneeze At
12/29/2008 5:59 AM ET
(RTTNews) - Shares of biotech company ViroPharma Inc. (VPHM: News ) appear to be bucking the market downturn, and have gained nearly 55% over the past year. In contrast, the Amex Biotechnology Index (^BTK), the benchmark index for the biotechnology sector, has tumbled 20% during the same time period.
ViroPharma's stock traded as high as $111.62 on March 6, 2000. The disappointing initial trial results of the company's investigational drug Picovir (pleconaril) against viral meningitis and cold, released in April 2000, sent the stock into a tailspin. Since then, it has been a rollercoaster ride for the stock.
In the last twelve months, ViroPharma stock has traded in the range of $7.94 - $15.16. As on December 26, the company had a market cap of $1.02 billion based on the stock's closing price of $13.
Company Overview
ViroPharma, based in the State of Delaware was founded in 1994 by Claude Nash and Mark McKinlay. The company focuses its drug development activities on viral diseases including cytomegalovirus and hepatitis C.
ViroPharma went public on November 19, 1996 offering 2.25 million shares at $7 each. Up till now, the company has been deriving all its sales from its only marketed drug -- Vancocin, which is used to treat deadly bacterial infections. The recent acquisition of Lev Pharmaceuticals Inc. offers an opportunity for ViroPharma to diversify its sales base. With the FDA approving Lev Pharma's Cinryze, a drug to treat hereditary angioedema as recently as October, ViroPharma will no longer be a one-trick pony
Vancocin - A Financial Lifesaver
ViroPharma, which was counting on its experimental common-cold drug Picovir as a source of revenue, met with disappointment following the FDA's rejection of the drug over safety concerns in June 2002.
In November 2004, ViroPharma acquired the American rights to antibiotic Vancocin from Eli Lilly & Co. (LLY), which had developed the drug, for $116 million. Vancocin is the oral capsule formulation of antibiotic Vancomycin hydrochloride. (Vancomycin is available as a powder for injection and as capsules or solution for oral use).
ViroPharma began marketing its flagship product, Vancocin in 2004. Eli Lilly sells the drug outside of the United States. ViroPharma also pays 35% of the net sales of Vancocin to Eli Lilly.
As on date, Vancocin is the only FDA-approved product to treat severe Clostridium difficile infection, also referred to as CDI. Clostridium difficile infection is one of the most common and devastating hospital-acquired infections. The bacterial infection occurs when patients are treated with antibiotics for other diseases. A prolonged treatment with antibiotics has a negative impact on beneficial bacteria that live in gastrointestinal tract, making the patients prone to bacterial infection. Closteridium difficile can cause diarrhea, ranging from a mild disturbance to a very severe illness with ulceration and bleeding from the colon. In severe conditions, the bacterium can cause perforation of the intestine leading to peritonitis, which can be fatal.
The Clostridium difficile infection is one of the most serious problems facing the U.S. healthcare system. The most commonly used drug to treat Clostridium difficile infection is the generic antibiotic Metronidazole. However, if the infection is severe, Vancocin is the only approved treatment.
Since its commercialization, sales of Vancocin have had an impressive growth. The patent covering Vancocin expired long back in 1996.
So .....Why doesn't the drug have an approved generic yet?
Vancocin works by targeting the Clostridium difficile bacterium in the gastrointestinal tract. Because of the complexities in the way the drug works, in order for a generic copy of Vancocin to be approved by the FDA, it has to prove itself to be truly bioequivalent to Vancocin in comparative clinical trials.
Usually generic drugs do not have to go through clinical trials and they are approved based on the bioequivalence data from the laboratory. No generic drug company has come forward to conduct clinical trials for generic Vancocin to establish the bioequivalence because of the huge expenses involved in conducting clinical trials.
According to experts, "an ineffective generic version of Vancocin will prove fatal because patients treated with ineffective generic Vancocin will have no second chances if it fails".
FDA's Changing Stance
In March 2006, the FDA's Office of Generic Drugs, or OGD revised the standard for the approval of generic copies of Vancocin. The OGD claimed that Vancocin is eligible for a BCS-based (Biopharmaceutics Classification System) biowaiver and that a clinical trial for generic Vancomycin is not necessary if it exhibits rapid dissolution in 'in vitro' test (lab setting). Rapid dissolution in 'in vitro' testing is one of the criteria for granting BCS-based biowaiver.
Since then, speculation has been rife that the FDA might approve a generic Vancocin based on the in vitro dissolution testing. On March 17, 2006 ViroPharma filed its citizen petition, asking the FDA to stay approval of generic Vancocin based solely on in vitro bioequivalence testing.
The BCS, a tool for classifying drugs based upon their aqueous solubility and intestinal permeability, is used by regulatory agencies to grant waivers from in vivo bioavailability and/or bioequivalence studies.
ViroPharma came to know of the new bioequivalence standard for generic Vancocin through a report issued by a trading firm Infinium Capital. Though ViroPharma eventually received verbal confirmation from the OGD about the change in standards, the FDA refused to supply ViroPharma with copies of FDA's correspondence with Infinium or anyone else related to the revised standards.
In its SEC filing dated May 31, 2006, ViroPharma stated that a press agency conducted an email interview with the FDA, in which the FDA sent an email to the press agency on March 17, 2006 stating that "[OGD] has recently revised the bioequivalence recommendations for oral Vancomycin from a clinical trial with bioequivalence endpoints to an in vitro method involving dissolution testing"
After ViroPharma's request to the FDA for records relating to the regulatory agency's decision on the bioequivalence standards for generic copies of Vancocin went unanswered, the company appealed to the Department of Health and Human Services, or HHS in November 2007. This time too, the HHS acknowledged receipt of ViroPharma's administrative appeal, but never responded.
As recently as December 15, 2008 the FDA revealed its draft guidance for establishing bioequivalence to Vancocin. Under the proposed draft guidance, apart from dissolution testing in a lab setting, a generic version of Vancocin should significantly contain the same inactive ingredients (Qualitative sameness - Q1) in substantially the same quantities (Quantitative sameness - Q2 ) as branded Vancocin.
According to the FDA's new guidance, if the generic versions do not have 'qualitative sameness' and 'quantitative sameness' as that of Vancocin, they need to prove their 'bioequivalence to Vancocin' in comparative clinical trials. The proposed draft guidance is open for public comment.
Meanwhile, on December 18, ViroPharma filed a suit against the Department of Health and Human Services and the FDA under the Freedom of Information Act, seeking administrative records related to the FDA's decision in March 2006 revised bioequivalence standard for generic Vancocin. The company has also requested the FDA for the administrative record relating to the regulatory agency's Vancocin bioequivalence guidance of December 15, 2008.
Thomas Doyle, ViroPharma's vice president, strategic initiatives is of the view that the FDA's Vancocin bioequivalence guidance of December 15, 2008, represents a deviation from the proposed OGD's (Office of Generic Drugs) March 2006 guidance.
.....How? All along, the OGD has asserted that Vancocin capsules are highly soluble and rapidly dissolving, and hence eligible for a BCS-based biowaiver. However, according to ViroPharma, Vancocin is not a BCS drug. The information released by the FDA as recently as December 15, also confirms that Vancocin is not rapidly dissolving. The confirmation was based on a Vancocin dissolution study completed by the FDA in February 2008.
With the new study not supporting OGD's March 2006 bioequivalence approach, the FDA has now proposed that a generic copy of Vancocin does not need to demonstrate rapid dissolution and that it requires "comparable" dissolution and Q1/Q2 sameness with respect to inactive ingredients.
The draft guidance has a 60 day public comment period to review. So a clear picture about generic competition for Vancocin will emerge once the draft guidance is finalized.
Last year, Vancocin's sales totaled $203.7 million, up 22% over 2006. For the first nine months of 2008, the drug raked in sales of $182.3 million, reflecting an increase of 17% from the comparable year-ago period. For full year of 2008, ViroPharma expects Vancocin sales to range between $235 million and $245 million. Analysts estimate sales of $233.3 million.
Shedding The One-Trick Pony Image
Seeking to diversify its sales base, in July 2008, ViroPharma agreed to acquire Lev Pharmaceuticals for $442.9 million. Under the terms of the agreement, ViroPharma paid $2.25 in cash plus $0.50 in stock for each Lev share. The purchase price represented a premium of 49% over Lev's share price prior to the deal announcement. On achievement of certain regulatory and commercial milestones, Lev shareholders will also receive an additional $1.00 per share. Including the additional payments, the deal has a potential value of $617.5 million.
Lev Pharmaceuticals is a little-known drug company, which was trading on the Over the Counter Bulletin Board Exchange when the deal was announced. Following the announcement of the deal on July 15, ViroPharma stock fell 15% to $10.62 as investors felt the acquisition was pricey and bid down the stock.
However, the acquisition seems to be paying off for ViroPharma. In October, the FDA approved Lev Pharma's Cinryze as a prophylactic (preventive) treatment for hereditary angioedema, or HAE, a rare and potentially life-threatening genetic disease. Lev Pharma had sought FDA approval of Cinryze, both as prophylactic and acute treatments of HAE attack. But the FDA approved Cinryze only as a prophylactic treatment and sought additional data to approve the drug in acute treatment of HAE.
Cinryze is the first product to be approved in the U.S. for HAE, which affects about 6,000 to 10,000 Americans. HAE is characterized by rapid swelling of the hands, feet, limbs, face, intestinal tract or airway.
Being an orphan drug, Cinryze will get seven years of marketing exclusivity. Cowen and Co. analyst Rachel McMinn estimates Cinryze to cost $3,400 per dose, or about $350,000 per year. Susquehanna analysts project annual sales of Cinryze to peak at about $127 million. According to analysts, an HAE drug approved as a prophylactic treatment brings in more revenue per patient, compared to an acute treatment setting.
ViroPharma, which has acquired Lev Pharma, has sought an expanded label indication for Cinryze for acute treatment of HAE. However, in the market for acute treatment of HAE, Cinryze has a number of possible contenders like CSL Behring's Berinert, Dyax Corp's DX-88 and Jerini AG's Firazyr.
While CSL Behring's Berinert is already under review by the FDA, Dyax Corp. completed the submission of Biologics License Application for DX-88 on September 24. DX-88 is expected to be approved by the FDA for acute HAE around mid-2009. The German company Jerini was issued a not approvable letter for Firazyr in April by the FDA. (A not approvable letter lists the deficiencies in the application and explains why the application cannot be approved).
However, for the prevention of HAE, ViroPharma's Cinryze remains the only FDA-approved drug till date.
Product Pipeline
The most promising candidate in ViroPharma's pipeline is Camvia (maribavir), a Phase 3 antiviral compound for the prevention of CMV (cytomegalovirus) disease in stem cell and solid organ transplant patients. ViroPharma expects to file a New Drug Application for Camvia with the FDA next year.
ViroPharma acquired worldwide rights (excluding Japan) to Camvia from GlaxoSmithKline in August 2003 for an up-front cash payment of $3.5 million, and will make additional milestone payments upon regulatory clearance.
The approved drugs for CMV disease, which are currently in the market carry black box warnings and have associated toxicities. Roche's Valganciclovir, the market leader for prevention of CMV disease, is associated with serious adverse effects and the drug's labeling carries a black box warning. In phase III trials, ViroPharma's Camvia has been found to be safe and well tolerated.
Camvia was granted orphan drug status in February 2007. According to ViroPharma, peak global sales of Camvia could be between $400 million and $500 million annually. The patents covering Camvia expire in 2015.
The Drugs That Never Made It To Market
Picovir
After data from clinical trials that evaluated Picovir (pleconaril) against meningitis proved unimpressive, ViroPharma began testing the drug for respiratory infections. But those results also turned out negative. It was only then that ViroPharma started trying Picovir against common cold. The compound used in Picovir was licensed by ViroPharma from Sanofi-Synthelabo, known as Sanofi-Aventis since 2004.
According to ViroPharma, Picovir in clinical trials, shortened the duration of cold to 6.3 days from 7.3 days. The company submitted its new drug application for Picovir in July 2001. However, in June 2002, an FDA panel unanimously rejected the drug, saying the potential risks far outweighed the small beneficial effect.
After Picovir was rejected by the FDA, ViroPharma re-licensed the drug to Schering Plough in 2003. Schering Plough is developing pleconaril in an intranasal formulation for common cold and asthma exacerbations and the drug has completed phase II trials.
HCV-796
HCV-796, a drug co-developed by Wyeth and ViroPharma was in a phase II trial when the companies abandoned the development of the drug in April 2008 due to safety concerns. In August 2007, the companies halted the trial as some patients treated with HCV-796 developed elevated liver enzymes, indicating liver damage.
Financials
The company has been profitable since 2005. Prior to the 2004 acquisition of Vancocin, the company incurred losses.
Net income for the third-quarter ended September 30, 2008 increased to $27.07 million or $0.33 per share from $21.28 million or $0.26 per share in the year-ago quarter. Quarterly revenue climbed to $65.9 million, up from $50.9 million in the comparable quarter a year before. Thus far, the company has posted 15 consecutive quarters of positive cash flow and profitability.
Free cash flow at the end of the recent third quarter was $75.53 million. Net debt at the end of the quarter was $250 million.
Upward Revision Of Estimates
Analysts are bullish on the company's earnings growth prospects and over the last 90 days, they have revised their estimates for 2008 by 10 cents to $0.99 per share.
Stock Performance
After trading in a range for almost a year, the stock broke out of the range and gained almost 50% in middle of 2008, and has managed to remain above its 200-day MA. The stock has some resistance around the $15 level.
Closing Thoughts
The threat of generic competition has continued to remain an overhang on ViroPharma, ever since the Office of Generic Drugs revealed the revised bioequivalence standard for generic Vancocin in 2006. The FDA's recent proposed draft guidance for establishing bioequivalence to Vancocin only strengthens the generic risk.
In addition to generic competition, Vancocin also faces a threat from Optimer Pharmaceuticals' (OPTR) OPT-80, which is currently under late-stage testing. In a phase III study, OPT-80 was found to have higher cure and lower relapse rates than Vancocin in the treatment of Closteridium difficile infection.
But that said, unlike in the past, ViroPharma has new sources of revenue to offset decline in Vancocin sales. The long-term growth potential of the company now rests on the success of Cinryze, which was recently approved by the FDA, and Camvia, which is expected to reach the market around 2010 or early 2011.
by RTT Staff Writer[/i]
mfg ipollit
Wie steht du atm zu dem schwächelnden Patienten Evotec?
Antwort auf Beitrag Nr.: 36.350.858 von ipollit am 11.01.09 15:53:11Meine Favoriten für 2009 sind:US-Kürzel IDMI,ITMN,SVNT,MNKD,IMGN,DVAX
Antwort auf Beitrag Nr.: 36.350.858 von ipollit am 11.01.09 15:53:11Ich würde noch eine Cytotools dazunehmen...
grüße
bikefan
grüße
bikefan
10 Future Blockbusters...
http://www.cel-sci.com/media_7.pdf
Welche Companies aufgrund dieses Berichtes interessant erscheinen, kann dann jeder fuer sich selber entscheiden...
Meiner verbirgt sich hinter dem Link...
Gruss,
Sil
http://www.cel-sci.com/media_7.pdf
Welche Companies aufgrund dieses Berichtes interessant erscheinen, kann dann jeder fuer sich selber entscheiden...
Meiner verbirgt sich hinter dem Link...
Gruss,
Sil
Genmab... wie schon so oft Übernahmespekulationen
Market Report: GSK 'lining up bid for Danish biotech firm'
By Nikhil Kumar
Tuesday, 13 January 2009
The drugs giant GlaxoSmithKline was the focus of market chatter last night, suggesting it was considering an offer for Genmab, the Danish biotechnology firm.
Speculation linking the two has done the rounds before, with analysts often mentioning GSK, which in 2006 signed an agreement to co-develop and commercialise a key Genmab drug, as the most likely predator for the Danish firm.
It was said that GSK may have made an informal approach and was contemplating an offer in the range of 450-500 kroner per share (£54-£61), more than double the level at which Genmab closed last night. Both companies declined to comment.
The chatter follows a recent note from Credit Suisse, in which the broker highlighted GSK as the most likely buyer, saying that Genmab had "all the 'tactical indicators' of M&A ticked".
Samir Devani, analyst at Nomura Code Securities in London, agreed that GSK was the most probable buyer but said: "We would be surprised if an offer is imminent in light of the sale of the co-promotion rights of ofatumumab [which is the subject of the partnership agreement between the two] back to GSK and believe a purchase is now more likely post-launch of the product. Irrespective, we believe at current levels Genmab is attractively valued and have a buy rating on the company."
Commenting on the speculated offer range, Mr Devani said: "It's not untypical to see acquisition premiums in the sector of 100 per cent, and GSK has previously bought shares in Genmab at 454 kroner per share." GSK closed up 16p at 1,305p last night.
mfg ipollit
Market Report: GSK 'lining up bid for Danish biotech firm'
By Nikhil Kumar
Tuesday, 13 January 2009
The drugs giant GlaxoSmithKline was the focus of market chatter last night, suggesting it was considering an offer for Genmab, the Danish biotechnology firm.
Speculation linking the two has done the rounds before, with analysts often mentioning GSK, which in 2006 signed an agreement to co-develop and commercialise a key Genmab drug, as the most likely predator for the Danish firm.
It was said that GSK may have made an informal approach and was contemplating an offer in the range of 450-500 kroner per share (£54-£61), more than double the level at which Genmab closed last night. Both companies declined to comment.
The chatter follows a recent note from Credit Suisse, in which the broker highlighted GSK as the most likely buyer, saying that Genmab had "all the 'tactical indicators' of M&A ticked".
Samir Devani, analyst at Nomura Code Securities in London, agreed that GSK was the most probable buyer but said: "We would be surprised if an offer is imminent in light of the sale of the co-promotion rights of ofatumumab [which is the subject of the partnership agreement between the two] back to GSK and believe a purchase is now more likely post-launch of the product. Irrespective, we believe at current levels Genmab is attractively valued and have a buy rating on the company."
Commenting on the speculated offer range, Mr Devani said: "It's not untypical to see acquisition premiums in the sector of 100 per cent, and GSK has previously bought shares in Genmab at 454 kroner per share." GSK closed up 16p at 1,305p last night.
mfg ipollit
Antwort auf Beitrag Nr.: 36.350.858 von ipollit am 11.01.09 15:53:11Sehr interessanter Thread.
Ich kannte zumindest viele Biotechs nicht und hab sie jetzt auf der Liste...
Ich würde mir auch noch Ligand anschauen. Die haben ein interessantes Chance/Risikoverhältnis.
Die Pipeline:
2 Medikamet am Markt bzw. grade zugelassen (King, Glaxosmithkline)
2 in Phase III (Pfizer, Wyeth)
4 in Phase II (Shering-Plough, Bristolmyers Squibb, Glaxosmthkline)
7 in Phase I (SheringPlough, Celgene)
6 Präklinisch
Ich kannte zumindest viele Biotechs nicht und hab sie jetzt auf der Liste...
Ich würde mir auch noch Ligand anschauen. Die haben ein interessantes Chance/Risikoverhältnis.
Die Pipeline:
2 Medikamet am Markt bzw. grade zugelassen (King, Glaxosmithkline)
2 in Phase III (Pfizer, Wyeth)
4 in Phase II (Shering-Plough, Bristolmyers Squibb, Glaxosmthkline)
7 in Phase I (SheringPlough, Celgene)
6 Präklinisch
Antwort auf Beitrag Nr.: 36.354.108 von Sillak am 12.01.09 11:55:53@Sillak: Wie kommst du denn an das Mag? Hast du das abonniert? Hab mir das mal auf deren Homepage angesehen, is leider sehr teuer, aber sicher interessant.
Grüße
blb
Grüße
blb
Hier mal meine Favoriten für 2009:
Deutschland konservativ: Morphosys
Deutschland spekulativ: Medigene, Wilex, Paion
USA konservativ: Celgene, Cephalon, Genzyme
USA spekulativ: Amag Ph., Sequenom, Spectrum (mehr zum Zocken), Vertex
Daneben: Actelion, Intercell
Teilweise hab ich die Titel selbst im Depot
Grüße
blb
Deutschland konservativ: Morphosys
Deutschland spekulativ: Medigene, Wilex, Paion
USA konservativ: Celgene, Cephalon, Genzyme
USA spekulativ: Amag Ph., Sequenom, Spectrum (mehr zum Zocken), Vertex
Daneben: Actelion, Intercell
Teilweise hab ich die Titel selbst im Depot
Grüße
blb
Antwort auf Beitrag Nr.: 36.375.273 von blb am 14.01.09 19:29:19Ne Myriad Genetics sollte man auch auf der Watchlist haben, neues MehrJahreshoch vergangene Woche. Steigern Gewinn um die 50 Prozent jährlich...

GSK und Genmab haben nun die US-Zulassung von Arzerra gegen CLL bei der FDA beantragt... wenn alles glatt läuft könnte in 6 Monaten das OK der FDA kommen. In Kürze müsste auch der Zulassungsantrag in Europa erfolgen.
GlaxoSmithKline and Genmab Submit Arzerra(TM) (Ofatumumab) Application to FDA for the Treatment of Advanced Stage Blood Cancer
Three New Studies Initiated in Other Oncology Settings
LONDON and COPENHAGEN, Denmark, Jan. 30 /PRNewswire-USNewswire/ -- GlaxoSmithKline (GSK) and Genmab A/S (OMX: GEN) announced today the submission of a Biologics License Application (BLA) to the U.S. Food and Drug Administration (FDA) for Arzerra(TM) (ofatumumab) to treat patients whose chronic lymphocytic leukemia (CLL) is resistant (refractory) to previous therapies. If approved, ofatumumab would be the first anti-CD20 monoclonal antibody available for this patient population.
CLL is the most common form of adult leukemia in the Western world,(1,2) affecting more than 90,000 Americans.(3) Patients with refractory CLL need new therapies since less than 25 percent respond to most current treatments while still having to cope with adverse effects.(4)
"The submission of the BLA for ofatumumab brings us closer to the possibility of providing a new treatment to patients with refractory CLL," said Lisa N. Drakeman, Ph.D., Chief Executive Officer of Genmab. "This is the first BLA ever filed for an antibody produced by Genmab and is a significant achievement in our partnership with GSK."
The submission is based on an analysis that included 138 patients with CLL who showed limited or no response to both fludarabine and alemtuzumab treatment (fludarabine alemtuzumab refractory) and patients who were refractory to fludarabine and considered inappropriate candidates for alemtuzumab due to bulky tumor masses (>5 cm) in their lymph nodes (bulky fludarabine refractory). The primary endpoint of the study was assessment of response. The overall response rate seen in these patient groups treated with single-agent ofatumumab was 58 percent for the fludarabine alemtuzumab refractory group (n=59) and 47 percent for the bulky fludarabine refractory group (n=79).(5,6)
The most common adverse events (AEs) seen with ofatumumab were related to infusion reactions and infections. AEs seen in at least 10 percent of patients included fever, cough, diarrhea, rash, low white blood cell counts, fatigue, pneumonia, anemia, shortness of breath and nausea. In clinical trials to date, infusion reactions that were serious yet manageable were seen in three percent of patients. One case of progressive multifocal leukoencephalopathy (PML), a rare brain infection resulting in death or causing severe disability, and one case of tumor lysis syndrome were reported.(5,6) These data were presented at the 50th Annual Meeting of the American Society of Hematology (ASH) in December 2008.
Potential to Combat CLL in Earlier Stages
The companies also announced today the initiation of an additional Phase III study of ofatumumab in combination with fludarabine and cyclophosphamide (FC) for patients with CLL when initial treatment no longer works (second-line treatment). The open-label study will randomize 352 patients to evaluate progression-free survival (PFS) of ofatumumab in combination with FC therapy versus FC therapy alone for the treatment of relapsed CLL.(7) Enrollment for this study will begin shortly.
"Bolstered by the positive results of ofatumumab for advanced stage CLL, we're now investigating ofatumumab in earlier stages in the treatment continuum," said Paolo Paoletti, M.D., Senior Vice President of Oncology R&D, GSK. "The clues we're seeing from ofatumumab also suggest possible activity in other oncology settings, which we're currently evaluating through several clinical trials."
GSK and Genmab will conduct additional studies of ofatumumab in CLL and non-Hodgkin's lymphoma (NHL) settings. In CLL, a Phase III front-line study is evaluating ofatumumab combined with chlorambucil in patients with previously untreated CLL. In NHL, an ongoing Phase II study will assess ofatumumab in patients with Waldenstrom's Macroglobulinemia - a rare type of slow-growing NHL.(8) Finally, a Phase II study is evaluating ofatumumab plus ICE or DHAP chemotherapy regimen in relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
About ofatumumab
Ofatumumab is an investigational monoclonal antibody that targets a membrane-proximal (close to the cell surface), small loop epitope (a portion of a molecule to which an antibody binds) on the CD20 molecule on B-cells.(9) This epitope is different from the binding sites targeted by other CD20 antibodies currently available.(10) The CD20 molecule is a key target in CLL therapy because it is highly expressed in most B-cell malignancies.(11)
Ofatumumab is being developed to treat chronic lymphocytic leukemia, non-Hodgkin's lymphoma, diffuse large B-cell lymphoma, rheumatoid arthritis, and relapsing-remitting multiple sclerosis under a co-development and commercialization agreement between Genmab and GlaxoSmithKline. It is not yet approved in any country.
The companies intend to submit an application for marketing approval in Europe shortly.
mfg ipollit
GlaxoSmithKline and Genmab Submit Arzerra(TM) (Ofatumumab) Application to FDA for the Treatment of Advanced Stage Blood Cancer
Three New Studies Initiated in Other Oncology Settings
LONDON and COPENHAGEN, Denmark, Jan. 30 /PRNewswire-USNewswire/ -- GlaxoSmithKline (GSK) and Genmab A/S (OMX: GEN) announced today the submission of a Biologics License Application (BLA) to the U.S. Food and Drug Administration (FDA) for Arzerra(TM) (ofatumumab) to treat patients whose chronic lymphocytic leukemia (CLL) is resistant (refractory) to previous therapies. If approved, ofatumumab would be the first anti-CD20 monoclonal antibody available for this patient population.
CLL is the most common form of adult leukemia in the Western world,(1,2) affecting more than 90,000 Americans.(3) Patients with refractory CLL need new therapies since less than 25 percent respond to most current treatments while still having to cope with adverse effects.(4)
"The submission of the BLA for ofatumumab brings us closer to the possibility of providing a new treatment to patients with refractory CLL," said Lisa N. Drakeman, Ph.D., Chief Executive Officer of Genmab. "This is the first BLA ever filed for an antibody produced by Genmab and is a significant achievement in our partnership with GSK."
The submission is based on an analysis that included 138 patients with CLL who showed limited or no response to both fludarabine and alemtuzumab treatment (fludarabine alemtuzumab refractory) and patients who were refractory to fludarabine and considered inappropriate candidates for alemtuzumab due to bulky tumor masses (>5 cm) in their lymph nodes (bulky fludarabine refractory). The primary endpoint of the study was assessment of response. The overall response rate seen in these patient groups treated with single-agent ofatumumab was 58 percent for the fludarabine alemtuzumab refractory group (n=59) and 47 percent for the bulky fludarabine refractory group (n=79).(5,6)
The most common adverse events (AEs) seen with ofatumumab were related to infusion reactions and infections. AEs seen in at least 10 percent of patients included fever, cough, diarrhea, rash, low white blood cell counts, fatigue, pneumonia, anemia, shortness of breath and nausea. In clinical trials to date, infusion reactions that were serious yet manageable were seen in three percent of patients. One case of progressive multifocal leukoencephalopathy (PML), a rare brain infection resulting in death or causing severe disability, and one case of tumor lysis syndrome were reported.(5,6) These data were presented at the 50th Annual Meeting of the American Society of Hematology (ASH) in December 2008.
Potential to Combat CLL in Earlier Stages
The companies also announced today the initiation of an additional Phase III study of ofatumumab in combination with fludarabine and cyclophosphamide (FC) for patients with CLL when initial treatment no longer works (second-line treatment). The open-label study will randomize 352 patients to evaluate progression-free survival (PFS) of ofatumumab in combination with FC therapy versus FC therapy alone for the treatment of relapsed CLL.(7) Enrollment for this study will begin shortly.
"Bolstered by the positive results of ofatumumab for advanced stage CLL, we're now investigating ofatumumab in earlier stages in the treatment continuum," said Paolo Paoletti, M.D., Senior Vice President of Oncology R&D, GSK. "The clues we're seeing from ofatumumab also suggest possible activity in other oncology settings, which we're currently evaluating through several clinical trials."
GSK and Genmab will conduct additional studies of ofatumumab in CLL and non-Hodgkin's lymphoma (NHL) settings. In CLL, a Phase III front-line study is evaluating ofatumumab combined with chlorambucil in patients with previously untreated CLL. In NHL, an ongoing Phase II study will assess ofatumumab in patients with Waldenstrom's Macroglobulinemia - a rare type of slow-growing NHL.(8) Finally, a Phase II study is evaluating ofatumumab plus ICE or DHAP chemotherapy regimen in relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
About ofatumumab
Ofatumumab is an investigational monoclonal antibody that targets a membrane-proximal (close to the cell surface), small loop epitope (a portion of a molecule to which an antibody binds) on the CD20 molecule on B-cells.(9) This epitope is different from the binding sites targeted by other CD20 antibodies currently available.(10) The CD20 molecule is a key target in CLL therapy because it is highly expressed in most B-cell malignancies.(11)
Ofatumumab is being developed to treat chronic lymphocytic leukemia, non-Hodgkin's lymphoma, diffuse large B-cell lymphoma, rheumatoid arthritis, and relapsing-remitting multiple sclerosis under a co-development and commercialization agreement between Genmab and GlaxoSmithKline. It is not yet approved in any country.
The companies intend to submit an application for marketing approval in Europe shortly.
mfg ipollit
ONXX habe ich ein wenig reduziert, RIGL etwas dazugekauft...
20,3% Genmab http://finance.yahoo.com/q?s=GEN.CO
7,9% Onyx http://finance.yahoo.com/q?s=ONXX
7,8% Regeneron http://finance.yahoo.com/q?s=REGN
6,9% Cubist http://finance.yahoo.com/q?s=CBST
5,6% Rigel http://finance.yahoo.com/q?s=RIGL
5,3% Isis http://finance.yahoo.com/q?s=ISIS
5,3% Exelixis http://finance.yahoo.com/q?s=EXEL
5,2% Evotec http://finance.yahoo.com/q?s=EVT.DE
4,9% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
4,8% Medigene http://finance.yahoo.com/q?s=MDG.DE
3,9% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,9% Micromet http://finance.yahoo.com/q?s=MITI
3,3% Arena http://finance.yahoo.com/q?s=ARNA
3,3% Addex http://finance.yahoo.com/q?s=ADXN.SW
3,0% Array http://finance.yahoo.com/q?s=ARRY
2,9% NicOx http://finance.yahoo.com/q?s=COX.PA
2,6% Progenics http://finance.yahoo.com/q?s=PGNX
1,6% Sucampo http://finance.yahoo.com/q?s=SCMP
1,4% Incyte http://finance.yahoo.com/q?s=INCY
mfg ipollit
20,3% Genmab http://finance.yahoo.com/q?s=GEN.CO
7,9% Onyx http://finance.yahoo.com/q?s=ONXX
7,8% Regeneron http://finance.yahoo.com/q?s=REGN
6,9% Cubist http://finance.yahoo.com/q?s=CBST
5,6% Rigel http://finance.yahoo.com/q?s=RIGL
5,3% Isis http://finance.yahoo.com/q?s=ISIS
5,3% Exelixis http://finance.yahoo.com/q?s=EXEL
5,2% Evotec http://finance.yahoo.com/q?s=EVT.DE
4,9% Seattle Genetics http://finance.yahoo.com/q?s=SGEN
4,8% Medigene http://finance.yahoo.com/q?s=MDG.DE
3,9% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,9% Micromet http://finance.yahoo.com/q?s=MITI
3,3% Arena http://finance.yahoo.com/q?s=ARNA
3,3% Addex http://finance.yahoo.com/q?s=ADXN.SW
3,0% Array http://finance.yahoo.com/q?s=ARRY
2,9% NicOx http://finance.yahoo.com/q?s=COX.PA
2,6% Progenics http://finance.yahoo.com/q?s=PGNX
1,6% Sucampo http://finance.yahoo.com/q?s=SCMP
1,4% Incyte http://finance.yahoo.com/q?s=INCY
mfg ipollit
Antwort auf Beitrag Nr.: 36.488.200 von ipollit am 31.01.09 18:21:26ein etwas längerer Report der Jyske Bank zu Genmab's Arzerra (Ofatumumab)...
"Is there a place for Ofatumumab?"
http://www.jyskebank.dk/_Jb/ASP/Apps/redirect.asp?ShadowID=2…
mfg ipollit
"Is there a place for Ofatumumab?"
http://www.jyskebank.dk/_Jb/ASP/Apps/redirect.asp?ShadowID=2…
mfg ipollit
nochmal etwas zu Micromet...
http://www.journalonko.de/aktuellview.php?id=1656
BiTE®-Antikörper: Durch Bispezifität T-Lymphozyten gegen Tumorzellen richten
D. Rüttinger, G. Zugmaier, D. Nagorsen, C. Reinhardt, P. A. Baeuerle, Micromet AG, München.
Im Kampf gegen Tumorzellen besitzen zytotoxische T-Zellen das wirksamste Arsenal an Effektormechanismen aller Immunzellen. Dennoch entstehen Malignome, wachsen und metastasieren häufig sogar trotz einer gegen den Tumor gerichteten T-Zellantwort. Verantwortlich dafür sind so genannte „Tumor- Escape“-Mechanismen, die die Tumorzellen für das Immunsystem unsichtbar machen oder auf die Aktivität von T-Zellen negativen Einfluss nehmen. Durch den Einsatz von bispezifischen Antikörpern gelingt es, solche Escape-Mechanismen zu umgehen und gleichzeitig das Potenzial der zytotoxischen T-Zellen im Kampf gegen den Tumor in ungewöhnlichem Ausmaß auszunutzen. BiTE®-Antikörper repräsentieren eine neue Klasse von bispezifischen Antikörpern, deren außerordentliche Wirksamkeit auf einer umfassenden aber gleichzeitig gut kontrollierten, zyototoxischen T-Zell-Antwort gegen Tumorzellen beruht. Ein erster Vertreter der BiTE®-Antikörper zeigte in Lymphom-Patienten eindrucksvolle klinische Ergebnisse.
Zytotoxische T-Lymphozyten haben sich im Immunsystem auf die rasche Eliminierung körpereigener Zellen spezialisiert, die zum Beispiel mit Viren befallen sind oder aber neuartige tumorassoziierte Antigene präsentieren. Tatsächlich gibt es Beispiele von Patienten, deren fortgeschrittene Tumore sich nach einer T-Zell-basierten Immuntherapie, z.B. mit Anti-CTLA4-Antikörpern oder nach einem adoptiven T-Zell-Transfer, vollständig zurückbildeten [1,2]. Auch bei der experimentellen therapeutischen Vakzinierung mit tumorassoziierten Antigenen gibt es immer wieder Beispiele von eindrucksvollem klinischen Ansprechen einzelner Patienten [3]. Diese Erfolgsbeispiele aus der klinischen Praxis sind jedoch eher selten und häufig korreliert eine messbare T-Zellantwort im Blut nicht mit einem klinischen Ansprechen der Patienten. Eine mögliche Ursache hierfür sind Tumor-Escape-Mechanismen, durch die sich maligne Zellen dem Zugriff des Immunsystems sehr effizient entziehen [4]. Zu diesen Escape-Mechanismen der Tumorzelle gehören vorwiegend Veränderungen in der Antigen-Präsentation, so zum Beispiel der Verlust oder die verminderte Expression von MHC-Klasse-I-Molekülen oder eine veränderte Prozessierung von Tumorantigenen im Proteasom [5]. Gleichzeitig sind Tumorzellen in der Lage, die sie umgebenden T-Zellen in ihrer Aktivität zu lähmen, so zum Beispiel durch die Ausschüttung immunsupprimierender Zytokine wie TGF-b oder IL-10, oder durch die Depletion der für T-Zellen essentiellen Aminosäure Tryptophan mittels des Enzyms IDO.
Die Wirkungsweise herkömmlicher T-Zelltherapien in der Tumortherapie beruht auf der Induktion einer spezifischen T-Zellantwort gegen den Tumor. Diese bedarf in der Regel der initialen Antigenpräsentation durch dendritische Zellen, der folgenden Differenzierung tumorspezifischer T-Zellklone aus naiven T-Zellen, und schließlich der Antigenpräsentation durch die Tumorzellen mittels MHC-Klasse-I-Molekülen, die von den T-Zellen spezifisch erkannt werden müssen. Eine Lyse von Tumorzellen durch T-Zellen kann letztlich nur dann stattfinden, wenn ein Komplex aus spezifischem T-Zell-Rezeptor, Antigen (Peptid) und MHC-Molekül im Kontakt mit der zytotoxischen T-Zelle entsteht. Die hohe Komplexizität der T-Zellreifung und Zielerkennung gibt einer Tumorzelle viele mögliche Angriffspunkte für Escape-Mechanismen. Mit bestimmten bispezifischen Antikörpern lässt sich allerdings diese Komplexität überwinden und im Prinzip alle bereits im Körper schon vorhandenen zytotoxischen T-Zellen gegen den Tumor zum Einsatz bringen.
Bispezifische Antikörper können gleichzeitig T-Zellen durch CD3, eine invariante Untereinheit des T-Zellrezeptors, erkennen sowie Tumorzellen auf eine Art und Weise wie es konventionelle Antikörper tun, nämlich durch Bindung eines Oberflächenantigens, z.B. CD19 oder HER-2/neu. Diese neuartige Verknüpfung zwischen T-Zelle und Tumorzelle kann den Komplex aus T-Zell-Rezeptor, Peptidantigen und MHC-Molekül funktionell ersetzen und macht somit eine zytotoxische T-Zellantwort einzig von der Anwesenheit des bispezifischen Antikörpers abhängig. Bedeutende Escape-Mechanismen der Tumorzellen können somit ignoriert werden.
Bis heute war die klinische Entwicklung von bispezifischen Antikörpern häufig durch Probleme bei der Herstellung und aufgrund ihrer geringen klinischen Aktivität limitiert. Dieser Kurzbericht beschreibt die bislang erfolgreiche Entwicklung einer neuartigen Klasse von bispezifischen Antikörper, den so genannten BiTE®-Antikörpern, die nun in der klinischen Erprobung erste Hinweise auf eine hohe Wirksamkeit bei der Tumorbehandlung gezeigt haben.
Struktur und Wirkungsweise
BiTE®-Antikörper bestehen aus zwei unterschiedlichen Einzelketten-Antikörpern (scFvs), die über ein nicht-immunogenes Peptid miteinander verbunden sind und, wie in
Abbildung 1 gezeigt, von zwei verschiedenen monoklonalen Antikörpern abstammen können. Ein Einzelketten-Antikörper setzt sich aus jeweils einer variablen schweren und einer variablen leichten Kette zusammen. BiTE®-Antikörper sind monomere Polypeptide mit einem Molekulargewicht zwischen 55 und 60 kDa und zeichnen sich durch eine hohe Stabilität aus. Alle BiTE®-Antikörper werden derzeit durch Fermentation mit eukaryotischen Zellkultursystemen hergestellt.

Abb.1: Generierung von BiTE®-Antikörpern.

Abb.2: Konditionale T-Zellaktivierung durch BiTE®-Antikörper.
Während konventionelle monoklonale Antikörper nur Immuneffektorzellen mit Fc-gamma Rezeptoren (z.B. NK-Zellen, Granulozyten und Makrophagen) binden können, gelingt es, mit dem CD3-spezifischen Arm der BiTE®-Antikörper direkt T-Lymphozyten zu binden und auch zu aktivieren. Die erwünschte T-Zell-Aktivierung findet allerdings ausschließlich in Gegenwart einer Tumorzelle statt (Abbildung 2). Eine einarmige Bindung an CD3 per se reicht selbst bei beliebig hoher Dosierung der BiTE®-Antikörper nicht aus, um T-Zellen zu aktivieren. T-Lymphozyten müssen also durch den BiTE®-Antikörper erst mit Tumorzellen zusammengebracht werden, was somit eine unkontrollierte Aktivierung der T-Zellen verhindert. Die dann einsetzende Lyse der Tumorzellen gelingt durch die typischen Effektormechanismen der aktivierten T-Zellen, so durch das Poren-bildende Protein Perforin und durch Granzyme, die programmierten Zelltod auslösen. Lokal freigesetzte Zytokine wie Interferon-gamma unterstützen vermutlich diese Wirkungen durch die Anlockung weiterer Immunzellen. Kürzlich erschienene Übersichtsartikel besprechen im Detail die Wirkungsweise der BiTE®-Antikörper und diskutieren Unterschiede und Übereinstimmungen mit einer regulären T-Zellantwort [6,7].
Anti-Tumor-Aktivität in präklinischen Modellen
In präklinischen Modellen wurden bislang BiTE®-Antikörper mit einer Spezifität gegen CD19, EpCAM (CD326), Rezeptor-Tyrosinkinase ECK (EphA2) und CEA untersucht [8-11]. In diesen Studien gelang es in einigen Fällen etablierte Tumore humanen Ursprungs in immundefizienten Versuchstieren (NOD/SCID Mäuse) zur vollständigen Rückbildung zu bringen. Aufgrund der CD3-Spezifität der BiTE®-Antikörper mussten hierzu humane T-Lymphozyten bzw. periphere mononukleäre Blutzellen aus Spendern ko-appliziert werden. In Kontrollexperimenten mit BiTE®-Antikörpern nicht relevanter Spezifität oder in der Abwesenheit von menschlichen T-Zellen blieb die Tumorrückbildung aus. Durch Verwendung von BiTE®-Antikörpern mit einer Spezifität für murines CD3 konnte auch in immunkompetenten Versuchstieren mit Hilfe der endogenen T-Zellen der Mäuse eine deutliche Hemmung des Tumorwachstums in Mamma- und Lungenkarzinom-Modellen nachgewiesen werden [12,13].
Klinische Erprobung
MT103 (MEDI-538) ist ein CD19/CD3-spezifischer BiTE®-Antikörper, der eine zytolytische Reaktion von T-Zellen gegen CD19-exprimierende Lymphom-Zellen bei Wirkstoffkonzentration im picomolaren Bereich auslöst. MT103 erwies sich in vitro, ex vivo und in Tiermodellen gegenüber menschlichen Lymphomzelllinien als außerordentlich wirksam [8]. Beim Jahreskongress 2007 der US-amerikanischen Society of Hematology (ASH) wurden neue klinische Daten einer derzeit laufenden Phase-I-Studie mit MT103 bei Patienten mit mehrfach Chemotherapie- und Rituximab-vorbehandeltem, rezidiviertem Non-Hodgkin-Lymphom (NHL) vorgestellt [14]. Die meisten Patienten in der Studie waren an Mantelzell- bzw. follikulärem Lymphom erkrankt. Insgesamt beeindruckte MT103 als Monotherapie mit hohen klinischen Ansprechraten bei insgesamt guter Verträglichkeit. MT103 wird außerdem derzeit in einer klinischen Phase-II-Studie bei Patienten mit residualer, akuter lymphoblastischer Leukämie (ALL) erprobt.
Ein neuartiger BiTE®-Antikörper, genannt MT110, der gegen das epitheliale Zelladhäsionsmolekül (EpCAM; CD326) gerichtet ist, wurde für die Therapie solider Tumore entwick-elt [15]. MT110 wird derzeit im Rahmen einer Phase-I-Studie bei Patienten mit EpCAM-positiven, lokal fortgeschrittenen, rezidivierten oder metastasierten Karzinomen auf Verträglichkeit und Antitumoraktivität hin untersucht.
Fazit
BiTE®-Antikörper sind durch ihre Bispezifität gegen CD3 auf T-Lymphozyten und gegen validierte Tumorantigene (wie CD19, EpCAM, CEA) in der Lage, T-Lymphozyten kurzzeitig mit Zielzellen zu verbinden und dadurch eine zytolytische Reaktion der in großer Zahl vorhandenen T-Zellen gegen die Tumorzellen auszulösen. Theoretisch ist diese BiTE®-vermittelte T-Zell-basierte Immunantwort wesentlich weniger anfällig gegenüber Tumor Escape-Mechanismen, wie sie bei anderen immuntherapeutischen Behandlungsansätzen (z.B. therapeutische Impfung oder adoptiver T-Zell-Transfer) limitierend erscheinen. Die vielversprechenden Ergebnisse aus zahlreichen präklinischen Modellen scheinen sich nun im Rahmen erster klinischer Studien zu bestätigen.
mfg ipollit
http://www.journalonko.de/aktuellview.php?id=1656
BiTE®-Antikörper: Durch Bispezifität T-Lymphozyten gegen Tumorzellen richten
D. Rüttinger, G. Zugmaier, D. Nagorsen, C. Reinhardt, P. A. Baeuerle, Micromet AG, München.
Im Kampf gegen Tumorzellen besitzen zytotoxische T-Zellen das wirksamste Arsenal an Effektormechanismen aller Immunzellen. Dennoch entstehen Malignome, wachsen und metastasieren häufig sogar trotz einer gegen den Tumor gerichteten T-Zellantwort. Verantwortlich dafür sind so genannte „Tumor- Escape“-Mechanismen, die die Tumorzellen für das Immunsystem unsichtbar machen oder auf die Aktivität von T-Zellen negativen Einfluss nehmen. Durch den Einsatz von bispezifischen Antikörpern gelingt es, solche Escape-Mechanismen zu umgehen und gleichzeitig das Potenzial der zytotoxischen T-Zellen im Kampf gegen den Tumor in ungewöhnlichem Ausmaß auszunutzen. BiTE®-Antikörper repräsentieren eine neue Klasse von bispezifischen Antikörpern, deren außerordentliche Wirksamkeit auf einer umfassenden aber gleichzeitig gut kontrollierten, zyototoxischen T-Zell-Antwort gegen Tumorzellen beruht. Ein erster Vertreter der BiTE®-Antikörper zeigte in Lymphom-Patienten eindrucksvolle klinische Ergebnisse.
Zytotoxische T-Lymphozyten haben sich im Immunsystem auf die rasche Eliminierung körpereigener Zellen spezialisiert, die zum Beispiel mit Viren befallen sind oder aber neuartige tumorassoziierte Antigene präsentieren. Tatsächlich gibt es Beispiele von Patienten, deren fortgeschrittene Tumore sich nach einer T-Zell-basierten Immuntherapie, z.B. mit Anti-CTLA4-Antikörpern oder nach einem adoptiven T-Zell-Transfer, vollständig zurückbildeten [1,2]. Auch bei der experimentellen therapeutischen Vakzinierung mit tumorassoziierten Antigenen gibt es immer wieder Beispiele von eindrucksvollem klinischen Ansprechen einzelner Patienten [3]. Diese Erfolgsbeispiele aus der klinischen Praxis sind jedoch eher selten und häufig korreliert eine messbare T-Zellantwort im Blut nicht mit einem klinischen Ansprechen der Patienten. Eine mögliche Ursache hierfür sind Tumor-Escape-Mechanismen, durch die sich maligne Zellen dem Zugriff des Immunsystems sehr effizient entziehen [4]. Zu diesen Escape-Mechanismen der Tumorzelle gehören vorwiegend Veränderungen in der Antigen-Präsentation, so zum Beispiel der Verlust oder die verminderte Expression von MHC-Klasse-I-Molekülen oder eine veränderte Prozessierung von Tumorantigenen im Proteasom [5]. Gleichzeitig sind Tumorzellen in der Lage, die sie umgebenden T-Zellen in ihrer Aktivität zu lähmen, so zum Beispiel durch die Ausschüttung immunsupprimierender Zytokine wie TGF-b oder IL-10, oder durch die Depletion der für T-Zellen essentiellen Aminosäure Tryptophan mittels des Enzyms IDO.
Die Wirkungsweise herkömmlicher T-Zelltherapien in der Tumortherapie beruht auf der Induktion einer spezifischen T-Zellantwort gegen den Tumor. Diese bedarf in der Regel der initialen Antigenpräsentation durch dendritische Zellen, der folgenden Differenzierung tumorspezifischer T-Zellklone aus naiven T-Zellen, und schließlich der Antigenpräsentation durch die Tumorzellen mittels MHC-Klasse-I-Molekülen, die von den T-Zellen spezifisch erkannt werden müssen. Eine Lyse von Tumorzellen durch T-Zellen kann letztlich nur dann stattfinden, wenn ein Komplex aus spezifischem T-Zell-Rezeptor, Antigen (Peptid) und MHC-Molekül im Kontakt mit der zytotoxischen T-Zelle entsteht. Die hohe Komplexizität der T-Zellreifung und Zielerkennung gibt einer Tumorzelle viele mögliche Angriffspunkte für Escape-Mechanismen. Mit bestimmten bispezifischen Antikörpern lässt sich allerdings diese Komplexität überwinden und im Prinzip alle bereits im Körper schon vorhandenen zytotoxischen T-Zellen gegen den Tumor zum Einsatz bringen.
Bispezifische Antikörper können gleichzeitig T-Zellen durch CD3, eine invariante Untereinheit des T-Zellrezeptors, erkennen sowie Tumorzellen auf eine Art und Weise wie es konventionelle Antikörper tun, nämlich durch Bindung eines Oberflächenantigens, z.B. CD19 oder HER-2/neu. Diese neuartige Verknüpfung zwischen T-Zelle und Tumorzelle kann den Komplex aus T-Zell-Rezeptor, Peptidantigen und MHC-Molekül funktionell ersetzen und macht somit eine zytotoxische T-Zellantwort einzig von der Anwesenheit des bispezifischen Antikörpers abhängig. Bedeutende Escape-Mechanismen der Tumorzellen können somit ignoriert werden.
Bis heute war die klinische Entwicklung von bispezifischen Antikörpern häufig durch Probleme bei der Herstellung und aufgrund ihrer geringen klinischen Aktivität limitiert. Dieser Kurzbericht beschreibt die bislang erfolgreiche Entwicklung einer neuartigen Klasse von bispezifischen Antikörper, den so genannten BiTE®-Antikörpern, die nun in der klinischen Erprobung erste Hinweise auf eine hohe Wirksamkeit bei der Tumorbehandlung gezeigt haben.
Struktur und Wirkungsweise
BiTE®-Antikörper bestehen aus zwei unterschiedlichen Einzelketten-Antikörpern (scFvs), die über ein nicht-immunogenes Peptid miteinander verbunden sind und, wie in
Abbildung 1 gezeigt, von zwei verschiedenen monoklonalen Antikörpern abstammen können. Ein Einzelketten-Antikörper setzt sich aus jeweils einer variablen schweren und einer variablen leichten Kette zusammen. BiTE®-Antikörper sind monomere Polypeptide mit einem Molekulargewicht zwischen 55 und 60 kDa und zeichnen sich durch eine hohe Stabilität aus. Alle BiTE®-Antikörper werden derzeit durch Fermentation mit eukaryotischen Zellkultursystemen hergestellt.
Abb.1: Generierung von BiTE®-Antikörpern.
Abb.2: Konditionale T-Zellaktivierung durch BiTE®-Antikörper.
Während konventionelle monoklonale Antikörper nur Immuneffektorzellen mit Fc-gamma Rezeptoren (z.B. NK-Zellen, Granulozyten und Makrophagen) binden können, gelingt es, mit dem CD3-spezifischen Arm der BiTE®-Antikörper direkt T-Lymphozyten zu binden und auch zu aktivieren. Die erwünschte T-Zell-Aktivierung findet allerdings ausschließlich in Gegenwart einer Tumorzelle statt (Abbildung 2). Eine einarmige Bindung an CD3 per se reicht selbst bei beliebig hoher Dosierung der BiTE®-Antikörper nicht aus, um T-Zellen zu aktivieren. T-Lymphozyten müssen also durch den BiTE®-Antikörper erst mit Tumorzellen zusammengebracht werden, was somit eine unkontrollierte Aktivierung der T-Zellen verhindert. Die dann einsetzende Lyse der Tumorzellen gelingt durch die typischen Effektormechanismen der aktivierten T-Zellen, so durch das Poren-bildende Protein Perforin und durch Granzyme, die programmierten Zelltod auslösen. Lokal freigesetzte Zytokine wie Interferon-gamma unterstützen vermutlich diese Wirkungen durch die Anlockung weiterer Immunzellen. Kürzlich erschienene Übersichtsartikel besprechen im Detail die Wirkungsweise der BiTE®-Antikörper und diskutieren Unterschiede und Übereinstimmungen mit einer regulären T-Zellantwort [6,7].
Anti-Tumor-Aktivität in präklinischen Modellen
In präklinischen Modellen wurden bislang BiTE®-Antikörper mit einer Spezifität gegen CD19, EpCAM (CD326), Rezeptor-Tyrosinkinase ECK (EphA2) und CEA untersucht [8-11]. In diesen Studien gelang es in einigen Fällen etablierte Tumore humanen Ursprungs in immundefizienten Versuchstieren (NOD/SCID Mäuse) zur vollständigen Rückbildung zu bringen. Aufgrund der CD3-Spezifität der BiTE®-Antikörper mussten hierzu humane T-Lymphozyten bzw. periphere mononukleäre Blutzellen aus Spendern ko-appliziert werden. In Kontrollexperimenten mit BiTE®-Antikörpern nicht relevanter Spezifität oder in der Abwesenheit von menschlichen T-Zellen blieb die Tumorrückbildung aus. Durch Verwendung von BiTE®-Antikörpern mit einer Spezifität für murines CD3 konnte auch in immunkompetenten Versuchstieren mit Hilfe der endogenen T-Zellen der Mäuse eine deutliche Hemmung des Tumorwachstums in Mamma- und Lungenkarzinom-Modellen nachgewiesen werden [12,13].
Klinische Erprobung
MT103 (MEDI-538) ist ein CD19/CD3-spezifischer BiTE®-Antikörper, der eine zytolytische Reaktion von T-Zellen gegen CD19-exprimierende Lymphom-Zellen bei Wirkstoffkonzentration im picomolaren Bereich auslöst. MT103 erwies sich in vitro, ex vivo und in Tiermodellen gegenüber menschlichen Lymphomzelllinien als außerordentlich wirksam [8]. Beim Jahreskongress 2007 der US-amerikanischen Society of Hematology (ASH) wurden neue klinische Daten einer derzeit laufenden Phase-I-Studie mit MT103 bei Patienten mit mehrfach Chemotherapie- und Rituximab-vorbehandeltem, rezidiviertem Non-Hodgkin-Lymphom (NHL) vorgestellt [14]. Die meisten Patienten in der Studie waren an Mantelzell- bzw. follikulärem Lymphom erkrankt. Insgesamt beeindruckte MT103 als Monotherapie mit hohen klinischen Ansprechraten bei insgesamt guter Verträglichkeit. MT103 wird außerdem derzeit in einer klinischen Phase-II-Studie bei Patienten mit residualer, akuter lymphoblastischer Leukämie (ALL) erprobt.
Ein neuartiger BiTE®-Antikörper, genannt MT110, der gegen das epitheliale Zelladhäsionsmolekül (EpCAM; CD326) gerichtet ist, wurde für die Therapie solider Tumore entwick-elt [15]. MT110 wird derzeit im Rahmen einer Phase-I-Studie bei Patienten mit EpCAM-positiven, lokal fortgeschrittenen, rezidivierten oder metastasierten Karzinomen auf Verträglichkeit und Antitumoraktivität hin untersucht.
Fazit
BiTE®-Antikörper sind durch ihre Bispezifität gegen CD3 auf T-Lymphozyten und gegen validierte Tumorantigene (wie CD19, EpCAM, CEA) in der Lage, T-Lymphozyten kurzzeitig mit Zielzellen zu verbinden und dadurch eine zytolytische Reaktion der in großer Zahl vorhandenen T-Zellen gegen die Tumorzellen auszulösen. Theoretisch ist diese BiTE®-vermittelte T-Zell-basierte Immunantwort wesentlich weniger anfällig gegenüber Tumor Escape-Mechanismen, wie sie bei anderen immuntherapeutischen Behandlungsansätzen (z.B. therapeutische Impfung oder adoptiver T-Zell-Transfer) limitierend erscheinen. Die vielversprechenden Ergebnisse aus zahlreichen präklinischen Modellen scheinen sich nun im Rahmen erster klinischer Studien zu bestätigen.
mfg ipollit
Antwort auf Beitrag Nr.: 36.352.406 von altf4 am 12.01.09 00:23:21"schwächelnder Patient Evotec"
In der Tat ist Evotec zurzeit sehr schwach. Dass der CEO sich einfach so davon macht, ist kein gutes Zeichen. Ich würde eher reduzieren als jetzt hinzuzukaufen, solange alles so in der Schwebe ist. Im Bereich CNS habe ich Neurosearch unter Beobachtung... vielleicht werde ich irgendwann tauschen.
mfg ipollit
In der Tat ist Evotec zurzeit sehr schwach. Dass der CEO sich einfach so davon macht, ist kein gutes Zeichen. Ich würde eher reduzieren als jetzt hinzuzukaufen, solange alles so in der Schwebe ist. Im Bereich CNS habe ich Neurosearch unter Beobachtung... vielleicht werde ich irgendwann tauschen.
mfg ipollit
nochmal eine Zusammenfassung der Ofatumumab-Ergebnisse in CLL...
http://www.journalonko.de/newsview.php?id=2997
19.12.2008
Positive Phase-III-Studiendaten für Ofatumumab bei CLL
Der monoklonale CD20-Antikörper Ofatumumab zeigte positive Ergebnisse in einer klinischen Phase-III-Studie zur Behandlung von Patienten mit stark vorbehandelter chronischer lymphatischer Leukämie (CLL), die auf die Standard-Therapie nicht angesprochen haben. Dies zeigt die geplante Zwischenauswertung einer aktuellen Phase-III-Studie zu dem Wirkstoff, die beim 50. Jahreskongress der American Society of Hematology, 6. bis 9. Dezember 2008 in San Francisco, vorgestellt wurde. (1)
Positive Ergebnisse bei Fludarabin-/Alemtuzumab-refraktären Patienten
In der Studie wurden Patienten mit vorbehandelter CLL aufgenommenen, die entweder nur begrenzt oder überhaupt nicht auf die Standardtherapien mit Fludarabin als auch Alemtuzumab angesprochen haben (doppelt refraktär, DR) oder die nicht auf Fludarabin angesprochen haben und für eine Behandlung mit Alemtuzumab als ungeeignet betrachtet werden, da sie Lymphknoten von mehr als 5 cm im Durchmesser aufwiesen, so genannte „Bulky Disease“ (bulky Fludarabin-refraktär; BFR).
Insgesamt erhielten die 138 Studienteilnehmer acht Wochen lang eine Ofatumumab-Infusion pro Woche, gefolgt von vier Infusionen in monatlichen Abständen. Die Dosierung betrug 300 mg Ofatumumab bei der ersten und 2000 mg bei jeder folgenden Infusion. Der Erkrankungszustand wird bis Woche 28 alle 4 Wochen beurteilt, danach alle 3 Monate bis zum Fortschreiten der Erkrankung (Progression) bis zu zwei Jahren. Die Studie nimmt weiter Patienten auf. Die endgültige Auswertung erfolgt, sobald jede Gruppe 100 Patienten umfasst.
Primärer Endpunkt der Studie ist die objektive Gesamtansprechrate auf die Ofatumumab-Monotherapie über einen Zeitraum von 24 Wochen, beurteilt durch eine unabhängige Expertengruppe. Sie lag bei 58 % für die Gruppe der doppel-refraktären Patienten DR (n=59) und bei 47 % für die Gruppe der Fludarabin-refraktären Patienten mit bulky disease (BFR) (n=79). Dabei handelte es sich meist um ein partielles Ansprechen (PR), ein Patient zeigte sogar ein komplettes Ansprechen (CR). (1)
Die mediane Gesamtüberlebenszeit betrug 13,7 Monate für die DR-Gruppe und 15,4 Monate für die BFR-Gruppe.1 Dabei war das Ansprechen auf Ofatumumab signifikant mit einer längeren Überlebenszeit verbunden. Die mediane progressionsfreie Überlebenszeit betrug 5,7 Monate für die DR-Gruppe und 5,9 Monate für die BFR-Gruppe. (1)
Das häufigste unerwünschte Ereignis war eine meist leicht bis mäßig ausgeprägte Reaktion auf die Infusion (Fieber). Die häufigsten schweren unerwünschten Ereignisse (Schweregrade 3 oder 4) waren Infektionen (25% in der DR-Gruppe; 25% in der BFR-Gruppe), darunter eine progressive multifokale Leukoenzephalopathie (PML) bei einem Patienten mit progredienter Erkrankung. Frühtodesfälle (innerhalb von 8 Wochen nach Behandlungsbeginn) traten bei vier Patienten (7%) in der DR-Gruppe und bei zwei Patienten (3%) in der BFR-Gruppe auf. Kein Patient entwickelte Antikörper gegen Ofatumumab. (1)
Über CLL
CLL ist die häufigste Form der Leukämie (30 % aller Leukämien) und eine der häufigsten bösartigen Erkrankungen des lymphatischen Systems in der westlichen Welt. (2) In Deutschland wird jedes Jahr bei etwa drei von 100.000 Personen eine CLL neu diagnostiziert. Männer erkranken häufiger daran als Frauen (Verhältnis = 1,7:1). (3)
Die Behandlung von Patienten mit refraktärer Erkrankung bleibt eine Herausforderung. Spricht die CLL nicht auf derzeitig verfügbare Standardbehandlungen an, ist die Prognose schlecht: Die mediane Gesamtüberlebenszeit reicht von 8 bis 14 Monaten. (4) Derzeit gibt es kein zugelassenes Medikament zur Behandlung dieser Patienten.
Ofatumumab ist ein neuartiger, vollständig humanisierter monoklonaler Antikörper. Er bindet an einen bestimmten, kleinen, nahe der Zelloberfläche gelegenen Bereich des CD20-Moleküls der B-Lymphozyten (eine Untergruppe der weißen Blutkörperchen). (5) Diese Bindungsstelle unterscheidet sich von der, die von anderen verfügbaren oder gerade in der Entwicklung befindlichen CD20-Antikörpern angesprochen wird.6 Das CD20-Molekül ist bei den meisten bösartigen
B-Zell-Erkrankungen vorhanden und damit ein Hauptziel neuer Behandlungsansätze. (2)
Ofatumumab wird im Rahmen einer gemeinsamen Entwicklungs- und Vermarktungsvereinbarung zwischen Genmab und GlaxoSmithKline zur Behandlung von CLL und anderer bösartiger Erkrankungen des Lymphsystems, der rheumatoiden Arthritis und schubförmig remittierender multipler Sklerose (RRMS) entwickelt. Der Wirkstoff ist bisher in noch keinem Land zugelassen.[/i]
mfg ipollit
http://www.journalonko.de/newsview.php?id=2997
19.12.2008
Positive Phase-III-Studiendaten für Ofatumumab bei CLL
Der monoklonale CD20-Antikörper Ofatumumab zeigte positive Ergebnisse in einer klinischen Phase-III-Studie zur Behandlung von Patienten mit stark vorbehandelter chronischer lymphatischer Leukämie (CLL), die auf die Standard-Therapie nicht angesprochen haben. Dies zeigt die geplante Zwischenauswertung einer aktuellen Phase-III-Studie zu dem Wirkstoff, die beim 50. Jahreskongress der American Society of Hematology, 6. bis 9. Dezember 2008 in San Francisco, vorgestellt wurde. (1)
Positive Ergebnisse bei Fludarabin-/Alemtuzumab-refraktären Patienten
In der Studie wurden Patienten mit vorbehandelter CLL aufgenommenen, die entweder nur begrenzt oder überhaupt nicht auf die Standardtherapien mit Fludarabin als auch Alemtuzumab angesprochen haben (doppelt refraktär, DR) oder die nicht auf Fludarabin angesprochen haben und für eine Behandlung mit Alemtuzumab als ungeeignet betrachtet werden, da sie Lymphknoten von mehr als 5 cm im Durchmesser aufwiesen, so genannte „Bulky Disease“ (bulky Fludarabin-refraktär; BFR).
Insgesamt erhielten die 138 Studienteilnehmer acht Wochen lang eine Ofatumumab-Infusion pro Woche, gefolgt von vier Infusionen in monatlichen Abständen. Die Dosierung betrug 300 mg Ofatumumab bei der ersten und 2000 mg bei jeder folgenden Infusion. Der Erkrankungszustand wird bis Woche 28 alle 4 Wochen beurteilt, danach alle 3 Monate bis zum Fortschreiten der Erkrankung (Progression) bis zu zwei Jahren. Die Studie nimmt weiter Patienten auf. Die endgültige Auswertung erfolgt, sobald jede Gruppe 100 Patienten umfasst.
Primärer Endpunkt der Studie ist die objektive Gesamtansprechrate auf die Ofatumumab-Monotherapie über einen Zeitraum von 24 Wochen, beurteilt durch eine unabhängige Expertengruppe. Sie lag bei 58 % für die Gruppe der doppel-refraktären Patienten DR (n=59) und bei 47 % für die Gruppe der Fludarabin-refraktären Patienten mit bulky disease (BFR) (n=79). Dabei handelte es sich meist um ein partielles Ansprechen (PR), ein Patient zeigte sogar ein komplettes Ansprechen (CR). (1)
Die mediane Gesamtüberlebenszeit betrug 13,7 Monate für die DR-Gruppe und 15,4 Monate für die BFR-Gruppe.1 Dabei war das Ansprechen auf Ofatumumab signifikant mit einer längeren Überlebenszeit verbunden. Die mediane progressionsfreie Überlebenszeit betrug 5,7 Monate für die DR-Gruppe und 5,9 Monate für die BFR-Gruppe. (1)
Das häufigste unerwünschte Ereignis war eine meist leicht bis mäßig ausgeprägte Reaktion auf die Infusion (Fieber). Die häufigsten schweren unerwünschten Ereignisse (Schweregrade 3 oder 4) waren Infektionen (25% in der DR-Gruppe; 25% in der BFR-Gruppe), darunter eine progressive multifokale Leukoenzephalopathie (PML) bei einem Patienten mit progredienter Erkrankung. Frühtodesfälle (innerhalb von 8 Wochen nach Behandlungsbeginn) traten bei vier Patienten (7%) in der DR-Gruppe und bei zwei Patienten (3%) in der BFR-Gruppe auf. Kein Patient entwickelte Antikörper gegen Ofatumumab. (1)
Über CLL
CLL ist die häufigste Form der Leukämie (30 % aller Leukämien) und eine der häufigsten bösartigen Erkrankungen des lymphatischen Systems in der westlichen Welt. (2) In Deutschland wird jedes Jahr bei etwa drei von 100.000 Personen eine CLL neu diagnostiziert. Männer erkranken häufiger daran als Frauen (Verhältnis = 1,7:1). (3)
Die Behandlung von Patienten mit refraktärer Erkrankung bleibt eine Herausforderung. Spricht die CLL nicht auf derzeitig verfügbare Standardbehandlungen an, ist die Prognose schlecht: Die mediane Gesamtüberlebenszeit reicht von 8 bis 14 Monaten. (4) Derzeit gibt es kein zugelassenes Medikament zur Behandlung dieser Patienten.
Ofatumumab ist ein neuartiger, vollständig humanisierter monoklonaler Antikörper. Er bindet an einen bestimmten, kleinen, nahe der Zelloberfläche gelegenen Bereich des CD20-Moleküls der B-Lymphozyten (eine Untergruppe der weißen Blutkörperchen). (5) Diese Bindungsstelle unterscheidet sich von der, die von anderen verfügbaren oder gerade in der Entwicklung befindlichen CD20-Antikörpern angesprochen wird.6 Das CD20-Molekül ist bei den meisten bösartigen
B-Zell-Erkrankungen vorhanden und damit ein Hauptziel neuer Behandlungsansätze. (2)
Ofatumumab wird im Rahmen einer gemeinsamen Entwicklungs- und Vermarktungsvereinbarung zwischen Genmab und GlaxoSmithKline zur Behandlung von CLL und anderer bösartiger Erkrankungen des Lymphsystems, der rheumatoiden Arthritis und schubförmig remittierender multipler Sklerose (RRMS) entwickelt. Der Wirkstoff ist bisher in noch keinem Land zugelassen.[/i]
mfg ipollit
Cubist... aktuelle MK 1.23 Mrd USD
die eps-Schätzungen liegen laut yahoo aktuell bei eps09e $1.34 une eps10e $1.84, was einem KGV09e von 15,9 bzw. KGV10e von 11,6 entspricht. Der Umsatz soll bei 573 bzw 675 Mio USD liegen.
January 23, 2009 11:06 AM EST
Needham & Co reiterates a 'Buy' on Cubist Pharmaceuticals Inc. (Nasdaq: CBST). Price target $27.
Needham analyst says, "Management highlighted the potential negative effects of the economy in 2009 on the number of elective surgeries and hence hospital infections. As a result, management has provided what they view as conservative guidance of $520-540MM in 2009 U.S. Cubicin sales. We believe long term trends in favor of Cubicin, including declining vancomycin sensitivity and delays in the development and launch of potential competitors, will be prevailing factors this year. We estimate $546MM in 2009 U.S. Cubicin sales with an EPS of $1.38, reflecting the higher tax rate. We are maintaining our 2010 revenue and EPS estimates."
********
Auf dem letzten CC wurde das US-peak sale Ziel vom Unternehmen selbst auf 1 Mrd USD angehoben...
http://seekingalpha.com/article/116053-cubist-pharmaceutical…
We've previously talked about our expectations for more than $750 million in peak sales in the US for CUBICIN. Based on our updated assessment of market drivers and the very large opportunities still ahead of us, we are announcing today that we now estimate peak sales for CUBICIN in the US of at least $1 billion.
Our increased peak sales estimate for CUBICIN is based on our expectations for continued erosion in the dominant market position of vancomycin, as well other factors including: the continued growth of our MRSA fueled in part by the growing problem of community acquired MRSA, the growth opportunity in the out-patient market, due to the interest of hospitals in getting patients discharged as quickly as possible, changes in our view of the competitive environment for the foreseeable future, and our existing US patent protection for CUBICIN extending until September of 2019.
Now let's move to 2009. First revenues; total revenues are expected to be in a range of $557.5 million to $577.5 million. The midpoint $567.5 million would be an increase of 31% from total 2008 revenues.
Finally for the cash position. We expect to generate around $130 million of free cash in 2009, and end the year with around $550 million of cash; subject of course to our success in business development.
Our expected milestones in 2009 include the completion of enrollment in the Ecallantide Conserve 1 trial, the completion of the Phase I studies for our gram-negative and CDAD programs and data due later in the year from the RSV lung transplant and dose fractionation studies.
We continue to be very active on the business development front and have a goal of bringing in a late stage product pipeline program this year. As we assess the additional opportunities for acquiring assets in today's market, we will continue to make strategically prudent, scientifically well-informed and financially disciplined decisions.
In 2009 for CUBICIN, our expected milestones include completing enrollment in ongoing Phase II studies of CUBICIN in prosthetic joint infections and in children with complicated skin infections. Continued progress for international partners with market launches and regulatory milestones and some important publications focused on pharmacoeconomics and out-patient use of CUBICIN.
mfg ipollit
die eps-Schätzungen liegen laut yahoo aktuell bei eps09e $1.34 une eps10e $1.84, was einem KGV09e von 15,9 bzw. KGV10e von 11,6 entspricht. Der Umsatz soll bei 573 bzw 675 Mio USD liegen.
January 23, 2009 11:06 AM EST
Needham & Co reiterates a 'Buy' on Cubist Pharmaceuticals Inc. (Nasdaq: CBST). Price target $27.
Needham analyst says, "Management highlighted the potential negative effects of the economy in 2009 on the number of elective surgeries and hence hospital infections. As a result, management has provided what they view as conservative guidance of $520-540MM in 2009 U.S. Cubicin sales. We believe long term trends in favor of Cubicin, including declining vancomycin sensitivity and delays in the development and launch of potential competitors, will be prevailing factors this year. We estimate $546MM in 2009 U.S. Cubicin sales with an EPS of $1.38, reflecting the higher tax rate. We are maintaining our 2010 revenue and EPS estimates."
********
Auf dem letzten CC wurde das US-peak sale Ziel vom Unternehmen selbst auf 1 Mrd USD angehoben...
http://seekingalpha.com/article/116053-cubist-pharmaceutical…
We've previously talked about our expectations for more than $750 million in peak sales in the US for CUBICIN. Based on our updated assessment of market drivers and the very large opportunities still ahead of us, we are announcing today that we now estimate peak sales for CUBICIN in the US of at least $1 billion.
Our increased peak sales estimate for CUBICIN is based on our expectations for continued erosion in the dominant market position of vancomycin, as well other factors including: the continued growth of our MRSA fueled in part by the growing problem of community acquired MRSA, the growth opportunity in the out-patient market, due to the interest of hospitals in getting patients discharged as quickly as possible, changes in our view of the competitive environment for the foreseeable future, and our existing US patent protection for CUBICIN extending until September of 2019.
Now let's move to 2009. First revenues; total revenues are expected to be in a range of $557.5 million to $577.5 million. The midpoint $567.5 million would be an increase of 31% from total 2008 revenues.
Finally for the cash position. We expect to generate around $130 million of free cash in 2009, and end the year with around $550 million of cash; subject of course to our success in business development.
Our expected milestones in 2009 include the completion of enrollment in the Ecallantide Conserve 1 trial, the completion of the Phase I studies for our gram-negative and CDAD programs and data due later in the year from the RSV lung transplant and dose fractionation studies.
We continue to be very active on the business development front and have a goal of bringing in a late stage product pipeline program this year. As we assess the additional opportunities for acquiring assets in today's market, we will continue to make strategically prudent, scientifically well-informed and financially disciplined decisions.
In 2009 for CUBICIN, our expected milestones include completing enrollment in ongoing Phase II studies of CUBICIN in prosthetic joint infections and in children with complicated skin infections. Continued progress for international partners with market launches and regulatory milestones and some important publications focused on pharmacoeconomics and out-patient use of CUBICIN.
mfg ipollit
MITI - Events in 2009
Micromet Key Events for 2009
Key Presentations and Milestones Expected for 2009
BETHESDA, Md., Jan. 5 /PRNewswire-FirstCall/ -- Micromet, Inc. (Nasdaq: MITI - News), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, provides an outlook on key events anticipated for the year 2009.
Micromet intends to provide an update on its research and development programs at the following events:
-- April 12 to 16, 2009: Update on several preclinical BiTE antibodies at the American Association for Cancer Research (AACR) in Denver, CO;
-- April 24, 2009: The company will hold an R&D meeting in New York, NY for investors and analysts to provide a review of its product pipeline;
-- May 29 to June 5, 2009: Update on a phase 1b clinical trial combining adecatumumab with docetaxel in late stage metastatic breast cancer patients at the annual meeting of the American Society for Clinical Oncology (ASCO) in San Francisco, CA;
-- June 4 to 7, 2009: Update on phase 2 clinical trial with blinatumomab in acute lymphoblastic leukemia (ALL) patients at the 14th Congress of the European Hematology Association (EHA) in Berlin, Germany;
-- September 20 to 24, 2009: First interim data from a phase 1 clinical trial with MT110, an EpCAM-targeting BiTE antibody, in patients with metastatic gastro-intestinal and lung cancers at the joint 15th Congress of the European Cancer Organisation (ECCO) and 34th Congress of the European Society for Medical Oncology (ESMO) in Berlin, Germany;
-- December 5 to 8, 2009: Update on a phase 2 clinical trial with blinatumomab in ALL patients and final results from a phase 1 clinical trial in late stage NHL at the annual meeting of the American Society for Hematology (ASH) in New Orleans, LA.
Additional expected milestones in 2009:
-- Adecatumumab, an EpCAM-targeting human monoclonal antibody in development with Merck-Serono is expected to start a randomized, controlled, multicenter phase 2 clinical trial in relapsed colorectal cancer patients after complete resection of liver metastasis in Q1 2009;
-- Blinatumomab, a CD19-targeting BiTE antibody in development with MedImmune, is expected to start an additional phase 2 clinical trial in patients with non-Hodgkin's lymphoma (NHL) towards the end of 2009;
-- MT203, a GM-CSF neutralizing human monoclonal antibody in development with Nycomed for the treatment of chronic inflammatory and autoimmune diseases, is expected to start a first clinical phase 1 trial in mid 2009;
-- MT228, a glycolipid binding human antibody in development by Eisai for the treatment of melanoma, is expected to start a first phase 1 clinical trial in 2009;
-- MT293, a humanized monoclonal antibody targeting denatured collagen, in development by Tracon Pharmaceuticals, Inc. for the treatment of solid tumors is expected to complete a clinical phase 1 trial.
Micromet intends to publish results from the research and development of its programs in peer-reviewed scientific and clinical journals throughout the year. Micromet also intends to enter into one to two new collaborations with corporate partners in 2009.
mfg ipollit
Micromet Key Events for 2009
Key Presentations and Milestones Expected for 2009
BETHESDA, Md., Jan. 5 /PRNewswire-FirstCall/ -- Micromet, Inc. (Nasdaq: MITI - News), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, provides an outlook on key events anticipated for the year 2009.
Micromet intends to provide an update on its research and development programs at the following events:
-- April 12 to 16, 2009: Update on several preclinical BiTE antibodies at the American Association for Cancer Research (AACR) in Denver, CO;
-- April 24, 2009: The company will hold an R&D meeting in New York, NY for investors and analysts to provide a review of its product pipeline;
-- May 29 to June 5, 2009: Update on a phase 1b clinical trial combining adecatumumab with docetaxel in late stage metastatic breast cancer patients at the annual meeting of the American Society for Clinical Oncology (ASCO) in San Francisco, CA;
-- June 4 to 7, 2009: Update on phase 2 clinical trial with blinatumomab in acute lymphoblastic leukemia (ALL) patients at the 14th Congress of the European Hematology Association (EHA) in Berlin, Germany;
-- September 20 to 24, 2009: First interim data from a phase 1 clinical trial with MT110, an EpCAM-targeting BiTE antibody, in patients with metastatic gastro-intestinal and lung cancers at the joint 15th Congress of the European Cancer Organisation (ECCO) and 34th Congress of the European Society for Medical Oncology (ESMO) in Berlin, Germany;
-- December 5 to 8, 2009: Update on a phase 2 clinical trial with blinatumomab in ALL patients and final results from a phase 1 clinical trial in late stage NHL at the annual meeting of the American Society for Hematology (ASH) in New Orleans, LA.
Additional expected milestones in 2009:
-- Adecatumumab, an EpCAM-targeting human monoclonal antibody in development with Merck-Serono is expected to start a randomized, controlled, multicenter phase 2 clinical trial in relapsed colorectal cancer patients after complete resection of liver metastasis in Q1 2009;
-- Blinatumomab, a CD19-targeting BiTE antibody in development with MedImmune, is expected to start an additional phase 2 clinical trial in patients with non-Hodgkin's lymphoma (NHL) towards the end of 2009;
-- MT203, a GM-CSF neutralizing human monoclonal antibody in development with Nycomed for the treatment of chronic inflammatory and autoimmune diseases, is expected to start a first clinical phase 1 trial in mid 2009;
-- MT228, a glycolipid binding human antibody in development by Eisai for the treatment of melanoma, is expected to start a first phase 1 clinical trial in 2009;
-- MT293, a humanized monoclonal antibody targeting denatured collagen, in development by Tracon Pharmaceuticals, Inc. for the treatment of solid tumors is expected to complete a clinical phase 1 trial.
Micromet intends to publish results from the research and development of its programs in peer-reviewed scientific and clinical journals throughout the year. Micromet also intends to enter into one to two new collaborations with corporate partners in 2009.
mfg ipollit
zu RIGL und SGEN...
http://www.hammerstockblog.com/the-delicate-art-of-expectati…
Rigel And Seattle Genetics -The Delicate Art of Expectation Management
In the previous article, I discussed the pharmaceutical industry’s race after approved drugs and late stage agents with proof of concept in humans. I mentioned Rigel’s (RIGL) lead drug, R788, as a likely target for collaboration due to its impressive activity, the huge addressable market and the fact it is an oral drug. For the past year, Rigel’s management has been consistently and rigorously claiming it will have a partnership in place during the first quarter of 2009. Although the company has had more than one opportunity to change this forecast, it stuck by its original statement. For example, when new safety data got published last year and worried investors sent the stock down 50% in two trading sessions, many believed that the imminent deal was not going to materialize. To their surprise, Rigel reassured investors the time frame for a partnership remains intact, explaining that none of the recently published data was actually new to potential partners. Then, Rigel appeared in countless investor conferences, the last of which was only last month, promising investors a licensing deal is forthcoming.
Last week, the company announced it no longer expects to have a deal by the end of March. Instead, it intends to wait until it has results from two ongoing trials, due this summer. Deciding to wait until more data is available makes a lot of sense, providing the data is good. Typically, the further a drug gets in clinical development, the higher its value in the eyes of potential partners. The problem is not the decision itself, but its timing, as this kind of decision could have been made long ago. So what led Rigel’s management to suddenly change its mind after a year of expectations build up?
During last week’s conference call, Rigel mentioned several factors that played a role in the decision. First was the opportunity to get a more lucrative deal, if the data from the larger trials is as good as that of the previous one. Another factor was the better than expected enrollment rate, resulting in earlier data read out from the trials. Lastly, Rigel claims that several potential partners had indicated they prefer to wait for the new data before signing a deal. Some of these partners were described as “very large pharmas” by the company’s CEO, Jim Gower, during the call.
When asked about the terms of the deal, Gower admitted that as they got closer to signing a deal, the deal terms became less attractive, because partners preferred waiting for the updated data this summer. To me, this looks like the only reasonable excuse, as Rigel and potential partners were always aware of the ongoing trials, and truthfully, having data a couple of months earlier does not seem that dramatic. Apparently, Rigel simply did not get the deal it was hoping for.
The field of drug development is anything but expected, and in many cases, things do not turn out as planned. Rigel’s decision is understandable, as it may eventually lead to a better deal for R788. Investors must also realize that with the higher reward comes a higher risk level, and if the data is not as good as expected, R788’s value may plunge.
Financially, the deal is not urgent, as the company has enough cash to support its activity through the second quarter if 2010, almost a year after expected data from the two ongoing trials. But even if Rigel made the right decision, it certainly made all the possible mistakes on the way with respect to managing investors’ expectations. In this area, Rigel’s management could learn something from Seattle Genetics’ (SGEN) management team.
Seattle Genetics has three clinically validated drug candidates: dacetuzumab (SGN-40), SGN-35 and lintuzumab (SGN-33). All three might produce “registrational-quality” data in 2010, so the company could have as much as three approved drugs by early 2012. Dacetuzumab, has already been licensed out to Genentech in 2007, leaving the two other drugs as potential targets for licensing activity. SGN-35, Seattle Genetics’ lead antibody-drug conjugate represents the smallest market opportunity among the three, however, it also represents the highest likelihood for approval.
SGN-35
SGN-35 demonstrated remarkable activity in heavily pretreated Hodgkin’s lymphoma patients. Last month at the ASH meeting, investigators presented positive data from a dose escalation phase I study. The data included 41 Hodgkin’s lymphoma patients, 15 (36.6%) of whom had an objective response, including 7 complete responses. In the three highest cohorts, SGN-35’s activity was even more impressive, with a response rate of 50%. This data is in line with data presented at previous conferences, as described in this article. Importantly, responses were quite durable and even patients who did not have an objective response derived great benefit from SGN-35. The median progression free survival was about 6 months for all patients, 83% of whom experienced tumor shrinkage.
Based on these promising results, Seattle Genetics decided to initiate a registrational trial in Hodgkin’s lymphoma patients who are refractory or relapsed to standard first line treatments. Since there are no approved therapies for this setting, this trial is SGN-35’s fastest route to market. Last month, Seattle Genetics announced it had received a SPA for a pivotal trial, in which SGN-35 will be evaluated in refractory or relapsed Hodgkin’s lymphoma patients. Due to the highly unmet medical need and the small patient population (3000 patients in the U.S. annually), the trial will be a small (100 patients) single arm study, with objective response rate as the primary endpoint. The design is typical for pivotal trials for niche indications, just like the study Allos (ALTH) conducted for its drug, PDX, in another rare form of blood cancer. Seattle Genetics plans to launch a trial in another rare lymphoma, ALCL.
The company estimates the immediate market opportunity of SGN-35 to be $300-$400 million in the United States and Europe. I find these estimates over-optimistic, based on durability of response and the prevalence of the diseases, but SGN-35 could easily reach peak sales of $250, if the phase I results are replicated in the registration trial. A potential partner might be interested in expanding SGN-35’s use into first line treatment in addition to its potential utility for autoimmune diseases, which could substantially increase the potential market for the drug.
Lintuzumab
Seattle Genetics’ second licensing candidate seems to be even more promising from a commercial point of view. Similarly to SGN-35, lintuzumab addresses an indication with very limited treatment options and consequently very high demand for new drugs – Acute myeloid leukemia (AML) in patients 60 years of age and older. SGN-33 is currently in a randomized phase II trial for the treatment of elderly AML patients. The study, which evaluates a common treatment for these patients (low dose cytarabine) with or without lintuzumab is expected to have final data in the first half of 2010.
In my opinion, the lintuzumab program is the most attractive opportunity for Seattle Genetics because it represents a very large untapped market with a very low entry bar and virtually no real competition. Elderly AML patients are in dire need of new treatments with a good safety profile since they often cannot tolerate standard chemotherapy regimens. In fact, in many cases these patients die of treatment side effects and not of the disease itself, which is typically characterized by a survival of 6-9 months. The company has not disclosed any new data for lintuzumab in the past 12 months, but the clinical data it presented at the 2007ASH annual meeting generated a great deal of enthusiasm in the industry. I discussed these data and lintuzumab’s explosive potential here, here and here.
The potential of the AML market made it an area of intense activity, with several agents in the clinic. Two of these agents, Sunesis’ (SNSS) voreloxin and Genzyme’s (GNZM) Clolar demonstrated even slightly higher response rates than that of lintuzumab in different trials, but neither seem to threat lintuzumab. As chemotherapy agents, these drugs are likely to lead to severe side effects and potentially death in such frail patients. Lintuzumab, on the other hand, seems to enjoy a very clean safety profile, similarly to other antibodies for the treatment of cancer, such as Rituxan. As a result, lintuzumab can be easily combined with other agents and can be given for very long periods of time (perhaps even as maintenance therapy). These properties render lintuzumab the safest bet among all investigational agents for elderly AML because even in a scenario where multiple agents are approved, these agents’ manufacturers would aspire to combine their chemo drugs with lintuzumab, in order to achieve a better response with little or no added toxicity.
Seattle Genetics believes that if lintuzumab manages to prolong survival by two months or more, it could file for approval with the FDA. This implies a possible product launch in the 2011 and a very high adoption rate by physicians, who currently prefer putting elderly patients in clinical trials or even not treating them at all, due to lack of safe and effective treatments. More than 13,000 new cases of AML are expected in the U.S each year, two thirds of which will be diagnosed in elderly patients. Assuming a similar rate in Europe, the overall market potential in developed countries could reach 25,000 patients, which could translate into sales in the $700-900 million range, depending on durability of responses. Evidently, if lintuzumab proves active and safe in elderly AML patients, it will quickly find its way to younger patients as well. In addition, lintuzumab is currently being evaluated in MDS, an indication that is expected generate more than $500 million in sales in 2010 for Celgene’s (CELG) Vidaza.
On top of the registrational phase II study, lintuzumab is currently being evaluated as a single agent in elderly AML patients. The goal of this trial is to get a better sense of the single agent response rate of SGN-33 and more importantly, to address the issue of response durability, which is very short in patients who respond to chemotherapy. Because SGN-33 can be given for very long periods of time, it may do a better job maintaining responses, as opposed to chemotherapy which is given only for several weeks. In terms of response rate, it would be interesting to see if lintuzumab can achieve similar results to those reported at ASH 2007. There may even be a positive surprise with respect to the original 17 patients, as the lead investigator disclosed that one of the patients, who had been defined as a non-responder, was starting to respond after treatment discontinuation. The company is expected to publish data from this trial in the first half of 2009.
Partnership Discussions
Seattle Genetics’ management has never hidden its intentions to become a fully commercial company, with an independent sales force which will market its oncology products. The poster child of a small biotech that becomes a global company with worldwide sales infrastructure is Celgene, who leveraged the success of Revlimid and Thalomid to build one of the biggest hematology sales forces in the industry. Celgene’s approach resulted in huge financial gains in the long run, but also required a large amount of resources, both financial and managerial. Under current market conditions, with limited access to capital, it would be unrealistic for a company the size of Seattle Genetics to become a global company, so the next best thing it can do is retaining rights for its drugs in North America and license the non-U.S rights to a large partner. Therefore, it is likely that SGN-35 and perhaps lintuzumab will be licensed out during 2009.
Partnership discussions for SGN-35 started last year, with the goal of retaining all U.S. rights and licensing international rights for the drug. This is an unusual deal structure for a candidate with phase I data, but Seattle Genetics repeatedly stated that it would not have it any other way. In contrast to Rigel, Seattle Genetics was very vague regarding the timing of any potential deal and repeatedly cautioned investors that it would not do a deal unless the terms are attractive enough. Theoretically, this implies that Seattle Genetics may go all the way without a strategic partner, but the cost will be immense.
The company ended 2008 with $161 million in cash, which could support operations until the third quarter of 2010, barely enough for obtaining registration quality data for both SGN-35 and lintuzumab. Last month, Seattle Genetics stunned the market by announcing a $53 million secondary public offering, thus securing another two quarters of activity. This is the first public offering for a biotech company in six months, but even more impressive than the timing was the high pricing of the offering, 9.72$ per share. Amazingly, investors received this unexpected dilution well, sending shares higher following the announcement.
To me, the exact timing of the transaction implies that Seattle Genetics felt it did not have sufficient leverage at the negotiation table. Allegedly, Seattle Genetics could have waited for at least 9 months before going back to the equity markets, potentially benefiting from a recovery or positive company announcements. The ability to raise capital in such an environment is impressive, particularly at a price hardly representing any discount, but current price levels are far from ideal for dilution. Therefore, there was something urgent in Seattle Genetics’ decision, which can only be explained by the need to ink a licensing deal in the near term.
Summary
Rigel and Seattle Genetics have a lot in common. Both have mid stage clinical drugs that demonstrated impressive activity in relatively small trials, both are involved in ongoing discussions with potential partners, not willing to compromise. Of course, there are also differences between the two. Seattle Genetics’ SGN-35 addresses an orphan indication that requires a small and inexpensive clinical program, while Rigel’s R788 targets a multi-billion indication, requiring an expensive clinical program (several hundred millions of dollars). Thus, Seattle Genetics is under less pressure to find a partner, but although it could eventually support a worldwide global sales force, based on the recent investment round and active discussions the company is involved in, I find this scenario unlikely.
Both Rigel and Seattle Genetics hit unexpected bumps along the road to the ideal partnership, and preferred to take unpopular steps over compromising on the deal terms: Rigel broke its promise in order to get more clinical data, while Seattle Genetics chose to dilute its existing shareholders to improve its financial position. In retrospect, both companies did the right thing. The difference was in the way each company managed investors’ expectations. In Seattle Genetics’ case, investors cheered its decision whereas Rigel got a cold shoulder and a couple of potential lawsuits.
mfg ipollit
http://www.hammerstockblog.com/the-delicate-art-of-expectati…
Rigel And Seattle Genetics -The Delicate Art of Expectation Management
In the previous article, I discussed the pharmaceutical industry’s race after approved drugs and late stage agents with proof of concept in humans. I mentioned Rigel’s (RIGL) lead drug, R788, as a likely target for collaboration due to its impressive activity, the huge addressable market and the fact it is an oral drug. For the past year, Rigel’s management has been consistently and rigorously claiming it will have a partnership in place during the first quarter of 2009. Although the company has had more than one opportunity to change this forecast, it stuck by its original statement. For example, when new safety data got published last year and worried investors sent the stock down 50% in two trading sessions, many believed that the imminent deal was not going to materialize. To their surprise, Rigel reassured investors the time frame for a partnership remains intact, explaining that none of the recently published data was actually new to potential partners. Then, Rigel appeared in countless investor conferences, the last of which was only last month, promising investors a licensing deal is forthcoming.
Last week, the company announced it no longer expects to have a deal by the end of March. Instead, it intends to wait until it has results from two ongoing trials, due this summer. Deciding to wait until more data is available makes a lot of sense, providing the data is good. Typically, the further a drug gets in clinical development, the higher its value in the eyes of potential partners. The problem is not the decision itself, but its timing, as this kind of decision could have been made long ago. So what led Rigel’s management to suddenly change its mind after a year of expectations build up?
During last week’s conference call, Rigel mentioned several factors that played a role in the decision. First was the opportunity to get a more lucrative deal, if the data from the larger trials is as good as that of the previous one. Another factor was the better than expected enrollment rate, resulting in earlier data read out from the trials. Lastly, Rigel claims that several potential partners had indicated they prefer to wait for the new data before signing a deal. Some of these partners were described as “very large pharmas” by the company’s CEO, Jim Gower, during the call.
When asked about the terms of the deal, Gower admitted that as they got closer to signing a deal, the deal terms became less attractive, because partners preferred waiting for the updated data this summer. To me, this looks like the only reasonable excuse, as Rigel and potential partners were always aware of the ongoing trials, and truthfully, having data a couple of months earlier does not seem that dramatic. Apparently, Rigel simply did not get the deal it was hoping for.
The field of drug development is anything but expected, and in many cases, things do not turn out as planned. Rigel’s decision is understandable, as it may eventually lead to a better deal for R788. Investors must also realize that with the higher reward comes a higher risk level, and if the data is not as good as expected, R788’s value may plunge.
Financially, the deal is not urgent, as the company has enough cash to support its activity through the second quarter if 2010, almost a year after expected data from the two ongoing trials. But even if Rigel made the right decision, it certainly made all the possible mistakes on the way with respect to managing investors’ expectations. In this area, Rigel’s management could learn something from Seattle Genetics’ (SGEN) management team.
Seattle Genetics has three clinically validated drug candidates: dacetuzumab (SGN-40), SGN-35 and lintuzumab (SGN-33). All three might produce “registrational-quality” data in 2010, so the company could have as much as three approved drugs by early 2012. Dacetuzumab, has already been licensed out to Genentech in 2007, leaving the two other drugs as potential targets for licensing activity. SGN-35, Seattle Genetics’ lead antibody-drug conjugate represents the smallest market opportunity among the three, however, it also represents the highest likelihood for approval.
SGN-35
SGN-35 demonstrated remarkable activity in heavily pretreated Hodgkin’s lymphoma patients. Last month at the ASH meeting, investigators presented positive data from a dose escalation phase I study. The data included 41 Hodgkin’s lymphoma patients, 15 (36.6%) of whom had an objective response, including 7 complete responses. In the three highest cohorts, SGN-35’s activity was even more impressive, with a response rate of 50%. This data is in line with data presented at previous conferences, as described in this article. Importantly, responses were quite durable and even patients who did not have an objective response derived great benefit from SGN-35. The median progression free survival was about 6 months for all patients, 83% of whom experienced tumor shrinkage.
Based on these promising results, Seattle Genetics decided to initiate a registrational trial in Hodgkin’s lymphoma patients who are refractory or relapsed to standard first line treatments. Since there are no approved therapies for this setting, this trial is SGN-35’s fastest route to market. Last month, Seattle Genetics announced it had received a SPA for a pivotal trial, in which SGN-35 will be evaluated in refractory or relapsed Hodgkin’s lymphoma patients. Due to the highly unmet medical need and the small patient population (3000 patients in the U.S. annually), the trial will be a small (100 patients) single arm study, with objective response rate as the primary endpoint. The design is typical for pivotal trials for niche indications, just like the study Allos (ALTH) conducted for its drug, PDX, in another rare form of blood cancer. Seattle Genetics plans to launch a trial in another rare lymphoma, ALCL.
The company estimates the immediate market opportunity of SGN-35 to be $300-$400 million in the United States and Europe. I find these estimates over-optimistic, based on durability of response and the prevalence of the diseases, but SGN-35 could easily reach peak sales of $250, if the phase I results are replicated in the registration trial. A potential partner might be interested in expanding SGN-35’s use into first line treatment in addition to its potential utility for autoimmune diseases, which could substantially increase the potential market for the drug.
Lintuzumab
Seattle Genetics’ second licensing candidate seems to be even more promising from a commercial point of view. Similarly to SGN-35, lintuzumab addresses an indication with very limited treatment options and consequently very high demand for new drugs – Acute myeloid leukemia (AML) in patients 60 years of age and older. SGN-33 is currently in a randomized phase II trial for the treatment of elderly AML patients. The study, which evaluates a common treatment for these patients (low dose cytarabine) with or without lintuzumab is expected to have final data in the first half of 2010.
In my opinion, the lintuzumab program is the most attractive opportunity for Seattle Genetics because it represents a very large untapped market with a very low entry bar and virtually no real competition. Elderly AML patients are in dire need of new treatments with a good safety profile since they often cannot tolerate standard chemotherapy regimens. In fact, in many cases these patients die of treatment side effects and not of the disease itself, which is typically characterized by a survival of 6-9 months. The company has not disclosed any new data for lintuzumab in the past 12 months, but the clinical data it presented at the 2007ASH annual meeting generated a great deal of enthusiasm in the industry. I discussed these data and lintuzumab’s explosive potential here, here and here.
The potential of the AML market made it an area of intense activity, with several agents in the clinic. Two of these agents, Sunesis’ (SNSS) voreloxin and Genzyme’s (GNZM) Clolar demonstrated even slightly higher response rates than that of lintuzumab in different trials, but neither seem to threat lintuzumab. As chemotherapy agents, these drugs are likely to lead to severe side effects and potentially death in such frail patients. Lintuzumab, on the other hand, seems to enjoy a very clean safety profile, similarly to other antibodies for the treatment of cancer, such as Rituxan. As a result, lintuzumab can be easily combined with other agents and can be given for very long periods of time (perhaps even as maintenance therapy). These properties render lintuzumab the safest bet among all investigational agents for elderly AML because even in a scenario where multiple agents are approved, these agents’ manufacturers would aspire to combine their chemo drugs with lintuzumab, in order to achieve a better response with little or no added toxicity.
Seattle Genetics believes that if lintuzumab manages to prolong survival by two months or more, it could file for approval with the FDA. This implies a possible product launch in the 2011 and a very high adoption rate by physicians, who currently prefer putting elderly patients in clinical trials or even not treating them at all, due to lack of safe and effective treatments. More than 13,000 new cases of AML are expected in the U.S each year, two thirds of which will be diagnosed in elderly patients. Assuming a similar rate in Europe, the overall market potential in developed countries could reach 25,000 patients, which could translate into sales in the $700-900 million range, depending on durability of responses. Evidently, if lintuzumab proves active and safe in elderly AML patients, it will quickly find its way to younger patients as well. In addition, lintuzumab is currently being evaluated in MDS, an indication that is expected generate more than $500 million in sales in 2010 for Celgene’s (CELG) Vidaza.
On top of the registrational phase II study, lintuzumab is currently being evaluated as a single agent in elderly AML patients. The goal of this trial is to get a better sense of the single agent response rate of SGN-33 and more importantly, to address the issue of response durability, which is very short in patients who respond to chemotherapy. Because SGN-33 can be given for very long periods of time, it may do a better job maintaining responses, as opposed to chemotherapy which is given only for several weeks. In terms of response rate, it would be interesting to see if lintuzumab can achieve similar results to those reported at ASH 2007. There may even be a positive surprise with respect to the original 17 patients, as the lead investigator disclosed that one of the patients, who had been defined as a non-responder, was starting to respond after treatment discontinuation. The company is expected to publish data from this trial in the first half of 2009.
Partnership Discussions
Seattle Genetics’ management has never hidden its intentions to become a fully commercial company, with an independent sales force which will market its oncology products. The poster child of a small biotech that becomes a global company with worldwide sales infrastructure is Celgene, who leveraged the success of Revlimid and Thalomid to build one of the biggest hematology sales forces in the industry. Celgene’s approach resulted in huge financial gains in the long run, but also required a large amount of resources, both financial and managerial. Under current market conditions, with limited access to capital, it would be unrealistic for a company the size of Seattle Genetics to become a global company, so the next best thing it can do is retaining rights for its drugs in North America and license the non-U.S rights to a large partner. Therefore, it is likely that SGN-35 and perhaps lintuzumab will be licensed out during 2009.
Partnership discussions for SGN-35 started last year, with the goal of retaining all U.S. rights and licensing international rights for the drug. This is an unusual deal structure for a candidate with phase I data, but Seattle Genetics repeatedly stated that it would not have it any other way. In contrast to Rigel, Seattle Genetics was very vague regarding the timing of any potential deal and repeatedly cautioned investors that it would not do a deal unless the terms are attractive enough. Theoretically, this implies that Seattle Genetics may go all the way without a strategic partner, but the cost will be immense.
The company ended 2008 with $161 million in cash, which could support operations until the third quarter of 2010, barely enough for obtaining registration quality data for both SGN-35 and lintuzumab. Last month, Seattle Genetics stunned the market by announcing a $53 million secondary public offering, thus securing another two quarters of activity. This is the first public offering for a biotech company in six months, but even more impressive than the timing was the high pricing of the offering, 9.72$ per share. Amazingly, investors received this unexpected dilution well, sending shares higher following the announcement.
To me, the exact timing of the transaction implies that Seattle Genetics felt it did not have sufficient leverage at the negotiation table. Allegedly, Seattle Genetics could have waited for at least 9 months before going back to the equity markets, potentially benefiting from a recovery or positive company announcements. The ability to raise capital in such an environment is impressive, particularly at a price hardly representing any discount, but current price levels are far from ideal for dilution. Therefore, there was something urgent in Seattle Genetics’ decision, which can only be explained by the need to ink a licensing deal in the near term.
Summary
Rigel and Seattle Genetics have a lot in common. Both have mid stage clinical drugs that demonstrated impressive activity in relatively small trials, both are involved in ongoing discussions with potential partners, not willing to compromise. Of course, there are also differences between the two. Seattle Genetics’ SGN-35 addresses an orphan indication that requires a small and inexpensive clinical program, while Rigel’s R788 targets a multi-billion indication, requiring an expensive clinical program (several hundred millions of dollars). Thus, Seattle Genetics is under less pressure to find a partner, but although it could eventually support a worldwide global sales force, based on the recent investment round and active discussions the company is involved in, I find this scenario unlikely.
Both Rigel and Seattle Genetics hit unexpected bumps along the road to the ideal partnership, and preferred to take unpopular steps over compromising on the deal terms: Rigel broke its promise in order to get more clinical data, while Seattle Genetics chose to dilute its existing shareholders to improve its financial position. In retrospect, both companies did the right thing. The difference was in the way each company managed investors’ expectations. In Seattle Genetics’ case, investors cheered its decision whereas Rigel got a cold shoulder and a couple of potential lawsuits.
mfg ipollit
Intercell muss absolut rein ins Depot:
Der Biotechsektor an sich weist eine geringe Korrelation mit anderen Aktien auf und ist damit zur Depotdiversifizierung geeignet; als einige der wenigen Werte hat dies Intercell auch während der noch andauernden Finanzkrise bewiesen. Die Firma sticht hier aus dem Sektor noch hervor, durch seine weit vorangeschrittenen Entwicklungen
Weitere Produkte sind in der Entwicklungsphase. Vier Wirkstoffe könnten sich bereits 2009 in der finalen dritten Phase und 3 in der Phase II der klinischen Studien befinden. Ein Wirkstoff gegen die in sich verbreitenden Krankenhausinfektionen wird mit Hochdruck durch die klinische zweite Phase gebracht.
Novartis hat bei seinem Einstieg 2007 bei Intercell mehr als 31 Euro/Aktie bezahlt.
Hier hab ich noch mehr zu dem Wert gefunden:
http://tinyurl.com/c8zw74
Der Biotechsektor an sich weist eine geringe Korrelation mit anderen Aktien auf und ist damit zur Depotdiversifizierung geeignet; als einige der wenigen Werte hat dies Intercell auch während der noch andauernden Finanzkrise bewiesen. Die Firma sticht hier aus dem Sektor noch hervor, durch seine weit vorangeschrittenen Entwicklungen
Weitere Produkte sind in der Entwicklungsphase. Vier Wirkstoffe könnten sich bereits 2009 in der finalen dritten Phase und 3 in der Phase II der klinischen Studien befinden. Ein Wirkstoff gegen die in sich verbreitenden Krankenhausinfektionen wird mit Hochdruck durch die klinische zweite Phase gebracht.
Novartis hat bei seinem Einstieg 2007 bei Intercell mehr als 31 Euro/Aktie bezahlt.
Hier hab ich noch mehr zu dem Wert gefunden:
http://tinyurl.com/c8zw74
Antwort auf Beitrag Nr.: 36.350.858 von ipollit am 11.01.09 15:53:11
Biotech-Aktien stabilisieren Aktienportfolios
http://aktien-boersen.blogspot.com/2009/05/biotech-aktien-st…
Biotech-Aktien stabilisieren Aktienportfolios
http://aktien-boersen.blogspot.com/2009/05/biotech-aktien-st…
nach einem halben Jahr hat sich unter dem Strich nicht viel getan. Im Vergleich zum Jahresanfang ergibt sich ein leichtes Minus von 1% und damit eine Performance leicht unter der von NBI und Dax.
Zum Jahresanfang... reduziert wurde ONXX, aufgestockt RIGL, INCY und CBST. ALTH ist neu dazu gekommen.
15,1% Genmab "http://finance.yahoo.com/q?s=GEN.CO"
9,8% Rigel "http://finance.yahoo.com/q?s=RIGL"
7,7% Cubist "http://finance.yahoo.com/q?s=CBST"
7,3% Evotec "http://finance.yahoo.com/q?s=EVT.DE"
6,9% Regeneron "http://finance.yahoo.com/q?s=REGN"
6,3% Onyx "http://finance.yahoo.com/q?s=ONXX"
5,3% Isis "http://finance.yahoo.com/q?s=ISIS"
4,8% Incyte "http://finance.yahoo.com/q?s=INCY"
4,6% Allos "http://finance.yahoo.com/q?s=ALTH"
4,5% Medigene "http://finance.yahoo.com/q?s=MDG.DE"
4,5% Exelixis "http://finance.yahoo.com/q?s=EXEL"
4,1% Micromet "http://finance.yahoo.com/q?s=MITI"
3,8% Seattle Genetics "http://finance.yahoo.com/q?s=SGEN"
2,9% NicOx "http://finance.yahoo.com/q?s=COX.PA"
2,8% Arena "http://finance.yahoo.com/q?s=ARNA"
2,6% Addex "http://finance.yahoo.com/q?s=ADXN.SW"
2,1% Array "http://finance.yahoo.com/q?s=ARRY"
1,8% Sucampo "http://finance.yahoo.com/q?s=SCMP"
1,7% ViroPharma "http://finance.yahoo.com/q?s=VPHM"
1,6% Progenics "http://finance.yahoo.com/q?s=PGNX"
Auf der Watch-Liste sind immer noch Basilea und Neurosearch. Evotec hat sich erstaunlich positiv entwickelt und könnte reduziert werden. NicOx, ARNA, VPHM und PRGNX sind Verkaufskandidaten.
Rückschläge...
- VPHM: zwar sieht der Marktstart con Cinryze bisher positiv aus, doch ist unerwartet Maribavir in PIII gescheitert, so dass zusammen mit dem drohenden Verlust des Patentschutzes von Vancocin, bald schon Verluste geschrieben werden können. Interessant bleibt nur, ob in den kommenden Wochen die FDA vielleicht doch nicht generisches Vancocin zulässt, sondern dafür klinische Studien verlangt
- CBST: Teva versucht den Patentschutz von Cubicin auszuhebeln, was jetzt Gerichte zu entscheiden haben. Diese Unsicherheit hat den Kurs massiv gedrückt. Im worst-case wird Cubicin (Patentschutz bis 2019) in 2 Jahren generisch.
- ONXX: Partner Bayer hat heimlich eine Variante von Nexavar ohne Wissen von ONXX in der Entwicklung. ONXX hat daraufhin Bayer verklagt.
- Genmab: FDA benötigt 3 Monate länger, um über die Zulassung von Arzerra zu entscheiden... statt Ende Juli nun Ende Ende Oktober. Der Expertenrat hat zwar der FDA die Zulassung empfohlen, doch tauchten in den Unterlagen der FDA einige unerwartete Fragen auf: so ist es z.B. wahrscheinlich, dass Arzerra nur eine Zulassung bekommt bei CLL-Patienten, bei denen bereits 2 Therapien versagt haben und nicht nur eine. Zudem wurde zwei Studien (nicht die Zulassungsstudie) mit Zalutumumab kurzzeitig von der FDA angehalten... inzwischen laufen aber alle wieder.
- Rigel: Partnersuche für R788 wurde auf Ende des Jahres verschoben; dafür gab es kürzlich positive TASKi2-Daten... die TASKi3-Daten, die wahrscheinlich etwas schlechter ausfallen, sollten in den nächsten Tagen folgen
- Evotec: EVT-302 ist in der PII gescheitert. EVT-101 wird nicht gegen Schmerzen oder Alzheimer entwickelt, sondern wurde an Roche auslizensiert, wo es nun gegen Depressionen entwickelt wird.
- Incyte: kein Rückschlag, aber die hohen Schulden drücken den Kurs
- Medigene: es konnte entgegen der bisherigen Planung noch kein Partner für EndoTag-1 gefunden werden... wahrscheinlich zieht sich das noch bis ins nächste Jahr hin
- NicOx: die Partnersuche für Naproxcinod zieht sich hin
- Arena: die ersten PIII-Daten zu Lorcaserin sind zwar nicht direkt negativ gewesen, wurden aber negativ vom Markt aufgenommen. Die Sicherheit scheint gut zu sein, es bleibt aber offen, ob die Wirksamkeit ausreicht, um erfolgreich zu sein.
- Progenics: Relistor verkauft sich nur sehr schlecht... Partner Wyeth könnte nach der Übernahme durch Pfizer abspringen
NBI
 [/url]
[/url]
Genmab

RIGL
CBST

Evotec
 [/url]
[/url]
REGN
ONXX
ISIS
INCY
ALTH
Medigene
 [/url]
[/url]
EXEL
MITI

SGEN
NiCox

ARNA
Addex
 [/url]
[/url]
ARRY
SCMP
VPHM
PGNX

mfg ipollit
Zum Jahresanfang... reduziert wurde ONXX, aufgestockt RIGL, INCY und CBST. ALTH ist neu dazu gekommen.
15,1% Genmab "http://finance.yahoo.com/q?s=GEN.CO"
9,8% Rigel "http://finance.yahoo.com/q?s=RIGL"
7,7% Cubist "http://finance.yahoo.com/q?s=CBST"
7,3% Evotec "http://finance.yahoo.com/q?s=EVT.DE"
6,9% Regeneron "http://finance.yahoo.com/q?s=REGN"
6,3% Onyx "http://finance.yahoo.com/q?s=ONXX"
5,3% Isis "http://finance.yahoo.com/q?s=ISIS"
4,8% Incyte "http://finance.yahoo.com/q?s=INCY"
4,6% Allos "http://finance.yahoo.com/q?s=ALTH"
4,5% Medigene "http://finance.yahoo.com/q?s=MDG.DE"
4,5% Exelixis "http://finance.yahoo.com/q?s=EXEL"
4,1% Micromet "http://finance.yahoo.com/q?s=MITI"
3,8% Seattle Genetics "http://finance.yahoo.com/q?s=SGEN"
2,9% NicOx "http://finance.yahoo.com/q?s=COX.PA"
2,8% Arena "http://finance.yahoo.com/q?s=ARNA"
2,6% Addex "http://finance.yahoo.com/q?s=ADXN.SW"
2,1% Array "http://finance.yahoo.com/q?s=ARRY"
1,8% Sucampo "http://finance.yahoo.com/q?s=SCMP"
1,7% ViroPharma "http://finance.yahoo.com/q?s=VPHM"
1,6% Progenics "http://finance.yahoo.com/q?s=PGNX"
Auf der Watch-Liste sind immer noch Basilea und Neurosearch. Evotec hat sich erstaunlich positiv entwickelt und könnte reduziert werden. NicOx, ARNA, VPHM und PRGNX sind Verkaufskandidaten.
Rückschläge...
- VPHM: zwar sieht der Marktstart con Cinryze bisher positiv aus, doch ist unerwartet Maribavir in PIII gescheitert, so dass zusammen mit dem drohenden Verlust des Patentschutzes von Vancocin, bald schon Verluste geschrieben werden können. Interessant bleibt nur, ob in den kommenden Wochen die FDA vielleicht doch nicht generisches Vancocin zulässt, sondern dafür klinische Studien verlangt
- CBST: Teva versucht den Patentschutz von Cubicin auszuhebeln, was jetzt Gerichte zu entscheiden haben. Diese Unsicherheit hat den Kurs massiv gedrückt. Im worst-case wird Cubicin (Patentschutz bis 2019) in 2 Jahren generisch.
- ONXX: Partner Bayer hat heimlich eine Variante von Nexavar ohne Wissen von ONXX in der Entwicklung. ONXX hat daraufhin Bayer verklagt.
- Genmab: FDA benötigt 3 Monate länger, um über die Zulassung von Arzerra zu entscheiden... statt Ende Juli nun Ende Ende Oktober. Der Expertenrat hat zwar der FDA die Zulassung empfohlen, doch tauchten in den Unterlagen der FDA einige unerwartete Fragen auf: so ist es z.B. wahrscheinlich, dass Arzerra nur eine Zulassung bekommt bei CLL-Patienten, bei denen bereits 2 Therapien versagt haben und nicht nur eine. Zudem wurde zwei Studien (nicht die Zulassungsstudie) mit Zalutumumab kurzzeitig von der FDA angehalten... inzwischen laufen aber alle wieder.
- Rigel: Partnersuche für R788 wurde auf Ende des Jahres verschoben; dafür gab es kürzlich positive TASKi2-Daten... die TASKi3-Daten, die wahrscheinlich etwas schlechter ausfallen, sollten in den nächsten Tagen folgen
- Evotec: EVT-302 ist in der PII gescheitert. EVT-101 wird nicht gegen Schmerzen oder Alzheimer entwickelt, sondern wurde an Roche auslizensiert, wo es nun gegen Depressionen entwickelt wird.
- Incyte: kein Rückschlag, aber die hohen Schulden drücken den Kurs
- Medigene: es konnte entgegen der bisherigen Planung noch kein Partner für EndoTag-1 gefunden werden... wahrscheinlich zieht sich das noch bis ins nächste Jahr hin
- NicOx: die Partnersuche für Naproxcinod zieht sich hin
- Arena: die ersten PIII-Daten zu Lorcaserin sind zwar nicht direkt negativ gewesen, wurden aber negativ vom Markt aufgenommen. Die Sicherheit scheint gut zu sein, es bleibt aber offen, ob die Wirksamkeit ausreicht, um erfolgreich zu sein.
- Progenics: Relistor verkauft sich nur sehr schlecht... Partner Wyeth könnte nach der Übernahme durch Pfizer abspringen
NBI
Genmab
RIGL
CBST
Evotec
REGN
ONXX
ISIS
INCY
ALTH
Medigene
EXEL
MITI
SGEN
NiCox
ARNA
Addex
[/url]ARRY
SCMP
VPHM
PGNX
mfg ipollit
Antwort auf Beitrag Nr.: 37.601.913 von ipollit am 19.07.09 16:21:19
ipollit, schön, wieder etwas von Dir zu lesen. Hast Du eigentlich eine Meinung zu Medarex? Allgemein und vielleicht auch konkret bezogen auf die Aussichten von Ipilimumab? Da wird es binnen der nächsten 6 Monate ja die entscheidenden P III-Daten geben.
ipollit, schön, wieder etwas von Dir zu lesen. Hast Du eigentlich eine Meinung zu Medarex? Allgemein und vielleicht auch konkret bezogen auf die Aussichten von Ipilimumab? Da wird es binnen der nächsten 6 Monate ja die entscheidenden P III-Daten geben.
Antwort auf Beitrag Nr.: 37.602.020 von SLGramann am 19.07.09 16:59:31hi SLGramann!
Genau weiß ich nicht, was MEDX alles macht. Die AK-Technologie scheint gut zu sein, inzwischen gibt es ja auch schon drei Partner-AKs auf dem Markt. Die Royalties für diese dürften aber wohl nur 2-3% betragen. Zu Ilaris (Royalties gehen auch an REGN, die ich im Depot habe) weiß ich nur, dass die aktuelle Indikation nicht viel einbringt. Zu Stelara und Simponi habe ich soweit ich mich erinnere mal gelesen, dass zumindest bei einem noch offen ist, ob es sich am Markt auch durchsetzen kann. Und selbst wenn diese alle recht erfolgreich werden, dann ist dies noch keine Garantie, dass man dies auch am Kurs sieht... z.B. ist aus PDLI trotz erfolgreicher Partner-Produkte nicht viel geworden.
Also hängt im Moment schon viel von Ipilimumab ab, zumal die MK mit etwa 1 Mrd USD schon nicht mehr so billig ist. An MEDX habe ich mich bisher wegen Ipilimumab nicht getraut... ob es bei MM erfolgreich ist, scheint ziemlich unsicher zu sein, soweit ich das immer verstanden habe. Welche Daten kommen denn genau... OS-Daten zu MM in 4Q09? Waren die bisherigen Ergebnisse von Ipilimumab nicht negativ? Das metastasierte Melanom ist eine extrem schwierige und riskante Indikation. Bisher hat dort alles versagt... zuletzt sind z.B. Synta damit abgestützt. Man sollte sich also des Risikos bewusst sein, dass Ipilimumab hier ebenfalls scheitert, was inzwischen wohl ein schwerer Rückschlag für Medarex wäre - oder habe ich etwas übersehen? Andererseits dürfte ein Erfolg für eine entsprechende Überraschung sorgen. Ich glaube zwar nicht, dass sich MEDX dann vervielfachen wird, aber was positive Überraschungen bewirken können, hat man ja heute an HGSI mit einem Plus von 277% gesehen.
Also viel Glück!
Gruß, ipollit
Genau weiß ich nicht, was MEDX alles macht. Die AK-Technologie scheint gut zu sein, inzwischen gibt es ja auch schon drei Partner-AKs auf dem Markt. Die Royalties für diese dürften aber wohl nur 2-3% betragen. Zu Ilaris (Royalties gehen auch an REGN, die ich im Depot habe) weiß ich nur, dass die aktuelle Indikation nicht viel einbringt. Zu Stelara und Simponi habe ich soweit ich mich erinnere mal gelesen, dass zumindest bei einem noch offen ist, ob es sich am Markt auch durchsetzen kann. Und selbst wenn diese alle recht erfolgreich werden, dann ist dies noch keine Garantie, dass man dies auch am Kurs sieht... z.B. ist aus PDLI trotz erfolgreicher Partner-Produkte nicht viel geworden.
Also hängt im Moment schon viel von Ipilimumab ab, zumal die MK mit etwa 1 Mrd USD schon nicht mehr so billig ist. An MEDX habe ich mich bisher wegen Ipilimumab nicht getraut... ob es bei MM erfolgreich ist, scheint ziemlich unsicher zu sein, soweit ich das immer verstanden habe. Welche Daten kommen denn genau... OS-Daten zu MM in 4Q09? Waren die bisherigen Ergebnisse von Ipilimumab nicht negativ? Das metastasierte Melanom ist eine extrem schwierige und riskante Indikation. Bisher hat dort alles versagt... zuletzt sind z.B. Synta damit abgestützt. Man sollte sich also des Risikos bewusst sein, dass Ipilimumab hier ebenfalls scheitert, was inzwischen wohl ein schwerer Rückschlag für Medarex wäre - oder habe ich etwas übersehen? Andererseits dürfte ein Erfolg für eine entsprechende Überraschung sorgen. Ich glaube zwar nicht, dass sich MEDX dann vervielfachen wird, aber was positive Überraschungen bewirken können, hat man ja heute an HGSI mit einem Plus von 277% gesehen.
Also viel Glück!
Gruß, ipollit
etwas zu ALTH... Kommentar von Leerink:
INITIATION - OUTPERFORM - Target $11
PDX: A Precious Hematology Commodity; Initiating on ALTH with an Outperform
• Bottom Line: We are initiating coverage on ALTH with an Outperform rating and an $11/share valuation based on our view that PDX is likely to gain FDA approval, garner an attractive Orphan Drug price, and penetrate effectively into the niche peripheral T-cell lymphoma (pTCL)
market to achieve $300M in sales in the U.S. PDX is an antifolate drug that has completed the pivotal PROPEL study in pTCL and is awaiting FDA approval with a September 24, 2009, PDUFA date. pTCL is a rare subtype of Non-Hodgkin's Lymphoma (NHL), and we estimate it accounts for over 6% of new lymphomas in the U.S. based on data from large SEER registries. With approximately 75K new lymphomas annually in the U.S., this translates into 4,500+ new patients eligible for therapy each year. Additionally, there is the potential for a much larger OUS market given the higher prevalence of pTCL in Asian territories.
• Hematology Assets Are a Precious Commodity. Following the
acquisitions in 2008 of Millennium and MGI Pharma, there are few unencumbered hematology programs remaining. We do not believe upside to ALTH's valuation depends on an acquisition, although the ability to leverage a hematology sales force to market PDX is an appealing proposition.
• OUS Market May Have To Wait. We currently assume the EMEA will require a placebo-controlled trial prior to approval of PDX, similar to its requirement for GENZ's Clolar in AML. We model an ex-U.S. launch in 2013, and note that there would be upside to this assumption if the EMEA were to approve PDX based on the PROPEL study.
• Pipeline Opportunities. ALTH is evaluating PDX in other hematology/oncology settings, including relapsed/refractory cutaneous T-cell lymphoma (CTCL), non-small cell lung cancer (NSCLC), and bladder cancer. Although these other PDX opportunities provide potential upside, our expectations are low and we do not include them in our valuation.
INVESTMENT THESIS
We rate ALTH Outperform.Data for antifolate drug PDX in the pivotal single-arm PROPEL inr efractory T-cell lymphoma (pTCL) should be sufficient to support FDA approval, and with an estimated 4,500 new pTCL cases in the U.S. each year combined with Orphan Drug pricing, we believe peak PDX sales could surpass $300M. Our caution on the size of the U.S. market is easing, as sizable SEER patient registries, MEDACorp consultant (“consultant”)ack and ALTH management combine to make a good case that there are enough patients each year to build a meaningful revenue position, particularly with a high price point. The PDUFA date for PDX is September 24, 2009. The OUS pTCL market may be considerably larger than that in the U.S. given a high incidence of pTCL inAsian territories. Approval in the EU is likely to require a placebo-controlled trial, in our view, and this may take the form of a frontline study in combination with chemotherapy or as a placebo-controlled study in a treatment-experienced population. If a controlled trial were required, response rate may be an adequate primary endpoint as long as PFS or OS were supportive. ALTH is also developing PDX for non-small cell lung cancer (Phase 2b data versus Tarceva should be available in 1H10) as well as other solid tumor indications; we do not include these opportunities in our valuation.
PROPEL Data Should Be Good Enough for Approval
The pivotal PROPEL study was a single-arm monotherapy trial of PDX in patients with relapsed/refractory pTCL that was conducted under the auspices of an SPA with the FDA.
PROPEL enrolled 115 patients and evaluated PDX at 30mg/m2 in 7-week cycles that included 1 week of rest. Results of this study were presented at the 2008 ASH conference, and updated data were presented at ASCO in May 2009. Among 109 evaluable patients, the CR+ CRu (unconfirmed CR) + PR rate by independent review was 28% with a median duration of response of 9.4 months.
Responses that occurred typically did so within the first cycle. Median overall survival was 14.7 months after 52 events. Grade 3/4 side effects occurring in >10% of patients included mucositis (22%), thrombocytopenia (33%), anemia (17%), and neutropenia (20%); most adverse events were grade 1 to 3 and were reversible and manageable. ALTH believes that a response rate (RR) >20% with duration of response >3 months represents clinically meaningful results, and we agree with these parameters given the absence of material offsetting drug-related toxicity.
Based on the PROPEL study data, we have made assumptions regarding the average duration of
therapy with PDX and conclude that, on average, in real-world use, patients are likely to receive
3.5 months of therapy, or roughly $115,000/patient, assuming a price point of $33,000/month.
VALUATION
Our $11 valuation on ALTH represents a 12x multiple on our 2012 EPS estimate of $0.89. This multiple is below the average 2011E P/E multiple of 16x for midcap biotech companies discounted back one additional year (to 14x) at 12%. We chose a lower-than-average multiple to reflect the uncertainty of the OUS commercial opportunity, our uncertainty on the size of the global pTCL market and PDX vulnerability to generic entrants as a small-molecule drug. Additionally, we use a DCF analysis through PDX patent expiration with a 12% discount rate to reach the same valuation.
***********

pTCL ist wohl nur als schneller Markteinstieg für PDX gedacht... die großen Indikationen kommen erst danach.
mfg ipollit
INITIATION - OUTPERFORM - Target $11
PDX: A Precious Hematology Commodity; Initiating on ALTH with an Outperform
• Bottom Line: We are initiating coverage on ALTH with an Outperform rating and an $11/share valuation based on our view that PDX is likely to gain FDA approval, garner an attractive Orphan Drug price, and penetrate effectively into the niche peripheral T-cell lymphoma (pTCL)
market to achieve $300M in sales in the U.S. PDX is an antifolate drug that has completed the pivotal PROPEL study in pTCL and is awaiting FDA approval with a September 24, 2009, PDUFA date. pTCL is a rare subtype of Non-Hodgkin's Lymphoma (NHL), and we estimate it accounts for over 6% of new lymphomas in the U.S. based on data from large SEER registries. With approximately 75K new lymphomas annually in the U.S., this translates into 4,500+ new patients eligible for therapy each year. Additionally, there is the potential for a much larger OUS market given the higher prevalence of pTCL in Asian territories.
• Hematology Assets Are a Precious Commodity. Following the
acquisitions in 2008 of Millennium and MGI Pharma, there are few unencumbered hematology programs remaining. We do not believe upside to ALTH's valuation depends on an acquisition, although the ability to leverage a hematology sales force to market PDX is an appealing proposition.
• OUS Market May Have To Wait. We currently assume the EMEA will require a placebo-controlled trial prior to approval of PDX, similar to its requirement for GENZ's Clolar in AML. We model an ex-U.S. launch in 2013, and note that there would be upside to this assumption if the EMEA were to approve PDX based on the PROPEL study.
• Pipeline Opportunities. ALTH is evaluating PDX in other hematology/oncology settings, including relapsed/refractory cutaneous T-cell lymphoma (CTCL), non-small cell lung cancer (NSCLC), and bladder cancer. Although these other PDX opportunities provide potential upside, our expectations are low and we do not include them in our valuation.
INVESTMENT THESIS
We rate ALTH Outperform.Data for antifolate drug PDX in the pivotal single-arm PROPEL inr efractory T-cell lymphoma (pTCL) should be sufficient to support FDA approval, and with an estimated 4,500 new pTCL cases in the U.S. each year combined with Orphan Drug pricing, we believe peak PDX sales could surpass $300M. Our caution on the size of the U.S. market is easing, as sizable SEER patient registries, MEDACorp consultant (“consultant”)ack and ALTH management combine to make a good case that there are enough patients each year to build a meaningful revenue position, particularly with a high price point. The PDUFA date for PDX is September 24, 2009. The OUS pTCL market may be considerably larger than that in the U.S. given a high incidence of pTCL inAsian territories. Approval in the EU is likely to require a placebo-controlled trial, in our view, and this may take the form of a frontline study in combination with chemotherapy or as a placebo-controlled study in a treatment-experienced population. If a controlled trial were required, response rate may be an adequate primary endpoint as long as PFS or OS were supportive. ALTH is also developing PDX for non-small cell lung cancer (Phase 2b data versus Tarceva should be available in 1H10) as well as other solid tumor indications; we do not include these opportunities in our valuation.
PROPEL Data Should Be Good Enough for Approval
The pivotal PROPEL study was a single-arm monotherapy trial of PDX in patients with relapsed/refractory pTCL that was conducted under the auspices of an SPA with the FDA.
PROPEL enrolled 115 patients and evaluated PDX at 30mg/m2 in 7-week cycles that included 1 week of rest. Results of this study were presented at the 2008 ASH conference, and updated data were presented at ASCO in May 2009. Among 109 evaluable patients, the CR+ CRu (unconfirmed CR) + PR rate by independent review was 28% with a median duration of response of 9.4 months.
Responses that occurred typically did so within the first cycle. Median overall survival was 14.7 months after 52 events. Grade 3/4 side effects occurring in >10% of patients included mucositis (22%), thrombocytopenia (33%), anemia (17%), and neutropenia (20%); most adverse events were grade 1 to 3 and were reversible and manageable. ALTH believes that a response rate (RR) >20% with duration of response >3 months represents clinically meaningful results, and we agree with these parameters given the absence of material offsetting drug-related toxicity.
Based on the PROPEL study data, we have made assumptions regarding the average duration of
therapy with PDX and conclude that, on average, in real-world use, patients are likely to receive
3.5 months of therapy, or roughly $115,000/patient, assuming a price point of $33,000/month.
VALUATION
Our $11 valuation on ALTH represents a 12x multiple on our 2012 EPS estimate of $0.89. This multiple is below the average 2011E P/E multiple of 16x for midcap biotech companies discounted back one additional year (to 14x) at 12%. We chose a lower-than-average multiple to reflect the uncertainty of the OUS commercial opportunity, our uncertainty on the size of the global pTCL market and PDX vulnerability to generic entrants as a small-molecule drug. Additionally, we use a DCF analysis through PDX patent expiration with a 12% discount rate to reach the same valuation.
***********
pTCL ist wohl nur als schneller Markteinstieg für PDX gedacht... die großen Indikationen kommen erst danach.
mfg ipollit
Antwort auf Beitrag Nr.: 37.610.690 von ipollit am 20.07.09 22:57:04
Hi ipollit,
vielen Dank für Deine Antwort.
Ja, die Daten zu ipilimumab, die Ende Q4 oder Anfang Q1/2010 veröffentlicht werden sollen, sind aus einer P III mit dem primären Endpunkt Overall Survival.
Hier findet sich die Studie: http://clinicaltrials.gov/ct2/show/NCT00324155?term=ipilimum…
Weitere Hintergründe finden sich in Villes Blog:
http://antibody-world.blogspot.com/2009/04/medarex-inc-eine-…
Bei meinem Erstinvestment in Medarex am Anfang diesen Jahres habe ich die Bedeutung von ipilimumab unterschätzt.
Ich habe gestern meine Medarex-Position um die Hälfte abgebaut und dafür Seattle Genetics und Immunogen und den Cash verstärkt. SGEN, IMGN, MITI und Medarex sind jetzt etwa gleich große - oder besser gleich kleine - Positionen.
Medarex ist eigentlich sehr attraktiv, weil durch die zugelassenen Partner-AKs ein nachhaltiger und wahrscheinlich in wenigen Jahren doch relevanter Tantiemen-Strom fließen wird, der es Medarex ermöglichen wird, seine wohl unbestritten sehr gute AK-Technologie, einschließlich der ADCs, in relativer Ruhe voranzubringen. Selbst wenn ipilimumab scheitert, hat Medarex weiter große - dann aber eben nicht mehr so unmittelbar greifbare - Chancen. Ich denke, die Position, die ich jetzt noch habe, kann ich mit großer Ruhe auch über ein mögliches Scheitern von ipilimumab hinweg halten. Sollte der Kurs dann wieder auf 4 Dollar oder so abstürzen, stocke ich wahrscheinlich sogar wieder auf. Aber ich wünsche mir, dass ich diese Möglichkeit nicht bekommen werde.
Hi ipollit,
vielen Dank für Deine Antwort.
Ja, die Daten zu ipilimumab, die Ende Q4 oder Anfang Q1/2010 veröffentlicht werden sollen, sind aus einer P III mit dem primären Endpunkt Overall Survival.
Hier findet sich die Studie: http://clinicaltrials.gov/ct2/show/NCT00324155?term=ipilimum…
Weitere Hintergründe finden sich in Villes Blog:
http://antibody-world.blogspot.com/2009/04/medarex-inc-eine-…
Bei meinem Erstinvestment in Medarex am Anfang diesen Jahres habe ich die Bedeutung von ipilimumab unterschätzt.
Ich habe gestern meine Medarex-Position um die Hälfte abgebaut und dafür Seattle Genetics und Immunogen und den Cash verstärkt. SGEN, IMGN, MITI und Medarex sind jetzt etwa gleich große - oder besser gleich kleine - Positionen.
Medarex ist eigentlich sehr attraktiv, weil durch die zugelassenen Partner-AKs ein nachhaltiger und wahrscheinlich in wenigen Jahren doch relevanter Tantiemen-Strom fließen wird, der es Medarex ermöglichen wird, seine wohl unbestritten sehr gute AK-Technologie, einschließlich der ADCs, in relativer Ruhe voranzubringen. Selbst wenn ipilimumab scheitert, hat Medarex weiter große - dann aber eben nicht mehr so unmittelbar greifbare - Chancen. Ich denke, die Position, die ich jetzt noch habe, kann ich mit großer Ruhe auch über ein mögliches Scheitern von ipilimumab hinweg halten. Sollte der Kurs dann wieder auf 4 Dollar oder so abstürzen, stocke ich wahrscheinlich sogar wieder auf. Aber ich wünsche mir, dass ich diese Möglichkeit nicht bekommen werde.
Antwort auf Beitrag Nr.: 37.615.887 von SLGramann am 21.07.09 17:18:31eine mittlere Position ist in so einem Fall wohl am besten... im Falle eines Scheiters ist der Verlust nicht ganz so schmerzlich und bei Erfolg kann man trotzdem noch schöne Gewinne einfahren.
Statt auf Medarex setze ich auf Genmab, die ja aus Medarex hervorgegangen sind und dementsprechend dieselbe AK-Technologie nutzen. Damals hat Genmab von Medarex mit HuMax-CD20 und HuMax-EGFr zwei schöne AKs übernommen. Erster könnte als Arzerra bereits im November die Zulassung erhalten. In den nächsten Monaten werden wichtige PIII-Daten gegen NHL erwartet und dann wird sich zeigen, ob es sich gegen Rituxan, das ja auch über CD20 wirkt, behaupten kann. Theoretisch sollte Arzerra der bessere AK sein und Genmab hätte damit einen guten Konkurrenten zu Rituxan, das jährlich Umsätze von ein paar Mrd USD erzielt. Die entscheidenden HuMax-EGFr PIII-Ergebnisse werden gegen Ende des Jahres erwartet. HuMax-EGFr ist vom Wirkmechanismus mit Erbitux vergleichbar. Zudem ist der Roche AK R1507 in einer Zulassungs-PII, so dass Genmab theoretisch in absehbarer Zeit 3 AKs auf dem Markt haben könnte.
SGEN und IMGN sind ganz gut, denke ich... beide sollten sich auf Dauer lohnen.
gruß, ipollit
Statt auf Medarex setze ich auf Genmab, die ja aus Medarex hervorgegangen sind und dementsprechend dieselbe AK-Technologie nutzen. Damals hat Genmab von Medarex mit HuMax-CD20 und HuMax-EGFr zwei schöne AKs übernommen. Erster könnte als Arzerra bereits im November die Zulassung erhalten. In den nächsten Monaten werden wichtige PIII-Daten gegen NHL erwartet und dann wird sich zeigen, ob es sich gegen Rituxan, das ja auch über CD20 wirkt, behaupten kann. Theoretisch sollte Arzerra der bessere AK sein und Genmab hätte damit einen guten Konkurrenten zu Rituxan, das jährlich Umsätze von ein paar Mrd USD erzielt. Die entscheidenden HuMax-EGFr PIII-Ergebnisse werden gegen Ende des Jahres erwartet. HuMax-EGFr ist vom Wirkmechanismus mit Erbitux vergleichbar. Zudem ist der Roche AK R1507 in einer Zulassungs-PII, so dass Genmab theoretisch in absehbarer Zeit 3 AKs auf dem Markt haben könnte.
SGEN und IMGN sind ganz gut, denke ich... beide sollten sich auf Dauer lohnen.
gruß, ipollit
INCY... PIII-Studie von INCB18424 kann beginnen...
http://www.forbes.com/feeds/afx/2009/07/21/afx6680683.html
UPDATE 2-Incyte gets FDA nod on trial design, shares rise
07.21.09, 01:44 PM EDT
By Esha Dey
BANGALORE, July 21 (Reuters) - Incyte Corp said it agreed with the U.S. health regulators, through a special protocol assessment (SPA), on the design of a late-stage trial of its experimental treatment for myelofibrosis, a bone marrow disorder.
Analysts said the agreement was a positive for the company as the drug was very likely to hit the trial goals that have been agreed upon by the company and the regulators.
The trial, named COMFORT-I, will compare the efficacy and safety of the drug, INCB18424, with a dummy drug in about 240 patients with different kinds of myelofibrosis.
Myelofibrosis is a disorder in which the bone marrow develops scar tissue and causes swelling of the liver and spleen.
The main goal of the trial, which is expected to begin in August, is the proportion of patients achieving a 35 percent or greater reduction in spleen volume as compared to patients receiving a dummy drug.
'A single primary endpoint evaluating comparative spleen reduction is deemed sufficient under the SPA is a decided positive, as this is the one measure which we know the drug substantially impacts and for which there is considerable data,' J.P. Morgan analyst Cory Kasimov said in a note.
Kasimov reiterated his 'overweight' rating on the stock and said the company remained a top small cap pick.
The company said that it expects to file for regulatory approval of the drug in myelofibrosis in late 2010 or early 2011.
'The FDA's concurrence on a single primary endpoint should shorten the overall duration of the COMFORT I study driven by easier statistical powering and a shorter follow-up period,' Cowen and Co analyst Eric Schmidt said.
However, Schmidt kept his 'neutral' rating on the stock, citing the company's substantial debt position and a requirement to access funding.
As of March 31, the company's total consolidated debt was $421.8 million, with cash and cash equivalents of $139.1 million.
Robert W. Baird & Co analyst Thomas Russo said the SPA should help Incyte secure a partner for the drug, which could be key to resolving the financial overhang.
Russo also said that the drug has the potential to drive a breakthrough to profitability by 2012, while Morgan Stanley analyst Sapna Srivastava said it could be a $500 million drug.
The special protocol assessment (SPA) provides the company with a written agreement that the design and analysis of the trial are adequate to support a marketing application submission. (da kann ich ja nur lachen, wenn ich an GPC denke, denen das SPA nichts gebracht hat )
)
Shares of the company were up 11 percent at $4.44 in midday trade on Nasdaq. They had earlier touched a high of $4.66.

mfg ipollit
http://www.forbes.com/feeds/afx/2009/07/21/afx6680683.html
UPDATE 2-Incyte gets FDA nod on trial design, shares rise
07.21.09, 01:44 PM EDT
By Esha Dey
BANGALORE, July 21 (Reuters) - Incyte Corp said it agreed with the U.S. health regulators, through a special protocol assessment (SPA), on the design of a late-stage trial of its experimental treatment for myelofibrosis, a bone marrow disorder.
Analysts said the agreement was a positive for the company as the drug was very likely to hit the trial goals that have been agreed upon by the company and the regulators.
The trial, named COMFORT-I, will compare the efficacy and safety of the drug, INCB18424, with a dummy drug in about 240 patients with different kinds of myelofibrosis.
Myelofibrosis is a disorder in which the bone marrow develops scar tissue and causes swelling of the liver and spleen.
The main goal of the trial, which is expected to begin in August, is the proportion of patients achieving a 35 percent or greater reduction in spleen volume as compared to patients receiving a dummy drug.
'A single primary endpoint evaluating comparative spleen reduction is deemed sufficient under the SPA is a decided positive, as this is the one measure which we know the drug substantially impacts and for which there is considerable data,' J.P. Morgan analyst Cory Kasimov said in a note.
Kasimov reiterated his 'overweight' rating on the stock and said the company remained a top small cap pick.
The company said that it expects to file for regulatory approval of the drug in myelofibrosis in late 2010 or early 2011.
'The FDA's concurrence on a single primary endpoint should shorten the overall duration of the COMFORT I study driven by easier statistical powering and a shorter follow-up period,' Cowen and Co analyst Eric Schmidt said.
However, Schmidt kept his 'neutral' rating on the stock, citing the company's substantial debt position and a requirement to access funding.
As of March 31, the company's total consolidated debt was $421.8 million, with cash and cash equivalents of $139.1 million.
Robert W. Baird & Co analyst Thomas Russo said the SPA should help Incyte secure a partner for the drug, which could be key to resolving the financial overhang.
Russo also said that the drug has the potential to drive a breakthrough to profitability by 2012, while Morgan Stanley analyst Sapna Srivastava said it could be a $500 million drug.
The special protocol assessment (SPA) provides the company with a written agreement that the design and analysis of the trial are adequate to support a marketing application submission. (da kann ich ja nur lachen, wenn ich an GPC denke, denen das SPA nichts gebracht hat
 )
) Shares of the company were up 11 percent at $4.44 in midday trade on Nasdaq. They had earlier touched a high of $4.66.
mfg ipollit
ONXX... Nexavar könnte bei Brustkrebs helfen
http://www.reuters.com/article/rbssHealthcareNews/idUSLM3500…
UPDATE 2-Bayer says Nexavar shows promise in breast cancer
Wed Jul 22, 2009 11:15am EDT
FRANKFURT, July 22 (Reuters) - Bayer (BAYG.DE) and its development partner Onyx Pharmaceuticals (ONXX.O) said their cancer pill Nexavar showed promise in treating breast tumours, the second-most common form of cancer, sending Onyx shares soaring.
Nexavar, when combined with standard chemotherapy Xeloda, helped keep tumours in check for longer than in a control group of patients receiving Xeloda only, the drugmakers said in a joint statement on Wednesday, citing data from a Phase II study.
Xeloda is sold by Switzerland's Roche (ROG.VX).
Shares in Onyx, which depends entirely on Nexavar for sales, surged 25.1 percent to $35.90 at 1427 GMT. Shares in Bayer were up 2.2 percent, outperforming the European DJ Stoxx Health Care Index's .SXDP 0.7 percent rise.
Christopher Raymond, an analyst at Baird, called the results "a possible game-changer" for Onyx.
"Combining the ubiquitous use of Xeloda in advanced breast cancer with the robustness of this 200+ patient trial, we would not be surprised to see measurable off-label use once the data are presented," the analyst said in a note to investors.
The trial involved 229 women with breast tumours that had started spreading, they added.
More details would be released in an upcoming scientific meeting, they said.
Nexavar is one of Bayer's most promising drugs, along with anti-blood clotting pill Xarelto. It is sold as a liver and kidney cancer treatment in more than 70 countries and Bayer is also pursuing approvals for use against lung tumours.
The German drugmaker aims to generate worldwide sales of more than 2 billion euros ($2.84 billion) a year from the drug.
Bayer and U.S. biotech company Onyx are testing Nexavar in a further three ongoing breast cancer studies in Phase II and are working on possible trial setups for the third and last phase of testing required for regulatory approval.

mfg ipollit
http://www.reuters.com/article/rbssHealthcareNews/idUSLM3500…
UPDATE 2-Bayer says Nexavar shows promise in breast cancer
Wed Jul 22, 2009 11:15am EDT
FRANKFURT, July 22 (Reuters) - Bayer (BAYG.DE) and its development partner Onyx Pharmaceuticals (ONXX.O) said their cancer pill Nexavar showed promise in treating breast tumours, the second-most common form of cancer, sending Onyx shares soaring.
Nexavar, when combined with standard chemotherapy Xeloda, helped keep tumours in check for longer than in a control group of patients receiving Xeloda only, the drugmakers said in a joint statement on Wednesday, citing data from a Phase II study.
Xeloda is sold by Switzerland's Roche (ROG.VX).
Shares in Onyx, which depends entirely on Nexavar for sales, surged 25.1 percent to $35.90 at 1427 GMT. Shares in Bayer were up 2.2 percent, outperforming the European DJ Stoxx Health Care Index's .SXDP 0.7 percent rise.
Christopher Raymond, an analyst at Baird, called the results "a possible game-changer" for Onyx.
"Combining the ubiquitous use of Xeloda in advanced breast cancer with the robustness of this 200+ patient trial, we would not be surprised to see measurable off-label use once the data are presented," the analyst said in a note to investors.
The trial involved 229 women with breast tumours that had started spreading, they added.
More details would be released in an upcoming scientific meeting, they said.
Nexavar is one of Bayer's most promising drugs, along with anti-blood clotting pill Xarelto. It is sold as a liver and kidney cancer treatment in more than 70 countries and Bayer is also pursuing approvals for use against lung tumours.
The German drugmaker aims to generate worldwide sales of more than 2 billion euros ($2.84 billion) a year from the drug.
Bayer and U.S. biotech company Onyx are testing Nexavar in a further three ongoing breast cancer studies in Phase II and are working on possible trial setups for the third and last phase of testing required for regulatory approval.
mfg ipollit
Antwort auf Beitrag Nr.: 37.624.736 von ipollit am 22.07.09 17:37:48
Die MEdarex-Story ist zu Ende erzählt:
Wednesday, 22 Jul 2009 07:16pm EDT
Bristol-Myers Squibb Co. and Medarex, Inc. announced that the companies have signed a definitive merger agreement providing for the acquisition of Medarex by Bristol-Myers Squibb, for $16.00 per share in cash.
Der Zeitpunkt, meine Position zu reduzieren, war mal wieder brilliant...
Die MEdarex-Story ist zu Ende erzählt:
Wednesday, 22 Jul 2009 07:16pm EDT
Bristol-Myers Squibb Co. and Medarex, Inc. announced that the companies have signed a definitive merger agreement providing for the acquisition of Medarex by Bristol-Myers Squibb, for $16.00 per share in cash.
Der Zeitpunkt, meine Position zu reduzieren, war mal wieder brilliant...

Antwort auf Beitrag Nr.: 37.628.038 von SLGramann am 23.07.09 08:08:15naja, das war wirklich Pech...
********
July 23, 2009
09:29 EDT SGEN theflyonthewall.com: Needham: Regeneron, Seattle Genetics rank high as takeout candidates
Needham believes four characteristics attracted Bristol-Myers (BMY) to acquire Medarex (MEDX): hematology/oncology products, late-stage development, existing relationship, and antibody technology. Needham notes that Regeneron (REGN) and Seattle Genetics (SGEN) possess all four of those characteristics, while companies that have two out of four are: Alexion Pharma (ALXN), Allos Therapeutics (ALTH), AMAG Pharma (AMAG), ArQule (ARQL), Celldex Therapeutics (CLDX), Dendreon (DNDN), and Poniard Pharma (PARD).
NBI ist heute auf Jahreshoch gestiegen...


mfg ipollit
********
July 23, 2009
09:29 EDT SGEN theflyonthewall.com: Needham: Regeneron, Seattle Genetics rank high as takeout candidates
Needham believes four characteristics attracted Bristol-Myers (BMY) to acquire Medarex (MEDX): hematology/oncology products, late-stage development, existing relationship, and antibody technology. Needham notes that Regeneron (REGN) and Seattle Genetics (SGEN) possess all four of those characteristics, while companies that have two out of four are: Alexion Pharma (ALXN), Allos Therapeutics (ALTH), AMAG Pharma (AMAG), ArQule (ARQL), Celldex Therapeutics (CLDX), Dendreon (DNDN), and Poniard Pharma (PARD).
NBI ist heute auf Jahreshoch gestiegen...
mfg ipollit
Regeneron...
Neben dem eigenen zugelassenen Produkt Arcalyst bekommt REGN auch Royalties von bis zu 15% auf Umsätze mit Novartis Ilaris, das vor Kurzen zugelassen wurde. Schön wäre es, wenn Arcalyst nächstes Jahr positive PIII-Daten gegen Gicht erzielen würde. Die aktuelle Indikation CAPS ist sehr selten und bringt daher nicht viel Umsatz. Ilaris befindet sich bei Novartis in entsprechenden Studien, die es zu einem Blockbuster machen können.

Novartis drug appears to compete with partner Regeneron (REGN)
June 19, 2009 · Filed Under Diabetes, General, fda
Novartis this morning said the FDA approved the sale of its Ilaris drug, one of the world’ s most expensive drug treatments, which targets a rare disease called cryopyrin-associated periodic syndrome (CAPS), and potentially some others.
That event may focus traders’ attention on Novartis development partner Regeneron Pharma (REGN), which has a competing treatment for the same CAPS disorder.
But what traders need to keep in perspective today is that CAPS is a tiny market; there are only about 7,000 known cases of it worldwide.
And CAPS is a gateway for both dugs to Novartis’ Ilaris and Regeneron’s Arcalyst to eventually target conditions that affect many more people . It’s very possible — if not likely — that the two drugs might not square off in the same large markets down the road, pending future FDA approvals.
Arcalyst is seen as a drug that can target acute illness such as gout. In contrast, Ilaris may be better suited to chronic diseases, such as diabetes and chronic obstructive pulmonary disorder.

mfg ipollit
Neben dem eigenen zugelassenen Produkt Arcalyst bekommt REGN auch Royalties von bis zu 15% auf Umsätze mit Novartis Ilaris, das vor Kurzen zugelassen wurde. Schön wäre es, wenn Arcalyst nächstes Jahr positive PIII-Daten gegen Gicht erzielen würde. Die aktuelle Indikation CAPS ist sehr selten und bringt daher nicht viel Umsatz. Ilaris befindet sich bei Novartis in entsprechenden Studien, die es zu einem Blockbuster machen können.
Novartis drug appears to compete with partner Regeneron (REGN)
June 19, 2009 · Filed Under Diabetes, General, fda
Novartis this morning said the FDA approved the sale of its Ilaris drug, one of the world’ s most expensive drug treatments, which targets a rare disease called cryopyrin-associated periodic syndrome (CAPS), and potentially some others.
That event may focus traders’ attention on Novartis development partner Regeneron Pharma (REGN), which has a competing treatment for the same CAPS disorder.
But what traders need to keep in perspective today is that CAPS is a tiny market; there are only about 7,000 known cases of it worldwide.
And CAPS is a gateway for both dugs to Novartis’ Ilaris and Regeneron’s Arcalyst to eventually target conditions that affect many more people . It’s very possible — if not likely — that the two drugs might not square off in the same large markets down the road, pending future FDA approvals.
Arcalyst is seen as a drug that can target acute illness such as gout. In contrast, Ilaris may be better suited to chronic diseases, such as diabetes and chronic obstructive pulmonary disorder.
mfg ipollit
Antwort auf Beitrag Nr.: 37.635.908 von ipollit am 23.07.09 21:23:58ein paar weitere Folien zu Regeneron von der letzten Needham Konferenz (11.6.2009)...
interessant sind die sehr guten Entwicklungspartnerschaften mit Sanofi/Aventis für das Avastin ähnliche Aflibercept und mit Bayer für das Lucentis ähnliche VEGF Trap-Eye. Bei den Traps handelt es sich um keine AKs, sondern um lösliche Rezeptoren, die die entsprechenden Stoffe binden. Nächstes Jahr wird es mehrere PIII-Ergebnisse geben. Zudem gibt es eine große AK-Kooperation mit Sonofi-Aventis, eine eigene AK-Technologie sowie eine eigene AK-Produktionsstätte.
http://investor.regeneron.com/phoenix.zhtml?c=119576&p=irol-…




mfg ipollit
interessant sind die sehr guten Entwicklungspartnerschaften mit Sanofi/Aventis für das Avastin ähnliche Aflibercept und mit Bayer für das Lucentis ähnliche VEGF Trap-Eye. Bei den Traps handelt es sich um keine AKs, sondern um lösliche Rezeptoren, die die entsprechenden Stoffe binden. Nächstes Jahr wird es mehrere PIII-Ergebnisse geben. Zudem gibt es eine große AK-Kooperation mit Sonofi-Aventis, eine eigene AK-Technologie sowie eine eigene AK-Produktionsstätte.
http://investor.regeneron.com/phoenix.zhtml?c=119576&p=irol-…
mfg ipollit
Antwort auf Beitrag Nr.: 37.636.008 von ipollit am 23.07.09 21:32:50First Patient Enrolled in Regeneron and Bayer HealthCare VEGF Trap-Eye Phase 3 Program in Central Retinal Vein Occlusion
Regeneron received $20 million milestone payment from Bayer HealthCare with dosing of first patient
Phase 2 study of VEGF Trap-Eye in diabetic macular edema fully enrolled
On Thursday July 23, 2009, 7:00 am EDT
TARRYTOWN, N.Y.--(BUSINESS WIRE)--Regeneron Pharmaceuticals, Inc. (NASDAQ:REGN - News) today announced that the first patient has been enrolled in the Phase 3 program of VEGF Trap-Eye for the treatment of central retinal vein occlusion (CRVO), a leading cause of blindness in adults. Regeneron received a $20 million milestone payment from Bayer Healthcare that was triggered by the dosing of the first patient in the CRVO program. Regeneron also announced that enrollment in the Phase 2 DA VINCI study of VEGF Trap-Eye in diabetic macular edema (DME) has been completed and data are expected during the first half of 2010.
VEGF Trap-Eye, an investigational drug, is being developed by Regeneron and Bayer HealthCare AG for the potential treatment of eye diseases, including the neovascular form of age-related macular degeneration (wet AMD), DME, and CRVO.
The Phase 3 program in CRVO consists of two multinational, one-year clinical studies. The COPERNICUS (COntrolled Phase 3 Evaluation of Repeated iNtravitreal administration of VEGF Trap-Eye In Central retinal vein occlusion: Utility and Safety) study is being led by Regeneron and the GALILEO (General Assessment Limiting InfiLtration of Exudates in central retinal vein Occlusion with VEGF Trap-Eye) study is being led by Bayer HealthCare. Patients in both studies will receive six monthly intravitreal injections of either VEGF Trap-Eye at a dose of 2 milligrams (mg) or sham control injections. The primary endpoint of both studies is improvement in visual acuity versus baseline after six months of treatment. At the end of the initial six months, patients will be dosed on a PRN (as needed) basis for another six months. All patients will be eligible for rescue laser treatment. Results from both CRVO studies are expected in 2011.
In wet AMD, Regeneron and Bayer Healthcare are evaluating VEGF Trap-Eye in two ongoing Phase 3 studies, known as VIEW 1 and VIEW 2 (VEGF Trap: Investigation of Efficacy and Safety in Wet age-related macular degeneration). Enrollment in these trials is expected to be completed by the end of this year, and data are expected in late 2010.
Regeneron maintains exclusive rights to VEGF Trap-Eye in the United States. Bayer HealthCare has exclusive rights to market VEGF Trap-Eye outside the United States, where the companies will share equally in profits from any future sales of VEGF Trap-Eye.
mfg ipollit
Regeneron received $20 million milestone payment from Bayer HealthCare with dosing of first patient
Phase 2 study of VEGF Trap-Eye in diabetic macular edema fully enrolled
On Thursday July 23, 2009, 7:00 am EDT
TARRYTOWN, N.Y.--(BUSINESS WIRE)--Regeneron Pharmaceuticals, Inc. (NASDAQ:REGN - News) today announced that the first patient has been enrolled in the Phase 3 program of VEGF Trap-Eye for the treatment of central retinal vein occlusion (CRVO), a leading cause of blindness in adults. Regeneron received a $20 million milestone payment from Bayer Healthcare that was triggered by the dosing of the first patient in the CRVO program. Regeneron also announced that enrollment in the Phase 2 DA VINCI study of VEGF Trap-Eye in diabetic macular edema (DME) has been completed and data are expected during the first half of 2010.
VEGF Trap-Eye, an investigational drug, is being developed by Regeneron and Bayer HealthCare AG for the potential treatment of eye diseases, including the neovascular form of age-related macular degeneration (wet AMD), DME, and CRVO.
The Phase 3 program in CRVO consists of two multinational, one-year clinical studies. The COPERNICUS (COntrolled Phase 3 Evaluation of Repeated iNtravitreal administration of VEGF Trap-Eye In Central retinal vein occlusion: Utility and Safety) study is being led by Regeneron and the GALILEO (General Assessment Limiting InfiLtration of Exudates in central retinal vein Occlusion with VEGF Trap-Eye) study is being led by Bayer HealthCare. Patients in both studies will receive six monthly intravitreal injections of either VEGF Trap-Eye at a dose of 2 milligrams (mg) or sham control injections. The primary endpoint of both studies is improvement in visual acuity versus baseline after six months of treatment. At the end of the initial six months, patients will be dosed on a PRN (as needed) basis for another six months. All patients will be eligible for rescue laser treatment. Results from both CRVO studies are expected in 2011.
In wet AMD, Regeneron and Bayer Healthcare are evaluating VEGF Trap-Eye in two ongoing Phase 3 studies, known as VIEW 1 and VIEW 2 (VEGF Trap: Investigation of Efficacy and Safety in Wet age-related macular degeneration). Enrollment in these trials is expected to be completed by the end of this year, and data are expected in late 2010.
Regeneron maintains exclusive rights to VEGF Trap-Eye in the United States. Bayer HealthCare has exclusive rights to market VEGF Trap-Eye outside the United States, where the companies will share equally in profits from any future sales of VEGF Trap-Eye.
mfg ipollit
Antwort auf Beitrag Nr.: 37.624.881 von ipollit am 22.07.09 17:49:57Kommentar zu ONXX Nexavar...
http://www.pharmastrategyblog.com/
July 22, 2009
Oncology R&D can be a wild ride
Earlier this year, Pfizer cancelled two major phase III trials in breast cancer involving their multi-kinase inhibitor, Sutent (sunitinib). SUN 1107 evaluated single-agent sunitinib versus single-agent capecitabine for the treatment of a broad range of patients with advanced breast cancer after failure of standard treatment. In addition, SUN 1094, a Phase 3 study, evaluated Sutent plus paclitaxel versus bevacizumab plus paclitaxel for the first line treatment of patients with advanced breast cancer.
In both cases, the independent Data Monitoring Committee (DMC) found that if treatment with sunitinib continued, it would be unable to meet the primary endpoint of superior progression-free survival (PFS). Imagine the surprise of many then, when a Reuters press release hit the wires today stating:
"Nexavar, when combined with standard chemotherapy Xeloda, helped keep tumours in check for longer than in a control group of patients receiving Xeloda only, the drugmakers said in a joint statement on Wednesday, citing data from a Phase II study."
Wow, what a wild ride R&D is sometimes.
What's common about these two drugs? Well, they both inhibit multiple kinases, including VEGF, and both were tested in combination with capecitabine in breast cancer. It should be noted however, that Sutent showed similar promise in phase II, only for the phase III interim analysis to be resoundingly negative. It will be interesting to see how Nexavar fares, because there are no guarantees that the large scale trials will support the earlier data, as Pfizer found.
It is possible that Bayer/Onyx chose a different population of women with breast cancer. My eye was drawn to a hidden snippet in the press release that said:
"The trial involved 229 women with breast tumours that had started spreading."
This is important to note because the mechanism of action for VEGF inhibitors is via anti-angiogenesis when the tumours are sizeable enough that they need to grow by increased blood vessels and vascularisation. In the recent adjuvant colorectal trials looking at FOLFOX plus Avastin, it was clear that while Avastin (another VEGF inhibitor) worked in metastatic disease, it has significantly less value in the earlier setting before the tumour begins to vascularise.
It is therefor possible that the women in the Bayer/Onyx trial may be at a slightly later stage of tumour growth than the women in the Pfizer trials, which would likely make a better target for a VEGF inhibitor. I wonder. We shall see if this hypothesis is correct as more details emerge over the next 18 months.
****
mfg ipollit
http://www.pharmastrategyblog.com/
July 22, 2009
Oncology R&D can be a wild ride
Earlier this year, Pfizer cancelled two major phase III trials in breast cancer involving their multi-kinase inhibitor, Sutent (sunitinib). SUN 1107 evaluated single-agent sunitinib versus single-agent capecitabine for the treatment of a broad range of patients with advanced breast cancer after failure of standard treatment. In addition, SUN 1094, a Phase 3 study, evaluated Sutent plus paclitaxel versus bevacizumab plus paclitaxel for the first line treatment of patients with advanced breast cancer.
In both cases, the independent Data Monitoring Committee (DMC) found that if treatment with sunitinib continued, it would be unable to meet the primary endpoint of superior progression-free survival (PFS). Imagine the surprise of many then, when a Reuters press release hit the wires today stating:
"Nexavar, when combined with standard chemotherapy Xeloda, helped keep tumours in check for longer than in a control group of patients receiving Xeloda only, the drugmakers said in a joint statement on Wednesday, citing data from a Phase II study."
Wow, what a wild ride R&D is sometimes.
What's common about these two drugs? Well, they both inhibit multiple kinases, including VEGF, and both were tested in combination with capecitabine in breast cancer. It should be noted however, that Sutent showed similar promise in phase II, only for the phase III interim analysis to be resoundingly negative. It will be interesting to see how Nexavar fares, because there are no guarantees that the large scale trials will support the earlier data, as Pfizer found.
It is possible that Bayer/Onyx chose a different population of women with breast cancer. My eye was drawn to a hidden snippet in the press release that said:
"The trial involved 229 women with breast tumours that had started spreading."
This is important to note because the mechanism of action for VEGF inhibitors is via anti-angiogenesis when the tumours are sizeable enough that they need to grow by increased blood vessels and vascularisation. In the recent adjuvant colorectal trials looking at FOLFOX plus Avastin, it was clear that while Avastin (another VEGF inhibitor) worked in metastatic disease, it has significantly less value in the earlier setting before the tumour begins to vascularise.
It is therefor possible that the women in the Bayer/Onyx trial may be at a slightly later stage of tumour growth than the women in the Pfizer trials, which would likely make a better target for a VEGF inhibitor. I wonder. We shall see if this hypothesis is correct as more details emerge over the next 18 months.
****
mfg ipollit
SGEN...
Seattle Genetics Reports Strong Pipeline Progress and Second Quarter 2009 Financial Results
Company expects to complete enrollment in pivotal trial of brentuximab vedotin (SGN-35) during the third quarter of 2009
On Thursday July 23, 2009, 4:15 pm EDT
BOTHELL, Wash.--(BUSINESS WIRE)--Seattle Genetics, Inc. (Nasdaq: SGEN - News) today reported financial results for the second quarter and six months ended June 30, 2009. The company also highlighted recent product development progress.
“The second quarter featured strong clinical data presentations with SGN-35, now named brentuximab vedotin, that continue to demonstrate a compelling objective response rate and tolerability profile in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma (ALCL),” said Clay B. Siegall, Ph.D., President and Chief Executive Officer of Seattle Genetics. “In both of our phase I clinical trials, brentuximab vedotin achieved complete or partial responses in greater than 50 percent of patients treated at the higher dose levels. These data reinforce the potential of brentuximab vedotin and are supportive of our aggressive development plans. We expect to complete accrual to the ongoing pivotal trial for Hodgkin lymphoma in the third quarter of 2009, and we also recently initiated a phase II trial in systemic ALCL. We ended the second quarter financially strong, with $190 million in cash and investments, including a $4 million upfront payment received under our new antibody-drug conjugate (ADC) collaboration with Millennium: The Takeda Oncology Company. We anticipate continued milestone momentum across our product pipeline and by our ADC collaborators over the remainder of 2009.”
Recent and Planned Pipeline and ADC Collaborator Highlights
Brentuximab vedotin (SGN-35)
- Given robust enrollment to the Hodgkin lymphoma pivotal trial, expect to complete accrual in the third quarter of 2009
- Reported a median duration of response of at least 7.3 months from an every three week dosing phase I trial (European Hematology Association (EHA) 14th Congress)
- Reported that the objective response rate for patients treated at doses of 1.2 milligrams per kilogram (mg/kg) and higher every three weeks was 54 percent based on investigator assessment, compared to 57 percent based on independent review, demonstrating high concordance between the two assessments (EHA 14th Congress)
- Presented data from an ongoing phase I weekly-dosing clinical trial demonstrating that, among 20 evaluable patients treated at doses of 0.8 mg/kg and higher, 60 percent achieved an objective response, including 50 percent with complete responses (American Society of Clinical Oncology (ASCO) 2009 Annual Meeting)
- In both phase I clinical trials, brentuximab vedotin has been generally well tolerated. The majority of adverse events have been Grade 1 and 2, with the most common being fatigue, fever, peripheral neuropathy, neutropenia, diarrhea and nausea
- Initiated a phase II clinical trial for patients with relapsed or refractory systemic ALCL
- Obtained brentuximab vedotin as the SGN-35 U.S. Adopted Name (USAN), a nonproprietary designation for the product candidate
Preparing to initiate a clinical trial to assess retreatment of patients who previously received brentuximab vedotin therapy
- Planning to present additional phase I data in the second half of 2009
- Evaluating and planning multiple trials of brentuximab vedotin both as a single agent and in combination with chemotherapy for additional relapsed and refractory therapeutic settings, front-line therapy and other CD30-positive malignancies
Dacetuzumab (SGN-40)
- Presented data showing a correlation between a diagnostic gene signature and sensitivity to treatment with dacetuzumab in patients with diffuse large B-cell lymphoma (ASCO 2009 Annual Meeting)
- Expect to report clinical data from multiple ongoing clinical trials of dacetuzumab for non-Hodgkin lymphoma and multiple myeloma later in 2009
Lintuzumab (SGN-33)
- Reported data from a single-agent dose-escalation phase I clinical trial demonstrating multiple objective responses at well-tolerated doses in patients with acute myeloid leukemia (AML) and that 47 percent of AML patients treated across all dose levels experienced reductions in tumor blasts compared to baseline (EHA 14th Congress)
- Continued patient treatment in a randomized phase IIb trial of lintuzumab plus low-dose chemotherapy for patients 60 years and older with AML to determine if the combination extends overall survival. The trial, which is event driven, is expected to yield data in the first half of 2010.
SGN-70
- Initiated treatment of patients with autoimmune disease in a phase I trial, following completion of the healthy volunteer portion of the study
SGN-75
Advanced investigational new drug (IND)-enabling activities towards a planned IND submission in the second half of 2009 for CD70-positive hematologic malignancies and solid tumors
ASG-5ME (formerly AGS-5 ADC)
In collaboration with Agensys, a wholly-owned subsidiary of Astellas Pharma, advanced a novel ADC for solid tumors towards a planned IND submission in the first half of 2010
ADC Collaborations
Entered into a new ADC collaboration with Millennium: The Takeda Oncology Company, under which Seattle Genetics received a $4 million upfront payment and is entitled to receive progress-dependent milestone payments and mid-single digit royalties on any resulting ADC products
Second Quarter and Six Month 2009 Financial Results
Revenues in the second quarter of 2009 were $9.4 million, compared to $10.0 million in the second quarter of 2008. For the first six months of 2009, revenues were $18.6 million, up from $17.1 million in the first six months of 2008. Revenues are primarily driven by the earned portion of the upfront fee, reimbursements and milestone payments received under the company’s dacetuzumab collaboration with Genentech. Revenues also reflect amounts earned under the company’s ADC collaborations.
Total operating expenses for the second quarter of 2009 were $32.7 million, compared to $27.6 million for the second quarter of 2008. For the first six months of 2009, total operating expenses were $70.1 million, compared to $53.7 million in the first six months of 2008. The planned increases in 2009 were primarily driven by clinical development and manufacturing activities for brentuximab vedotin. Non-cash, share-based compensation expense for the first six months of 2009 was $5.4 million, compared to $5.0 million for the same period in 2008.
Net loss for the second quarter of 2009 was $22.5 million, or $0.26 per share, compared to $16.0 million, or $0.20 per share, for the second quarter of 2008. For the six months ended June 30, 2009, net loss was $49.7 million, or $0.59 per share, compared to $33.1 million, or $0.43 per share, for the same period in 2008.
As of June 30, 2009, Seattle Genetics had $189.9 million in cash and investments, compared to $192.4 million as of March 31, 2009. Cash and investments as of June 30, 2009 reflect net proceeds of approximately $11.5 million from the company’s sale of 1,178,163 shares of common stock in a private placement to Baker Brothers Life Sciences, L.P., which was approved at the company’s annual stockholders meeting on May 15, 2009.

mfg ipollit
Seattle Genetics Reports Strong Pipeline Progress and Second Quarter 2009 Financial Results
Company expects to complete enrollment in pivotal trial of brentuximab vedotin (SGN-35) during the third quarter of 2009
On Thursday July 23, 2009, 4:15 pm EDT
BOTHELL, Wash.--(BUSINESS WIRE)--Seattle Genetics, Inc. (Nasdaq: SGEN - News) today reported financial results for the second quarter and six months ended June 30, 2009. The company also highlighted recent product development progress.
“The second quarter featured strong clinical data presentations with SGN-35, now named brentuximab vedotin, that continue to demonstrate a compelling objective response rate and tolerability profile in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma (ALCL),” said Clay B. Siegall, Ph.D., President and Chief Executive Officer of Seattle Genetics. “In both of our phase I clinical trials, brentuximab vedotin achieved complete or partial responses in greater than 50 percent of patients treated at the higher dose levels. These data reinforce the potential of brentuximab vedotin and are supportive of our aggressive development plans. We expect to complete accrual to the ongoing pivotal trial for Hodgkin lymphoma in the third quarter of 2009, and we also recently initiated a phase II trial in systemic ALCL. We ended the second quarter financially strong, with $190 million in cash and investments, including a $4 million upfront payment received under our new antibody-drug conjugate (ADC) collaboration with Millennium: The Takeda Oncology Company. We anticipate continued milestone momentum across our product pipeline and by our ADC collaborators over the remainder of 2009.”
Recent and Planned Pipeline and ADC Collaborator Highlights
Brentuximab vedotin (SGN-35)
- Given robust enrollment to the Hodgkin lymphoma pivotal trial, expect to complete accrual in the third quarter of 2009
- Reported a median duration of response of at least 7.3 months from an every three week dosing phase I trial (European Hematology Association (EHA) 14th Congress)
- Reported that the objective response rate for patients treated at doses of 1.2 milligrams per kilogram (mg/kg) and higher every three weeks was 54 percent based on investigator assessment, compared to 57 percent based on independent review, demonstrating high concordance between the two assessments (EHA 14th Congress)
- Presented data from an ongoing phase I weekly-dosing clinical trial demonstrating that, among 20 evaluable patients treated at doses of 0.8 mg/kg and higher, 60 percent achieved an objective response, including 50 percent with complete responses (American Society of Clinical Oncology (ASCO) 2009 Annual Meeting)
- In both phase I clinical trials, brentuximab vedotin has been generally well tolerated. The majority of adverse events have been Grade 1 and 2, with the most common being fatigue, fever, peripheral neuropathy, neutropenia, diarrhea and nausea
- Initiated a phase II clinical trial for patients with relapsed or refractory systemic ALCL
- Obtained brentuximab vedotin as the SGN-35 U.S. Adopted Name (USAN), a nonproprietary designation for the product candidate
Preparing to initiate a clinical trial to assess retreatment of patients who previously received brentuximab vedotin therapy
- Planning to present additional phase I data in the second half of 2009
- Evaluating and planning multiple trials of brentuximab vedotin both as a single agent and in combination with chemotherapy for additional relapsed and refractory therapeutic settings, front-line therapy and other CD30-positive malignancies
Dacetuzumab (SGN-40)
- Presented data showing a correlation between a diagnostic gene signature and sensitivity to treatment with dacetuzumab in patients with diffuse large B-cell lymphoma (ASCO 2009 Annual Meeting)
- Expect to report clinical data from multiple ongoing clinical trials of dacetuzumab for non-Hodgkin lymphoma and multiple myeloma later in 2009
Lintuzumab (SGN-33)
- Reported data from a single-agent dose-escalation phase I clinical trial demonstrating multiple objective responses at well-tolerated doses in patients with acute myeloid leukemia (AML) and that 47 percent of AML patients treated across all dose levels experienced reductions in tumor blasts compared to baseline (EHA 14th Congress)
- Continued patient treatment in a randomized phase IIb trial of lintuzumab plus low-dose chemotherapy for patients 60 years and older with AML to determine if the combination extends overall survival. The trial, which is event driven, is expected to yield data in the first half of 2010.
SGN-70
- Initiated treatment of patients with autoimmune disease in a phase I trial, following completion of the healthy volunteer portion of the study
SGN-75
Advanced investigational new drug (IND)-enabling activities towards a planned IND submission in the second half of 2009 for CD70-positive hematologic malignancies and solid tumors
ASG-5ME (formerly AGS-5 ADC)
In collaboration with Agensys, a wholly-owned subsidiary of Astellas Pharma, advanced a novel ADC for solid tumors towards a planned IND submission in the first half of 2010
ADC Collaborations
Entered into a new ADC collaboration with Millennium: The Takeda Oncology Company, under which Seattle Genetics received a $4 million upfront payment and is entitled to receive progress-dependent milestone payments and mid-single digit royalties on any resulting ADC products
Second Quarter and Six Month 2009 Financial Results
Revenues in the second quarter of 2009 were $9.4 million, compared to $10.0 million in the second quarter of 2008. For the first six months of 2009, revenues were $18.6 million, up from $17.1 million in the first six months of 2008. Revenues are primarily driven by the earned portion of the upfront fee, reimbursements and milestone payments received under the company’s dacetuzumab collaboration with Genentech. Revenues also reflect amounts earned under the company’s ADC collaborations.
Total operating expenses for the second quarter of 2009 were $32.7 million, compared to $27.6 million for the second quarter of 2008. For the first six months of 2009, total operating expenses were $70.1 million, compared to $53.7 million in the first six months of 2008. The planned increases in 2009 were primarily driven by clinical development and manufacturing activities for brentuximab vedotin. Non-cash, share-based compensation expense for the first six months of 2009 was $5.4 million, compared to $5.0 million for the same period in 2008.
Net loss for the second quarter of 2009 was $22.5 million, or $0.26 per share, compared to $16.0 million, or $0.20 per share, for the second quarter of 2008. For the six months ended June 30, 2009, net loss was $49.7 million, or $0.59 per share, compared to $33.1 million, or $0.43 per share, for the same period in 2008.
As of June 30, 2009, Seattle Genetics had $189.9 million in cash and investments, compared to $192.4 million as of March 31, 2009. Cash and investments as of June 30, 2009 reflect net proceeds of approximately $11.5 million from the company’s sale of 1,178,163 shares of common stock in a private placement to Baker Brothers Life Sciences, L.P., which was approved at the company’s annual stockholders meeting on May 15, 2009.
mfg ipollit
RIGL... leider ist die PII TASKi-3 fehlgeschlagen, was allerdings nicht völlig überraschend kommt. wichtiger waren TASKIi-1+2, da diese auf die eigentliche Zielgruppe ausgerichtet sind. Bei TASKi-3 wurde dagegen geprüft, ob R788 auch bei RA-Patienten wirksam ist, bei denen zuvor mindestens ein intravenöses Biologika nicht mehr wirkt. Eigentlich sollte das orale R788 aber eine Alternative zu den intravenösen Mitteln sein und wegen der leichteren Anwendung vor diesen zum Einsatz kommen. Pfizer hat hier CP-690550 erst garnicht getestet, so dass der Fehler meiner Meinung nach nicht so gravierend ist.
*********
R788 in TASKi3 Clinical Trial Does Not Meet Efficacy Endpoints in RA Patients Who Had Previously Failed Biologic Therapies - Results Incongruent
Bone MRI Scans Show Improvement; Safety Results Consistent with TASKi2
On Thursday July 23, 2009, 6:00 pm EDT
SOUTH SAN FRANCISCO, Calif., July 23 /PRNewswire-FirstCall/ -- Rigel Pharmaceuticals, Inc. (Nasdaq: RIGL - News) today announced that in the TASKi3 Phase 2b clinical trial in rheumatoid arthritis (RA) patients who had failed to respond to at least one biologic treatment, the group treated with R788 (fostamatinib disodium) did not report significantly higher ACR 20, ACR 50, ACR 70 and DAS28 response rates than the placebo group at three months, and therefore, the trial failed to meet its efficacy endpoints. The objective components (CRP and ESR)* of these ACR scores did show a statistically significant difference; however, the subjective reported response rate components did not as compared to placebo. Although the ACR scores for the R788 group were within the expected range in this patient population, the reported placebo response rates were considerably higher than seen in any other previous study of RA biologic failure patients and rose unaccountably between week 6 (at which point the reported response rates between R788 and placebo were significantly different) and month 3 (when such reported response rates were no longer significantly different).
TASKi3 was the first clinical trial evaluating R788 in which anatomical changes in the patients' wrist and hands were evaluated using Magnetic Resonance Imaging (MRI) and scored using the RAMRIS (Rheumatoid Arthritis Magnetic Resonance Imaging Scoring) system. Those results showed improvements in the treated group versus the placebo group in the Synovitis and Osteitis scores, while the Erosion scores, known to be the slowest to change, showed no significant effect at three months. The most frequent adverse events were as expected from the earlier TASKi trials and appear to be manageable.
Rigel will host a conference call today at 7PM EDT/ 4PM PDT to discuss these results (see conference call details below).
"Our objective with R788 in RA is to position the product after methotrexate and before biological therapies are used. We have shown excellent results in that patient population in our earlier TASKi1 and TASKi2 studies, and we believe that patient population represents the large market opportunity for this product," said James M. Gower, chairman and chief executive officer of Rigel. "In this TASKi3 patient population, biologic failures, we have seen divergent results as sometimes happens in studies with subjective components. However, we are pleased to see excellent results in the objective measures and in the Synovitis and Osteitis MRI scores," he added.
*blood measurements of C-Reactive Protein (CRP) and Erythrocyte Sedimentation Rate (ESR)
Efficacy Results
Treatment N of Pts ACR 20 ACR 50 ACR 70 DAS28<2.6
--------- -------- ------ ------ ------ ---------
Placebo 73 27 (37%) 9 (12%) 4 (6%) 6 (10%)
------- --- ------- ------ ----- ------
100 mg bid 146 56 (38%) 32 (22%) 13 (9%) 15 (12%)
p=0.84 p=0.09 p=0.37 p=0.15
---------- --- ------------ ------------ ------------ -----------
p=values compared to placebo
Note: At 3 months. All patients were on stable doses of methotrexate
throughout the clinical trial.
MRI Results
TASKi3 Mean Change from Baseline in RAMRIS* Scores at Month 3
Placebo 100 mg bid p=values
------- ---------- --------
Synovitis** +0.35 -0.52 p=0.038
---------- ----- ----- -------
Osteitis**
Score +1.17 -0.19 p=0.058
---------- ----- ----- -------
Erosion Score +0.94 +0.78 p=0.62
------------- ----- ----- ------
* RAMRIS is a rheumatoid arthritis scoring system utilizing magnetic
resonance imaging to evaluate abnormalities (synovitis, bone edema
and bone erosion) in the hands and wrists. The system was developed
by OMERACT, (Outcome Measurements in Rheumatology) in 2002, and has
become a global standard measurement of inflammation and destruction
in those joints. For these scores a lower value indicates a better
clinical condition.
** Synovitis: inflammation of the synovial membrane lining joints
Osteitis: inflammation of the bone
Safety Results
Similar to TASKi2, the most common clinically meaningful drug-related adverse events noted in TASKi3 were diarrhea and hypertension. Dose reduction options were pre-specified in the trial protocol and, in cases where doses were reduced, patients generally completed the clinical trial with minimal safety issues. The most common adverse events in the trial overall were related to infections, though these were generally evenly distributed among the placebo and active dose group.
The mean increase in blood pressure from baseline at 3 months, using a last observation carry forward methodology, was 3.2-3.6 mmHg for the 100 mg bid dose group. In TASKi3, approximately 17% of patients in the 100 mg bid dose group had blood pressure medication adjusted or in some cases initiated during the course of the clinical trial, compared to 8% of the placebo patients. For those patients who had their dose of blood pressure medications adjusted or initiated, their blood pressure was successfully reduced and their blood pressure was generally well controlled throughout the trial. The blood pressure medications were standard doses of common blood pressure medications such as ACE inhibitors or diuretics.
"For this patient population, patients who failed biologic therapies, their bones and joints appear to respond to R788, but the objective and subjective components of the ACR and DAS28 scores are incongruent, mainly because the reported subjective placebo response rates were higher than expected," said Elliott Grossbard, M.D., chief medical officer for Rigel. "Nonetheless, R788 is well tolerated and its side effects appear generally manageable, and we look forward to planning our Phase 3 program for R788 with a corporate partner," he added.
Trial Design
TASKi3 was a 3 month, multi-center, randomized, double blind, placebo controlled, parallel dose clinical trial involving 219 RA patients in the U.S. who had failed to respond to at least one biologic treatment (such as TNF inhibitors). The patients were randomly assigned to two cohorts and thus received R788 orally in a 100 mg bid (twice daily) dose or placebo for a period of up to 3 months. Patients were assigned on a 2:1 basis to R788 or placebo. Throughout the clinical trial period, all of the patients continued to receive their stable dose of methotrexate.
Efficacy assessments for each participant were based on the American College of Rheumatology criteria, which denotes at least a 20% (ACR 20) improvement, at least a 50% (ACR 50) improvement, or at least a 70% (ACR 70) improvement, from the baseline assessment at the end of the 3 month treatment period. The ACR measurement factors included reported physician and patient global assessment of disease activity, patient reported pain score, and any change in CRP in the patient's blood. The primary efficacy endpoint for the clinical trial was the percent of patients assigned to the R788 100 mg bid dose who were ACR 20 responders at the end of 3 months. Secondary efficacy endpoints included other ACR scores and, a comparison of response rates for the R788 100 mg bid dose versus placebo as determined by MRI using the modified RAMRIS scoring system of wrists and hands at baseline and at month 3.
R788 and RA
RA is a progressive, painful and potentially debilitating disease, that affects more than 2 million people in the U.S. It is a chronic inflammatory disease that puts the body's immune system into overdrive where it ultimately causes inflammation in the joints and destroys soft tissues, cartilage and bone. Rigel's R788 is a novel, orally available syk kinase inhibitor designed to interrupt the cellular signaling at the trigger point of inflammation, thereby stopping the progression of the disease. In July 2009, Rigel announced results from its Phase 2b TASKi2 clinical trial showing significant improvement in RA patients treated with R788 who had failed to respond to methotrexate treatment.
****
bereits vor Bekanntgabe der Ergebnisse ging es deutlich nach unten...

mfg ipollit
*********
R788 in TASKi3 Clinical Trial Does Not Meet Efficacy Endpoints in RA Patients Who Had Previously Failed Biologic Therapies - Results Incongruent
Bone MRI Scans Show Improvement; Safety Results Consistent with TASKi2
On Thursday July 23, 2009, 6:00 pm EDT
SOUTH SAN FRANCISCO, Calif., July 23 /PRNewswire-FirstCall/ -- Rigel Pharmaceuticals, Inc. (Nasdaq: RIGL - News) today announced that in the TASKi3 Phase 2b clinical trial in rheumatoid arthritis (RA) patients who had failed to respond to at least one biologic treatment, the group treated with R788 (fostamatinib disodium) did not report significantly higher ACR 20, ACR 50, ACR 70 and DAS28 response rates than the placebo group at three months, and therefore, the trial failed to meet its efficacy endpoints. The objective components (CRP and ESR)* of these ACR scores did show a statistically significant difference; however, the subjective reported response rate components did not as compared to placebo. Although the ACR scores for the R788 group were within the expected range in this patient population, the reported placebo response rates were considerably higher than seen in any other previous study of RA biologic failure patients and rose unaccountably between week 6 (at which point the reported response rates between R788 and placebo were significantly different) and month 3 (when such reported response rates were no longer significantly different).
TASKi3 was the first clinical trial evaluating R788 in which anatomical changes in the patients' wrist and hands were evaluated using Magnetic Resonance Imaging (MRI) and scored using the RAMRIS (Rheumatoid Arthritis Magnetic Resonance Imaging Scoring) system. Those results showed improvements in the treated group versus the placebo group in the Synovitis and Osteitis scores, while the Erosion scores, known to be the slowest to change, showed no significant effect at three months. The most frequent adverse events were as expected from the earlier TASKi trials and appear to be manageable.
Rigel will host a conference call today at 7PM EDT/ 4PM PDT to discuss these results (see conference call details below).
"Our objective with R788 in RA is to position the product after methotrexate and before biological therapies are used. We have shown excellent results in that patient population in our earlier TASKi1 and TASKi2 studies, and we believe that patient population represents the large market opportunity for this product," said James M. Gower, chairman and chief executive officer of Rigel. "In this TASKi3 patient population, biologic failures, we have seen divergent results as sometimes happens in studies with subjective components. However, we are pleased to see excellent results in the objective measures and in the Synovitis and Osteitis MRI scores," he added.
*blood measurements of C-Reactive Protein (CRP) and Erythrocyte Sedimentation Rate (ESR)
Efficacy Results
Treatment N of Pts ACR 20 ACR 50 ACR 70 DAS28<2.6
--------- -------- ------ ------ ------ ---------
Placebo 73 27 (37%) 9 (12%) 4 (6%) 6 (10%)
------- --- ------- ------ ----- ------
100 mg bid 146 56 (38%) 32 (22%) 13 (9%) 15 (12%)
p=0.84 p=0.09 p=0.37 p=0.15
---------- --- ------------ ------------ ------------ -----------
p=values compared to placebo
Note: At 3 months. All patients were on stable doses of methotrexate
throughout the clinical trial.
MRI Results
TASKi3 Mean Change from Baseline in RAMRIS* Scores at Month 3
Placebo 100 mg bid p=values
------- ---------- --------
Synovitis** +0.35 -0.52 p=0.038
---------- ----- ----- -------
Osteitis**
Score +1.17 -0.19 p=0.058
---------- ----- ----- -------
Erosion Score +0.94 +0.78 p=0.62
------------- ----- ----- ------
* RAMRIS is a rheumatoid arthritis scoring system utilizing magnetic
resonance imaging to evaluate abnormalities (synovitis, bone edema
and bone erosion) in the hands and wrists. The system was developed
by OMERACT, (Outcome Measurements in Rheumatology) in 2002, and has
become a global standard measurement of inflammation and destruction
in those joints. For these scores a lower value indicates a better
clinical condition.
** Synovitis: inflammation of the synovial membrane lining joints
Osteitis: inflammation of the bone
Safety Results
Similar to TASKi2, the most common clinically meaningful drug-related adverse events noted in TASKi3 were diarrhea and hypertension. Dose reduction options were pre-specified in the trial protocol and, in cases where doses were reduced, patients generally completed the clinical trial with minimal safety issues. The most common adverse events in the trial overall were related to infections, though these were generally evenly distributed among the placebo and active dose group.
The mean increase in blood pressure from baseline at 3 months, using a last observation carry forward methodology, was 3.2-3.6 mmHg for the 100 mg bid dose group. In TASKi3, approximately 17% of patients in the 100 mg bid dose group had blood pressure medication adjusted or in some cases initiated during the course of the clinical trial, compared to 8% of the placebo patients. For those patients who had their dose of blood pressure medications adjusted or initiated, their blood pressure was successfully reduced and their blood pressure was generally well controlled throughout the trial. The blood pressure medications were standard doses of common blood pressure medications such as ACE inhibitors or diuretics.
"For this patient population, patients who failed biologic therapies, their bones and joints appear to respond to R788, but the objective and subjective components of the ACR and DAS28 scores are incongruent, mainly because the reported subjective placebo response rates were higher than expected," said Elliott Grossbard, M.D., chief medical officer for Rigel. "Nonetheless, R788 is well tolerated and its side effects appear generally manageable, and we look forward to planning our Phase 3 program for R788 with a corporate partner," he added.
Trial Design
TASKi3 was a 3 month, multi-center, randomized, double blind, placebo controlled, parallel dose clinical trial involving 219 RA patients in the U.S. who had failed to respond to at least one biologic treatment (such as TNF inhibitors). The patients were randomly assigned to two cohorts and thus received R788 orally in a 100 mg bid (twice daily) dose or placebo for a period of up to 3 months. Patients were assigned on a 2:1 basis to R788 or placebo. Throughout the clinical trial period, all of the patients continued to receive their stable dose of methotrexate.
Efficacy assessments for each participant were based on the American College of Rheumatology criteria, which denotes at least a 20% (ACR 20) improvement, at least a 50% (ACR 50) improvement, or at least a 70% (ACR 70) improvement, from the baseline assessment at the end of the 3 month treatment period. The ACR measurement factors included reported physician and patient global assessment of disease activity, patient reported pain score, and any change in CRP in the patient's blood. The primary efficacy endpoint for the clinical trial was the percent of patients assigned to the R788 100 mg bid dose who were ACR 20 responders at the end of 3 months. Secondary efficacy endpoints included other ACR scores and, a comparison of response rates for the R788 100 mg bid dose versus placebo as determined by MRI using the modified RAMRIS scoring system of wrists and hands at baseline and at month 3.
R788 and RA
RA is a progressive, painful and potentially debilitating disease, that affects more than 2 million people in the U.S. It is a chronic inflammatory disease that puts the body's immune system into overdrive where it ultimately causes inflammation in the joints and destroys soft tissues, cartilage and bone. Rigel's R788 is a novel, orally available syk kinase inhibitor designed to interrupt the cellular signaling at the trigger point of inflammation, thereby stopping the progression of the disease. In July 2009, Rigel announced results from its Phase 2b TASKi2 clinical trial showing significant improvement in RA patients treated with R788 who had failed to respond to methotrexate treatment.
****
bereits vor Bekanntgabe der Ergebnisse ging es deutlich nach unten...
mfg ipollit
Antwort auf Beitrag Nr.: 37.639.086 von ipollit am 24.07.09 11:12:53hier ein Kommentar zur CC...
http://investorshub.advfn.com/boards/read_msg.aspx?message_i…
RIGL - Notes on TASKi3 conference call
I just finished listening to this conference call that was over an hour long and am ready for bed. But first, a few notes (opinions all IMHO):
1. Main point - This trial was in patients who had failed at least 1 biologic, which is a small part of the market. The drug failed its primary endpoint regarding ACR scores, which are subjective, but did succeed on several other biological marker endpoints. More importantly, the TASKi2 trial, which was in methotrexate failures only (the big slice of the market), was clearly a success with respect to the primary endpoint of ACR scores.
2. The trial failed because the placebo rate was so high and not because R788 didn't work as expected. It's still a trial failure but just pointing out the reason for failure. Also, the placebo patients significantly underperformed R788 patients all the way up until the 6 week mark (halfway point) but then caught up to R788 during the final 6 weeks due to dramatic improvement. One caller questioned if perhaps there were treatment protocol violations to account for the dramatic upswing during the last 6 weeks, although RIGL management doesn't expect that to be the case.
3. RIGL will have an end of Phase 2b meeting with the FDA in late October and expects a partner for R788 sometime after this event.
4. Safety - the patient hospitalized for high blood pressure was a placebo patient. Also, there was 1 death in this trial related to septic shock but that too was a placebo patient.
5. RIGL is not abandoning the TNF-failure population, still believes the drug works in this setting (as do the clinical investigators), and expects to further explore this in Phase 3 with a partner. They indicated that R788 was much more efficacious in patients who had just failed 1 biologic as opposed to those who had already failed multiple whereas the placebo was constant across both patient populations.
6. RIGL doesn't believe it will be required to do another TNF-failure study to get approval for R788 but, as previously mentioned, they still expect to pursue this patient population.
7. 85% of the patients who finished treatment with R788 in the trial have elected to continue on the treatment into the open label extension.
mfg ipollit
http://investorshub.advfn.com/boards/read_msg.aspx?message_i…
RIGL - Notes on TASKi3 conference call
I just finished listening to this conference call that was over an hour long and am ready for bed. But first, a few notes (opinions all IMHO):
1. Main point - This trial was in patients who had failed at least 1 biologic, which is a small part of the market. The drug failed its primary endpoint regarding ACR scores, which are subjective, but did succeed on several other biological marker endpoints. More importantly, the TASKi2 trial, which was in methotrexate failures only (the big slice of the market), was clearly a success with respect to the primary endpoint of ACR scores.
2. The trial failed because the placebo rate was so high and not because R788 didn't work as expected. It's still a trial failure but just pointing out the reason for failure. Also, the placebo patients significantly underperformed R788 patients all the way up until the 6 week mark (halfway point) but then caught up to R788 during the final 6 weeks due to dramatic improvement. One caller questioned if perhaps there were treatment protocol violations to account for the dramatic upswing during the last 6 weeks, although RIGL management doesn't expect that to be the case.
3. RIGL will have an end of Phase 2b meeting with the FDA in late October and expects a partner for R788 sometime after this event.
4. Safety - the patient hospitalized for high blood pressure was a placebo patient. Also, there was 1 death in this trial related to septic shock but that too was a placebo patient.
5. RIGL is not abandoning the TNF-failure population, still believes the drug works in this setting (as do the clinical investigators), and expects to further explore this in Phase 3 with a partner. They indicated that R788 was much more efficacious in patients who had just failed 1 biologic as opposed to those who had already failed multiple whereas the placebo was constant across both patient populations.
6. RIGL doesn't believe it will be required to do another TNF-failure study to get approval for R788 but, as previously mentioned, they still expect to pursue this patient population.
7. 85% of the patients who finished treatment with R788 in the trial have elected to continue on the treatment into the open label extension.
mfg ipollit
Antwort auf Beitrag Nr.: 37.635.908 von ipollit am 23.07.09 21:23:58REGN... Zulassung von Arcalyst bei CAPS in der EU empfohlen (CAPS ist eine kleine Indikation, interessanter ist Arcalyst bei Gicht, wozu nächstes Jahr die PIII-Daten kommen)
24.07.2009 13:19
Regeneron Receives European Medicines Agency Positive Opinion for Marketing Authorization of Rilonacept for the Treatment of Cryopyrin-Associated Periodic Syndromes (CAPS)
European marketing approval for this rare orphan condition anticipated in late 2009
Regeneron Pharmaceuticals, Inc. (NASDAQ:REGN) today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMEA) has issued a positive opinion for the marketing authorization in the European Union of rilonacept, an interleukin-1 blocker, for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS) with severe symptoms, including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS), in adults and children aged 12 years and older. The positive opinion recommends the granting of a marketing authorization for rilonacept under exceptional circumstances. Such authorizations are permissible for products for which a company can demonstrate that comprehensive data cannot be provided, for example because of the rarity of the condition. Each year, Regeneron will need to provide the EMEA with any new information that may become available for review.
”We are very pleased to receive the EMEA's positive opinion for rilonacept,” said Leonard S. Schleifer, M.D., Ph.D., Regeneron's president and chief executive officer. ”We recognize that rilonacept may help address a significant unmet medical need that exists among CAPS patients in the European Union and are therefore committed to helping these patients obtain access to this new treatment.”
CAPS are a group of rare, inherited, auto-inflammatory conditions characterized by life-long, recurrent symptoms of rash, fever/chills, joint pain, eye redness/pain, and fatigue. Intermittent, disruptive exacerbations or flares can be triggered at any time by exposure to cooling temperatures, stress, exercise, or other unknown stimuli.
Rilonacept is a targeted inhibitor of interleukin-1 (IL-1), the key driver of inflammation in CAPS. In the pivotal clinical development program, patients treated with rilonacept reported a greater improvement in overall symptom scores than patients treated with placebo. These improvements were sustained over time with continued rilonacept treatment. Patient-reported symptoms assessment, using a validated daily diary instrument, represents a critical measure of effectiveness in a disease characterized by frequent, unpredictable symptom flares of variable severity and duration. Unlike other agents used in the treatment of CAPS, rilonacept is supported by patient-reported symptoms data using a validated assessment instrument.
Rilonacept has been developed as a once-weekly injection which can be administered at home by the patient or their care giver following appropriate training. The most commonly reported adverse reactions with rilonacept were injection-site reaction and upper respiratory tract infection. IL-1 blockade may interfere with immune response to infections. Serious, life-threatening infections have been reported in patients taking rilonacept. Treatment should not be initiated in patients with active or chronic infections. Rilonacept should be discontinued if a patient develops a serious infection.
About Cryopyrin-Associated Periodic Syndromes (CAPS)
Recently, medical researchers have identified and described a group of rare, inherited, auto-inflammatory disorders, known as Cryopyrin-Associated Periodic Syndromes or CAPS. Three related conditions make up the broader disease known as CAPS: Familial Cold Auto-inflammatory Syndrome (FCAS), Muckle-Wells Syndrome (MWS), and Neonatal-Onset Multisystem Inflammatory Disease (NOMID). Rilonacept has not been studied in patients with NOMID.
CAPS are characterized by life-long, recurrent symptoms of rash, fever/chills, joint pain, eye redness/pain, and fatigue. Intermittent, disruptive exacerbations or flares can be triggered at any time by exposure to cooling temperatures, stress, exercise, or other unknown stimuli.
CAPS are generally caused by autosomal-dominant mutations (changes) in the NLRP-3 (previously known as CIAS1) gene and resultant alterations in the protein, cryopyrin, which it encodes. Cryopyrin, active in circulating, infection-fighting, white blood cells, controls the production of a protein called interleukin-1 (IL-1). As part of the body's infection-fighting defense system, IL-1 circulates throughout the body and can trigger inflammatory reactions when it binds to inflammatory cells. Researchers have found that alterations in the cryopyrin protein lead to over-production of IL-1, resulting in an inflammatory response and the symptoms of CAPS. Most, but not all, patients with CAPS have the NLRP-3 gene mutation.
The incidence of CAPS has been estimated to be approximately 1 in 1,000,000 people in the European Union.
About Rilonacept
Rilonacept is a targeted inhibitor of interleukin-1 (IL-1), the key driver of inflammation in Cryopyrin-Associated Periodic Syndromes (CAPS). In the pivotal clinical development program for rilonacept, change in disease activity was measured using a composite patient-reported symptom score composed of a daily evaluation of rash, feelings of fever/chills, joint pain, eye redness/pain, and fatigue. Patients treated with rilonacept experienced an improvement in overall symptom scores as compared with patients treated with placebo. These improvements were sustained over time with continued treatment with rilonacept. The most commonly reported adverse reactions with rilonacept were injection-site reaction and upper respiratory tract infection.
In the United States, rilonacept (ARCALYST®) is indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) in adults and children 12 and older. ARCALYST was approved by the U.S. Food and Drug Administration in February 2008 and has been available in the United States since March 2008.
IL-1 blockade may interfere with immune response to infections. Serious, life-threatening infections have been reported in patients taking ARCALYST. ARCALYST should be discontinued if a patient develops a serious infection. Treatment with ARCALYST should not be initiated in patients with active or chronic infections. Taking ARCALYST with tumor necrosis factor inhibitors is not recommended because this may increase the risk of serious infections. Patients should not receive a live vaccine while taking ARCALYST. It is recommended that patients receive all recommended vaccinations prior to initiation of treatment with ARCALYST. Patients should be monitored for changes in their lipid profiles and provided with medical treatment if warranted. Hypersensitivity reactions associated with ARCALYST® (rilonacept) administration have been rare. Please see the full U.S. Prescribing Information for ARCALYST, available online at www.regeneron.com/ARCALYST-fpi.pdf
mfg ipollit
24.07.2009 13:19
Regeneron Receives European Medicines Agency Positive Opinion for Marketing Authorization of Rilonacept for the Treatment of Cryopyrin-Associated Periodic Syndromes (CAPS)
European marketing approval for this rare orphan condition anticipated in late 2009
Regeneron Pharmaceuticals, Inc. (NASDAQ:REGN) today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMEA) has issued a positive opinion for the marketing authorization in the European Union of rilonacept, an interleukin-1 blocker, for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS) with severe symptoms, including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS), in adults and children aged 12 years and older. The positive opinion recommends the granting of a marketing authorization for rilonacept under exceptional circumstances. Such authorizations are permissible for products for which a company can demonstrate that comprehensive data cannot be provided, for example because of the rarity of the condition. Each year, Regeneron will need to provide the EMEA with any new information that may become available for review.
”We are very pleased to receive the EMEA's positive opinion for rilonacept,” said Leonard S. Schleifer, M.D., Ph.D., Regeneron's president and chief executive officer. ”We recognize that rilonacept may help address a significant unmet medical need that exists among CAPS patients in the European Union and are therefore committed to helping these patients obtain access to this new treatment.”
CAPS are a group of rare, inherited, auto-inflammatory conditions characterized by life-long, recurrent symptoms of rash, fever/chills, joint pain, eye redness/pain, and fatigue. Intermittent, disruptive exacerbations or flares can be triggered at any time by exposure to cooling temperatures, stress, exercise, or other unknown stimuli.
Rilonacept is a targeted inhibitor of interleukin-1 (IL-1), the key driver of inflammation in CAPS. In the pivotal clinical development program, patients treated with rilonacept reported a greater improvement in overall symptom scores than patients treated with placebo. These improvements were sustained over time with continued rilonacept treatment. Patient-reported symptoms assessment, using a validated daily diary instrument, represents a critical measure of effectiveness in a disease characterized by frequent, unpredictable symptom flares of variable severity and duration. Unlike other agents used in the treatment of CAPS, rilonacept is supported by patient-reported symptoms data using a validated assessment instrument.
Rilonacept has been developed as a once-weekly injection which can be administered at home by the patient or their care giver following appropriate training. The most commonly reported adverse reactions with rilonacept were injection-site reaction and upper respiratory tract infection. IL-1 blockade may interfere with immune response to infections. Serious, life-threatening infections have been reported in patients taking rilonacept. Treatment should not be initiated in patients with active or chronic infections. Rilonacept should be discontinued if a patient develops a serious infection.
About Cryopyrin-Associated Periodic Syndromes (CAPS)
Recently, medical researchers have identified and described a group of rare, inherited, auto-inflammatory disorders, known as Cryopyrin-Associated Periodic Syndromes or CAPS. Three related conditions make up the broader disease known as CAPS: Familial Cold Auto-inflammatory Syndrome (FCAS), Muckle-Wells Syndrome (MWS), and Neonatal-Onset Multisystem Inflammatory Disease (NOMID). Rilonacept has not been studied in patients with NOMID.
CAPS are characterized by life-long, recurrent symptoms of rash, fever/chills, joint pain, eye redness/pain, and fatigue. Intermittent, disruptive exacerbations or flares can be triggered at any time by exposure to cooling temperatures, stress, exercise, or other unknown stimuli.
CAPS are generally caused by autosomal-dominant mutations (changes) in the NLRP-3 (previously known as CIAS1) gene and resultant alterations in the protein, cryopyrin, which it encodes. Cryopyrin, active in circulating, infection-fighting, white blood cells, controls the production of a protein called interleukin-1 (IL-1). As part of the body's infection-fighting defense system, IL-1 circulates throughout the body and can trigger inflammatory reactions when it binds to inflammatory cells. Researchers have found that alterations in the cryopyrin protein lead to over-production of IL-1, resulting in an inflammatory response and the symptoms of CAPS. Most, but not all, patients with CAPS have the NLRP-3 gene mutation.
The incidence of CAPS has been estimated to be approximately 1 in 1,000,000 people in the European Union.
About Rilonacept
Rilonacept is a targeted inhibitor of interleukin-1 (IL-1), the key driver of inflammation in Cryopyrin-Associated Periodic Syndromes (CAPS). In the pivotal clinical development program for rilonacept, change in disease activity was measured using a composite patient-reported symptom score composed of a daily evaluation of rash, feelings of fever/chills, joint pain, eye redness/pain, and fatigue. Patients treated with rilonacept experienced an improvement in overall symptom scores as compared with patients treated with placebo. These improvements were sustained over time with continued treatment with rilonacept. The most commonly reported adverse reactions with rilonacept were injection-site reaction and upper respiratory tract infection.
In the United States, rilonacept (ARCALYST®) is indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) in adults and children 12 and older. ARCALYST was approved by the U.S. Food and Drug Administration in February 2008 and has been available in the United States since March 2008.
IL-1 blockade may interfere with immune response to infections. Serious, life-threatening infections have been reported in patients taking ARCALYST. ARCALYST should be discontinued if a patient develops a serious infection. Treatment with ARCALYST should not be initiated in patients with active or chronic infections. Taking ARCALYST with tumor necrosis factor inhibitors is not recommended because this may increase the risk of serious infections. Patients should not receive a live vaccine while taking ARCALYST. It is recommended that patients receive all recommended vaccinations prior to initiation of treatment with ARCALYST. Patients should be monitored for changes in their lipid profiles and provided with medical treatment if warranted. Hypersensitivity reactions associated with ARCALYST® (rilonacept) administration have been rare. Please see the full U.S. Prescribing Information for ARCALYST, available online at www.regeneron.com/ARCALYST-fpi.pdf
mfg ipollit
die Bedeutung von Biotech-Produkten nimmt gegenüber denen von traditionellen Pharmas immer weiter zu. Insbesondere AKs sollen zu der wichtigsten Medikamenten-Klasse für die Pharmas werden mit Avastin, Humira und Rituxan auf den ersten 3 Plätzen in 2014...
http://www.fiercebiotech.com/story/biologics-will-dominate-2…
http://www.evaluatepharma.com/Universal/View.aspx?type=Story…
Biologics will dominate in 2014
June 18, 2009 — 7:52am ET | By Maureen Martino
According to forecasting firm EvaluatePharma, six of the top ten drugs in 2014 will be biologics. Analysts predict that Roche's cancer antibody Avastin will lead the pack at $9.2 billion in annual sales, with Abbott and Eisai's Humira a close second at $9.1 billion in sales. Rituxan (Roche), Enbrel (Wyeth, Amgen and Takeda) Lantus (Sanofi), Herceptin (Roche) and Remicade (J&J and Schering Plough) make up the other biologics that will dominate the market in 2010. Of these seven, only three--Rituxan, Remicade and Avastin--were top 10 drugs in 2008. And not only will biotech drugs make up the majority of the bestsellers, Evaluate predicts that half of the top 100 drugs in 2014 will be biologics.
Noticeably absent from this list are current bestsellers Lipitor, Plavix, Advair, Diovan and Nexium, which will all see sales drop due to generic competition.
"Anti-cancer antibodies appear set to easily become the most valuable therapeutic class of drug, justifying a large proportion of Roche's recent move to acquire Genentech outright." It also validates the trend of Big Pharma players acquiring small and mid-sized biotech now, while valuations are down.
The report underscores how important it is for Big Pharma to invest in biotechnology. While companies like Roche may be sitting pretty, big-name companies like Pfizer and GlaxoSmithKline are missing from the top 10 list in 2014. The "...weight of evidence for a shift to biotech products as the industry's growth driver is overwhelming, making the recent moves by Big pharma to access biotech platforms, not only for generating innovative medicines but also to launch bio-similar products, all the more compelling," observed the firm in its report.

btw... Genmab's Arzerra ist ein potentieller Konkurrent von Rituxan und Regeneron's Aflibercept ein potentieller Konkurrent von Avastin.
mfg ipollit
http://www.fiercebiotech.com/story/biologics-will-dominate-2…
http://www.evaluatepharma.com/Universal/View.aspx?type=Story…
Biologics will dominate in 2014
June 18, 2009 — 7:52am ET | By Maureen Martino
According to forecasting firm EvaluatePharma, six of the top ten drugs in 2014 will be biologics. Analysts predict that Roche's cancer antibody Avastin will lead the pack at $9.2 billion in annual sales, with Abbott and Eisai's Humira a close second at $9.1 billion in sales. Rituxan (Roche), Enbrel (Wyeth, Amgen and Takeda) Lantus (Sanofi), Herceptin (Roche) and Remicade (J&J and Schering Plough) make up the other biologics that will dominate the market in 2010. Of these seven, only three--Rituxan, Remicade and Avastin--were top 10 drugs in 2008. And not only will biotech drugs make up the majority of the bestsellers, Evaluate predicts that half of the top 100 drugs in 2014 will be biologics.
Noticeably absent from this list are current bestsellers Lipitor, Plavix, Advair, Diovan and Nexium, which will all see sales drop due to generic competition.
"Anti-cancer antibodies appear set to easily become the most valuable therapeutic class of drug, justifying a large proportion of Roche's recent move to acquire Genentech outright." It also validates the trend of Big Pharma players acquiring small and mid-sized biotech now, while valuations are down.
The report underscores how important it is for Big Pharma to invest in biotechnology. While companies like Roche may be sitting pretty, big-name companies like Pfizer and GlaxoSmithKline are missing from the top 10 list in 2014. The "...weight of evidence for a shift to biotech products as the industry's growth driver is overwhelming, making the recent moves by Big pharma to access biotech platforms, not only for generating innovative medicines but also to launch bio-similar products, all the more compelling," observed the firm in its report.
btw... Genmab's Arzerra ist ein potentieller Konkurrent von Rituxan und Regeneron's Aflibercept ein potentieller Konkurrent von Avastin.
mfg ipollit
mal was zu Medigene...
http://aktien.wallstreet-online.de/nachricht/2774965-medigen…
MediGene: Ist Amgen der Partner?
Sehnsüchtig wartet die Börse auf einen Entwicklungs- und Vermarktungspartner für den potenziellen Blockbuster EndoTAG-1. Die Substanz hat die Phase II-Studie in der Indikation Bauchspeicheldrüsenkrebs positiv abgeschlossen und soll über eine Partnerschaft weiterentwickelt werden. „Wir befinden uns in den Verpartnerungsgesprächen auf einem guten Weg“, sagt Finanzvorstand Thomas Klaue im Gespräch mit TradeCentre. Wer allerdings darauf hofft, das Unternehmen erhält bei Vertragsabschluss einen hohen zweistelligen Millionenbetrag als Vorabzahlung, wird enttäuscht sein. „Pharmafirmen sind aufgrund der Finanzkrise mit hohen Vorabzahlungen zurückhaltender und teilen das Risiko vermehrt über Meilensteinzahlungen in der weiteren Entwicklungsphase“, erklärt der CFO. Die Deal-Struktur dürfte sich anders gestalten als erhofft. Somit wird MediGene (DE0005020903) bei Vertragsunterschrift schrift eine vergleichsweise geringe Vorabzahlung erhalten und verstärkt Meilensteinzahlungen in der weiteren Entwicklungsphase bis hin zur möglichen Zulassung. Dennoch könnte bei einem Gesamtdeal über Meilensteine aus unserer Sicht ein dreistelliger Millionenbetrag in die Kasse der Bayern fließen. Wunschpartner ist laut Klaue ein in der Biotechnologie tätiges und in der Onkologie starkes Unternehmen, das MediGene als Partner Co-Promotion-Rechte in Europa überlässt. . Das Profil passt zum US-Biotech-Konzern Amgen. Einen Bezug zu Amgen hat die Firma. MediGenes CEO Frank Mathias war einst Deutschland-Chef von Amgen.
Sehr zufrieden zeigt sich der Finanzchef mit der Entwicklung des Produktes Eligard. „Eligard zeigt ein robustes und starkes Wachstum. Der vom Vertriebspartner erzielte Produktumsatz von Eligard könnte in diesem Jahr die Marke von 100 Millionen Euro knacken nach 80 Millionen Euro im Vorjahr“, sagt Klaue. Der auf MediGene entfallende Umsatz könnte die Marke von 35 Millionen Euro überschreiten nach 30 Millionen Euro im Vorjahr. Die Peak-Sales für MediGenes Umsatz aus Eligard sieht der CFO zwischen 40 und 45 Millionen Euro. Mit Veregen hat das Unternehmen ein zweites Produkt am Markt. „Über unseren Partner in den USA erwarten wir ab dem dritten Quartal erste spürbare Effekte und ab 2010 steigende Umsätze“. Für Europa erschließt das Unternehmen einzelne Territorien über verschiedene Partner. „Wir sind in Verhandlungen und erwarten nach dem Abschluss in Spanien weitere Vertriebspartnerschaften“. Erste Erlöse aus europäischen Gefilden sind ab 2010 zu erwarten. Da sich MediGene auf Onkologie und Immunologie fokussiert, passt Veregen nicht mehr zur strategischen Ausrichtung. Klaue schloss eine Veräußerung der Rechte nicht kategorisch aus. „Derzeit ist dies jedoch kein Thema“. Die Rechte an Oracea hat MediGene bereits veräußert. Aus dieser Veräußerung fließen noch bis zu 24 Millionen Euro in die Kasse. Eine erste Tranche soll Anfang 2011 eintreffen. Weitere Meilensteine werden in 2011 und 2012 erwartet.
Auf der Zahlenseite wird das Unternehmen den Vorjahresumsatz von über 39 Millionen Euro aufgrund der Produktumsätze durch Eligard leicht übertreffen. „Das bezieht sich insbesondere auf Eligard. Erlöse aus kommenden Deals sind darin nicht eingerechnet“. Der Verlust beim EBITDA wird sich nach rund minus 25 Millionen Euro im Vorjahr verbessern. „Im ersten Quartal lagen wir bereinigt um Einmaleffekte bei 1,1 Millionen Cashburn pro Monat. In den verbleibenden neun Monaten wird der monatliche Barmittelverbrauch bei circa 1,5 Millionen Euro liegen“. Per Ende März lag der Cashbestand bei 16,6 Millionen Euro. MediGene hat weiteren Zugriff auf 25 Millionen Euro durch Ausgabe neuer Aktien. Damit ist die Firma zunächst finanziert. Immer wieder ist MediGene Opfer der Übernahmegerüchteküche. Alternativ soll seit Jahr und Tag eine Fusion mit dem Biotechunternehmen Wilex geplant sein. „Wir sprechen derzeit weder über Fusionen, noch sprechen wir mit Interessenten für eine komplette Übernahme“, kommentiert Klaue diese Spekulationen.
Der Börsenwert von MediGene beträgt rund 140 Millionen Euro. Im Bereich um vier Euro bildet die Aktie einen stabilen Boden. Bei erfolgreichem Abschluss von EndoTAG-1 dürfte das Papier wach geküsst werden. Spekulativ orientierte Anleger können sich ein paar Stücke ins Depot legen. Den Stoppkurs raten wir circa 15 Prozent unter Einstieg zu platzieren.
mfg ipollit
http://aktien.wallstreet-online.de/nachricht/2774965-medigen…
MediGene: Ist Amgen der Partner?
Sehnsüchtig wartet die Börse auf einen Entwicklungs- und Vermarktungspartner für den potenziellen Blockbuster EndoTAG-1. Die Substanz hat die Phase II-Studie in der Indikation Bauchspeicheldrüsenkrebs positiv abgeschlossen und soll über eine Partnerschaft weiterentwickelt werden. „Wir befinden uns in den Verpartnerungsgesprächen auf einem guten Weg“, sagt Finanzvorstand Thomas Klaue im Gespräch mit TradeCentre. Wer allerdings darauf hofft, das Unternehmen erhält bei Vertragsabschluss einen hohen zweistelligen Millionenbetrag als Vorabzahlung, wird enttäuscht sein. „Pharmafirmen sind aufgrund der Finanzkrise mit hohen Vorabzahlungen zurückhaltender und teilen das Risiko vermehrt über Meilensteinzahlungen in der weiteren Entwicklungsphase“, erklärt der CFO. Die Deal-Struktur dürfte sich anders gestalten als erhofft. Somit wird MediGene (DE0005020903) bei Vertragsunterschrift schrift eine vergleichsweise geringe Vorabzahlung erhalten und verstärkt Meilensteinzahlungen in der weiteren Entwicklungsphase bis hin zur möglichen Zulassung. Dennoch könnte bei einem Gesamtdeal über Meilensteine aus unserer Sicht ein dreistelliger Millionenbetrag in die Kasse der Bayern fließen. Wunschpartner ist laut Klaue ein in der Biotechnologie tätiges und in der Onkologie starkes Unternehmen, das MediGene als Partner Co-Promotion-Rechte in Europa überlässt. . Das Profil passt zum US-Biotech-Konzern Amgen. Einen Bezug zu Amgen hat die Firma. MediGenes CEO Frank Mathias war einst Deutschland-Chef von Amgen.
Sehr zufrieden zeigt sich der Finanzchef mit der Entwicklung des Produktes Eligard. „Eligard zeigt ein robustes und starkes Wachstum. Der vom Vertriebspartner erzielte Produktumsatz von Eligard könnte in diesem Jahr die Marke von 100 Millionen Euro knacken nach 80 Millionen Euro im Vorjahr“, sagt Klaue. Der auf MediGene entfallende Umsatz könnte die Marke von 35 Millionen Euro überschreiten nach 30 Millionen Euro im Vorjahr. Die Peak-Sales für MediGenes Umsatz aus Eligard sieht der CFO zwischen 40 und 45 Millionen Euro. Mit Veregen hat das Unternehmen ein zweites Produkt am Markt. „Über unseren Partner in den USA erwarten wir ab dem dritten Quartal erste spürbare Effekte und ab 2010 steigende Umsätze“. Für Europa erschließt das Unternehmen einzelne Territorien über verschiedene Partner. „Wir sind in Verhandlungen und erwarten nach dem Abschluss in Spanien weitere Vertriebspartnerschaften“. Erste Erlöse aus europäischen Gefilden sind ab 2010 zu erwarten. Da sich MediGene auf Onkologie und Immunologie fokussiert, passt Veregen nicht mehr zur strategischen Ausrichtung. Klaue schloss eine Veräußerung der Rechte nicht kategorisch aus. „Derzeit ist dies jedoch kein Thema“. Die Rechte an Oracea hat MediGene bereits veräußert. Aus dieser Veräußerung fließen noch bis zu 24 Millionen Euro in die Kasse. Eine erste Tranche soll Anfang 2011 eintreffen. Weitere Meilensteine werden in 2011 und 2012 erwartet.
Auf der Zahlenseite wird das Unternehmen den Vorjahresumsatz von über 39 Millionen Euro aufgrund der Produktumsätze durch Eligard leicht übertreffen. „Das bezieht sich insbesondere auf Eligard. Erlöse aus kommenden Deals sind darin nicht eingerechnet“. Der Verlust beim EBITDA wird sich nach rund minus 25 Millionen Euro im Vorjahr verbessern. „Im ersten Quartal lagen wir bereinigt um Einmaleffekte bei 1,1 Millionen Cashburn pro Monat. In den verbleibenden neun Monaten wird der monatliche Barmittelverbrauch bei circa 1,5 Millionen Euro liegen“. Per Ende März lag der Cashbestand bei 16,6 Millionen Euro. MediGene hat weiteren Zugriff auf 25 Millionen Euro durch Ausgabe neuer Aktien. Damit ist die Firma zunächst finanziert. Immer wieder ist MediGene Opfer der Übernahmegerüchteküche. Alternativ soll seit Jahr und Tag eine Fusion mit dem Biotechunternehmen Wilex geplant sein. „Wir sprechen derzeit weder über Fusionen, noch sprechen wir mit Interessenten für eine komplette Übernahme“, kommentiert Klaue diese Spekulationen.
Der Börsenwert von MediGene beträgt rund 140 Millionen Euro. Im Bereich um vier Euro bildet die Aktie einen stabilen Boden. Bei erfolgreichem Abschluss von EndoTAG-1 dürfte das Papier wach geküsst werden. Spekulativ orientierte Anleger können sich ein paar Stücke ins Depot legen. Den Stoppkurs raten wir circa 15 Prozent unter Einstieg zu platzieren.
mfg ipollit
Antwort auf Beitrag Nr.: 37.635.908 von ipollit am 23.07.09 21:23:58
Neben dem eigenen zugelassenen Produkt Arcalyst bekommt REGN auch Royalties von bis zu 15% auf Umsätze mit Novartis Ilaris, das vor Kurzen zugelassen wurde.
ipollit, die Fragen jetzt sind vermutlich dämlich, deshalb schon mal sorry, aber ich raff den Zusammenhang zwischen Regeneron und Novartis/Ilaris nicht. Der AK-Partner für Ilaris (ACZ885 bzw. canakinumab) war doch Medarex. Wieso hatte Regeneron hier opt-in-Rechte und bekommt jetzt royaltys?
Und wie hängt Arcalyst damit zusammen? Ist das ein anderer AK? Oder gar kein AK?
PS: Regeneron scheint ja wirklich spannend zu sein! Ich wusste bis eben nichts über die, bin aber über Deinen Beitrag gestolpert und aufmerksam geworden.
Neben dem eigenen zugelassenen Produkt Arcalyst bekommt REGN auch Royalties von bis zu 15% auf Umsätze mit Novartis Ilaris, das vor Kurzen zugelassen wurde.
ipollit, die Fragen jetzt sind vermutlich dämlich, deshalb schon mal sorry, aber ich raff den Zusammenhang zwischen Regeneron und Novartis/Ilaris nicht. Der AK-Partner für Ilaris (ACZ885 bzw. canakinumab) war doch Medarex. Wieso hatte Regeneron hier opt-in-Rechte und bekommt jetzt royaltys?
Und wie hängt Arcalyst damit zusammen? Ist das ein anderer AK? Oder gar kein AK?
PS: Regeneron scheint ja wirklich spannend zu sein! Ich wusste bis eben nichts über die, bin aber über Deinen Beitrag gestolpert und aufmerksam geworden.
Antwort auf Beitrag Nr.: 37.655.596 von SLGramann am 27.07.09 21:26:03Medarex hat den AK generiert... Novartis hatte aber mit Regeneron zusammen eine Kooperation, um Mittel, die auf IL-1 basieren, zu entwickeln. U.a. entstand daraus Ilaris (AK gegen IL-1). Regeneron hat zudem mit der eigenen Trap-Technolgie einen IL-1 Trap (Arcalyst) durch die Klinik gebracht, für das Regeneron die vollen Rechte besitzt. Traps sind anders als AKs quasi lösliche Rezeptoren, an die z.B. dann das IL-1 bindet, während es bei Ilaris ein AK ist, der IL-1 einfängt. Arcalyst ist bereits gegen CAPS zugelassen und die Umsätze sind relativ gering, da diese Krankheit extrem selten ist. Nun wurde zudem Ilaris ebenfalls gegen CAPS zugelassen, für das Regeneron eben Royalty-Zahlungen erhält. Entscheidend ist mehr die Zulassung als dass sich mit CAPS nennenswerte Umsätze generieren lassen. Interessant wird es erst, wenn nächstes Jahr die PIII-Daten von Arcalyst zu Gicht kommen... das wäre ein ungleich größeres Umsatzpotential, wenn es wirkt. Ilaris befindet sich auch in der klinischen Entwicklung für unterschiedliche Indikationen. In dem von mir zitierten Artikel wird ja gesagt, dass Arcalyst vielleicht besser gegen akute Sachen wie Gicht eingesetzt werden kann, während Ilaris besser gegen chronische Erkrankungen wäre.
Die Phantasie entsteht allerdings mehr durch VEGF-Trap (das analoge zum Avastin-AK) und VEGF-Trap Eye (was Lucentis wäre)... zu beiden gibt es nächstes Jahr wichtige PIII-Daten.
Gruß, ipollit
Die Phantasie entsteht allerdings mehr durch VEGF-Trap (das analoge zum Avastin-AK) und VEGF-Trap Eye (was Lucentis wäre)... zu beiden gibt es nächstes Jahr wichtige PIII-Daten.
Gruß, ipollit
Genmab...
Arzerra mit positiven PIII-Daten für RA-Patienten, bei denen die 1st-line Standard-Therapie Methotrexate versagt hat. Die Ergebnisse sind vergleichbar mit denen von Rituxan bei RA, vielleicht ist Rituxan eher etwas besser. Es ist offen, ob das voll-humane Arzerra Sicherheitsvorteile gegenüber dem Maus-AK Rituxan ausspielen kann, da es zusammen mit Methotrexate verabreicht wird. Methotrexate unterdrückt allerdings das Immunsystem, wodurch die Immunantwort gegenüber den Maus-AK geringer ausfallen dürfte. Eine zweite PIII läuft noch, die Arzerra bei RA-Patienten testet, bei denen TNF-Hemmer versagt haben. Rituxan kommt als einer der letzten Optionen bei RA zum Einsatz, nach Versagen von Methotrexate und TNF-Hemmern&Co.
ACR20:
- Arzerra + Methotrexate: 50%
- Placebo + Methotrexate: 27%
Die wichtigen ACR50- und ACR70-Werte wurden noch nicht veröffentlicht, nur dass sie signifikant über Placebo liegen.
GlaxoSmithKline and Genmab Announce Top-Line Results for Ofatumumab in Rheumatoid Arthritis
COPENHAGEN, Denmark, July 29, 2009 (GLOBE NEWSWIRE) -- Summary: GSK and Genmab announced preliminary top-line results from a Phase III study of ofatumumab in RA.
GlaxoSmithKline (NYSE:GSK) and Genmab A/S (Copenhagen:GEN) announced today preliminary top-line results from a Phase III study of ofatumumab administered intravenously for the treatment of rheumatoid arthritis (RA) in patients who had an inadequate response to methotrexate. The study met the primary endpoint, ACR20 at 24 weeks, which indicates a 20 percent or greater improvement in the number of swollen and tender joints, as well as improvements in other disease-activity measures.
In the study, 260 patients were treated and included in the analysis. At week 24, the ACR20 response rate was significantly greater for RA patients on ofatumumab (n=129) than on placebo (n=131) with a 50 percent response rate in the patients receiving ofatumumab, compared to 27 percent for patients on placebo (p-value less than 0.001). All key secondary endpoints were significant (p-value less than or equal to 0.001).
There were no unexpected safety findings. The most common adverse events in the ofatumumab treated patients (greater than 5 percent) were rash, urticaria, nasopharyngitis, pruritus, throat irritation and hypersensitivity. Other than nasopharyngitis, these events generally occurred within 24 hours of the first infusion. One death, judged by the investigator as unrelated to ofatumumab, was reported in the study during the 24-week study period.
"We have always believed in ofatumumab's potential to make a difference in patients' lives. We are pleased with the results of this study, supporting the further investigation of this antibody's promise in the treatment of RA," said Lisa N. Drakeman, Ph.D., Chief Executive Officer of Genmab.
"RA can be a highly debilitating disease. It is encouraging to see the reduction in disease symptoms achieved with intravenous ofatumumab, and we look forward to presenting the full study results," said Carlo Russo, M.D., Senior Vice President, Biopharm Development, GSK.
About the study
In this 24 week double-blind study, patients with active RA were randomized to receive two 700 mg doses of intravenous ofatumumab or placebo two weeks apart in addition to background methotrexate. Disease status was measured every 4 weeks. Patients for this non-IND study were recruited from Europe, South America and Australia.
The primary objective of the study was to determine the efficacy of intravenous ofatumumab in reducing the clinical signs and symptoms in RA patients after two 700 mg doses of ofatumumab compared to placebo. The primary endpoint of the study was ACR20 at 24 weeks. Other key secondary objectives included safety, patient reported outcomes, biomarkers and ACR 50 and ACR 70.
ACR Response
The ACR 20 response is defined as a 20 percent or greater improvement from baseline in tender and swollen joint counts, and 20 percent or greater improvement in 3 of the 5 following assessments: patient and physician global assessments, pain, disability, and an acute phase reactant (ESR or CRP).
About ofatumumab
Ofatumumab is a novel, investigational, fully human monoclonal antibody that targets a membrane-proximal (close to the cell surface) small loop epitope (a portion of a molecule to which an antibody binds) on the CD20 molecule of B-cells. This epitope is different from the binding sites targeted by other CD20 antibodies currently available.
mfg ipollit
Arzerra mit positiven PIII-Daten für RA-Patienten, bei denen die 1st-line Standard-Therapie Methotrexate versagt hat. Die Ergebnisse sind vergleichbar mit denen von Rituxan bei RA, vielleicht ist Rituxan eher etwas besser. Es ist offen, ob das voll-humane Arzerra Sicherheitsvorteile gegenüber dem Maus-AK Rituxan ausspielen kann, da es zusammen mit Methotrexate verabreicht wird. Methotrexate unterdrückt allerdings das Immunsystem, wodurch die Immunantwort gegenüber den Maus-AK geringer ausfallen dürfte. Eine zweite PIII läuft noch, die Arzerra bei RA-Patienten testet, bei denen TNF-Hemmer versagt haben. Rituxan kommt als einer der letzten Optionen bei RA zum Einsatz, nach Versagen von Methotrexate und TNF-Hemmern&Co.
ACR20:
- Arzerra + Methotrexate: 50%
- Placebo + Methotrexate: 27%
Die wichtigen ACR50- und ACR70-Werte wurden noch nicht veröffentlicht, nur dass sie signifikant über Placebo liegen.
GlaxoSmithKline and Genmab Announce Top-Line Results for Ofatumumab in Rheumatoid Arthritis
COPENHAGEN, Denmark, July 29, 2009 (GLOBE NEWSWIRE) -- Summary: GSK and Genmab announced preliminary top-line results from a Phase III study of ofatumumab in RA.
GlaxoSmithKline (NYSE:GSK) and Genmab A/S (Copenhagen:GEN) announced today preliminary top-line results from a Phase III study of ofatumumab administered intravenously for the treatment of rheumatoid arthritis (RA) in patients who had an inadequate response to methotrexate. The study met the primary endpoint, ACR20 at 24 weeks, which indicates a 20 percent or greater improvement in the number of swollen and tender joints, as well as improvements in other disease-activity measures.
In the study, 260 patients were treated and included in the analysis. At week 24, the ACR20 response rate was significantly greater for RA patients on ofatumumab (n=129) than on placebo (n=131) with a 50 percent response rate in the patients receiving ofatumumab, compared to 27 percent for patients on placebo (p-value less than 0.001). All key secondary endpoints were significant (p-value less than or equal to 0.001).
There were no unexpected safety findings. The most common adverse events in the ofatumumab treated patients (greater than 5 percent) were rash, urticaria, nasopharyngitis, pruritus, throat irritation and hypersensitivity. Other than nasopharyngitis, these events generally occurred within 24 hours of the first infusion. One death, judged by the investigator as unrelated to ofatumumab, was reported in the study during the 24-week study period.
"We have always believed in ofatumumab's potential to make a difference in patients' lives. We are pleased with the results of this study, supporting the further investigation of this antibody's promise in the treatment of RA," said Lisa N. Drakeman, Ph.D., Chief Executive Officer of Genmab.
"RA can be a highly debilitating disease. It is encouraging to see the reduction in disease symptoms achieved with intravenous ofatumumab, and we look forward to presenting the full study results," said Carlo Russo, M.D., Senior Vice President, Biopharm Development, GSK.
About the study
In this 24 week double-blind study, patients with active RA were randomized to receive two 700 mg doses of intravenous ofatumumab or placebo two weeks apart in addition to background methotrexate. Disease status was measured every 4 weeks. Patients for this non-IND study were recruited from Europe, South America and Australia.
The primary objective of the study was to determine the efficacy of intravenous ofatumumab in reducing the clinical signs and symptoms in RA patients after two 700 mg doses of ofatumumab compared to placebo. The primary endpoint of the study was ACR20 at 24 weeks. Other key secondary objectives included safety, patient reported outcomes, biomarkers and ACR 50 and ACR 70.
ACR Response
The ACR 20 response is defined as a 20 percent or greater improvement from baseline in tender and swollen joint counts, and 20 percent or greater improvement in 3 of the 5 following assessments: patient and physician global assessments, pain, disability, and an acute phase reactant (ESR or CRP).
About ofatumumab
Ofatumumab is a novel, investigational, fully human monoclonal antibody that targets a membrane-proximal (close to the cell surface) small loop epitope (a portion of a molecule to which an antibody binds) on the CD20 molecule of B-cells. This epitope is different from the binding sites targeted by other CD20 antibodies currently available.
mfg ipollit
Antwort auf Beitrag Nr.: 37.682.864 von ipollit am 30.07.09 22:50:37noch zu Genmab...
die veröffentlichten Daten sind vorläufig... die kompletten endgültigen Daten werden zu einem späteren Zeitpunkt auf einer Konferenz vorgestellt.
diese Studie für sich genommen ist nicht als Zulassungsstudie gedacht gewesen.... bezügl. RA laufen noch zwei weitere Studien. Eine PII-Studie mit nicht intravenös sondern subkutan verabreichten Arzerra könnte bis Ende des Jahres Daten liefern. Eine PIII bei Patienten, bei denen TNF-Hemmer unwirksam sind (die meiner Meinung nach wichtigere Studie, da diese auf die spätere Indikation abzielt), soll nächstes Jahr Ergebnisse liefern.
Innerhalb der nächsten beiden Monate soll es Daten von 2 PII-Studien bei CLL und NHL geben.
Ergebnisse der sehr bedeutsamen PIII-Studie, die Arzerra bei NHL-Patienten testet, bei denen Rituxan nicht mehr wirkt, sollen im August vorliegen. NHL ist die Hauptindikation von Rituxan... so weit ich weiß, ist bei 50% Rituxan unwirksam und bei den anderen 50% bildet sich im Laufe der Zeit eine Resistenz. Da Arzerra anders an CD20 bindet und anders auf die Zelle wirkt, kann es eine Rituxan-Resistenz überwinden, worauf auch bisherige Daten hinweisen.
mfg ipollit
die veröffentlichten Daten sind vorläufig... die kompletten endgültigen Daten werden zu einem späteren Zeitpunkt auf einer Konferenz vorgestellt.
diese Studie für sich genommen ist nicht als Zulassungsstudie gedacht gewesen.... bezügl. RA laufen noch zwei weitere Studien. Eine PII-Studie mit nicht intravenös sondern subkutan verabreichten Arzerra könnte bis Ende des Jahres Daten liefern. Eine PIII bei Patienten, bei denen TNF-Hemmer unwirksam sind (die meiner Meinung nach wichtigere Studie, da diese auf die spätere Indikation abzielt), soll nächstes Jahr Ergebnisse liefern.
Innerhalb der nächsten beiden Monate soll es Daten von 2 PII-Studien bei CLL und NHL geben.
Ergebnisse der sehr bedeutsamen PIII-Studie, die Arzerra bei NHL-Patienten testet, bei denen Rituxan nicht mehr wirkt, sollen im August vorliegen. NHL ist die Hauptindikation von Rituxan... so weit ich weiß, ist bei 50% Rituxan unwirksam und bei den anderen 50% bildet sich im Laufe der Zeit eine Resistenz. Da Arzerra anders an CD20 bindet und anders auf die Zelle wirkt, kann es eine Rituxan-Resistenz überwinden, worauf auch bisherige Daten hinweisen.
mfg ipollit
Viropharma...
Cinryze zur Prophylaxe von HAE scheint sich gut zu entwickeln... VPHM sieht für dieses Jahr Umsätze von 80-95 Mio USD. In Kürze (am 4.8.) wird die FDA etwas zur möglichen Zulassung von generischem Vancocin sagen... Generika werden hier schon seit langem erwartet, so dass es wohl eher zu einer positiven Überraschung kommt, falls diese sich weiter verzögern sollten.
http://www.reuters.com/article/marketsNews/idINBNG2288772009…
ViroPharma quantifies Cinryze sales view; shares rise
* Q2 EPS $0.20 vs $0.29 year ago
* Says Q2 Vancocin sales down 14 pct
* Says withdrawing FY Vancocin sales view
* Sees FY Cinryze sales $80-$95 mln
* Shares up 15 pct
By Vidya L Nathan
July 29 (Reuters) - ViroPharma Inc (VPHM.O) posted a quarterly profit that fell 31 percent from a year ago, hurt by a drop in sales of its top drug Vancocin, but gave a clearer view of expectations for another key drug, Cinryze, sending shares up 16 percent.
Annual projections for Cinryze made up for uncertainty regarding Vancocin, for which the company withdrew its annual sales forecast pending a regulatory decision on guidelines for generic versions of the drug.
The company, which calls Cinryze its "real engine for growth", said it now expects between $80 million to $95 million in Cinryze sales for the year.
"The numbers are more robust in terms of the Cinryze sales. They are starting to provide visibility," WBB Securities analyst Steve Brozak said.
Cinryze -- used to treat a form of rare genetic disease called hereditary angioedema -- had second-quarter sales of $25.6 million, up from $6.7 million in the first quarter, boosting analysts' confidence in the drug's capacity for growth.
"Today is the first time we really feel that this product definitely has the potential in terms of sales. When the first quarter sales came, people just did not know what to anticipate," Maxim Group analyst Yale Jen said.
Vancocin, a treatment for clostridium difficile infection -- a bacterial infection of the bowel -- saw sales fall 14 percent to $56.3 million during the quarter.
Sales from Vancocin, the company's biggest revenue driver, have been threatened by a possible entry of generics for over two years.
While WBB Securities' Brozak said it would be hard to predict which way a regulatory advisory panel would rule, Maxim Group's Jen sees a high probability of Vancocin copycats entering the market by the fourth quarter.
Still, analysts agree that any impact from the entry of generics is already priced into the stock. For the second quarter, the company posted a net profit of $16.1 million, or 20 cents a share, compared with a net income of $22.8 million, or 29 cents a share a year ago. Revenue for the quarter rose 25 percent to $81.9 million.
Shares of the company were trading up $1.16 at $8.06 Wednesday on Nasdaq. They touched a high of $8.22 earlier in the session.

mfg ipollit
Cinryze zur Prophylaxe von HAE scheint sich gut zu entwickeln... VPHM sieht für dieses Jahr Umsätze von 80-95 Mio USD. In Kürze (am 4.8.) wird die FDA etwas zur möglichen Zulassung von generischem Vancocin sagen... Generika werden hier schon seit langem erwartet, so dass es wohl eher zu einer positiven Überraschung kommt, falls diese sich weiter verzögern sollten.
http://www.reuters.com/article/marketsNews/idINBNG2288772009…
ViroPharma quantifies Cinryze sales view; shares rise
* Q2 EPS $0.20 vs $0.29 year ago
* Says Q2 Vancocin sales down 14 pct
* Says withdrawing FY Vancocin sales view
* Sees FY Cinryze sales $80-$95 mln
* Shares up 15 pct
By Vidya L Nathan
July 29 (Reuters) - ViroPharma Inc (VPHM.O) posted a quarterly profit that fell 31 percent from a year ago, hurt by a drop in sales of its top drug Vancocin, but gave a clearer view of expectations for another key drug, Cinryze, sending shares up 16 percent.
Annual projections for Cinryze made up for uncertainty regarding Vancocin, for which the company withdrew its annual sales forecast pending a regulatory decision on guidelines for generic versions of the drug.
The company, which calls Cinryze its "real engine for growth", said it now expects between $80 million to $95 million in Cinryze sales for the year.
"The numbers are more robust in terms of the Cinryze sales. They are starting to provide visibility," WBB Securities analyst Steve Brozak said.
Cinryze -- used to treat a form of rare genetic disease called hereditary angioedema -- had second-quarter sales of $25.6 million, up from $6.7 million in the first quarter, boosting analysts' confidence in the drug's capacity for growth.
"Today is the first time we really feel that this product definitely has the potential in terms of sales. When the first quarter sales came, people just did not know what to anticipate," Maxim Group analyst Yale Jen said.
Vancocin, a treatment for clostridium difficile infection -- a bacterial infection of the bowel -- saw sales fall 14 percent to $56.3 million during the quarter.
Sales from Vancocin, the company's biggest revenue driver, have been threatened by a possible entry of generics for over two years.
While WBB Securities' Brozak said it would be hard to predict which way a regulatory advisory panel would rule, Maxim Group's Jen sees a high probability of Vancocin copycats entering the market by the fourth quarter.
Still, analysts agree that any impact from the entry of generics is already priced into the stock. For the second quarter, the company posted a net profit of $16.1 million, or 20 cents a share, compared with a net income of $22.8 million, or 29 cents a share a year ago. Revenue for the quarter rose 25 percent to $81.9 million.
Shares of the company were trading up $1.16 at $8.06 Wednesday on Nasdaq. They touched a high of $8.22 earlier in the session.
mfg ipollit
Incyte...

Incyte Reports Second Quarter 2009 Financial Results and Provides Update on Drug Development Programs
On Thursday July 30, 2009, 7:00 am EDT
WILMINGTON, Del.--(BUSINESS WIRE)--Incyte Corporation (Nasdaq:INCY - News) today reported second quarter 2009 financial results and provided an update on its highest priority clinical programs
"With the Phase III registration trials for our lead JAK1/JAK2 inhibitor, INCB18424, now underway in both the U.S. and Europe; the launch of a six-month Phase II trial for our second JAK1/JAK2 inhibitor, INCB28050, in rheumatoid arthritis; the release of positive Phase IIb results for our 11beta-HSD1 inhibitor for type 2 diabetes; and the expected release of Phase IIb results for topical INCB18424 in psoriasis later this summer, we have made substantial progress building our pipeline. I believe we are in a strong position to create and capture value from these programs both on our own and through strategic partnerships," stated Paul A. Friedman, M.D., Incyte's President and Chief Executive Officer.
Below is a summary of recent developments for our most advanced product candidates:
Janus Kinase (JAK) Inhibitor Program
INCB18424: (oral formulation) Myelofibrosis, Polycythemia Vera and Essential Thrombocythemia
- Agreement reached with the U.S. Food and Drug Administration (FDA) for a Special Protocol Assessment (SPA) for INCB18424 as a treatment in myelofibrosis (MF).
- Announcement that the Phase III trial under the SPA, COMFORT-I, is expected to begin patient enrollment in August and involve 240 patients with primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (PPV-MF) and post essential thrombocythemia myelofibrosis (PET-MF). COMFORT-I is scheduled to include over 90 clinical sites in the U.S., Canada and Australia.
- Initiation of a second Phase III trial, COMFORT-II, in Europe began enrolling patients in July and is expected to enroll 150 patients in approximately 70 clinical sites.
INCB18424: (topical formulation) Psoriasis and Other Inflammatory Conditions of the Skin
- Completed a three-month multiple-dose Phase IIb trial in patients with mild to moderate psoriasis. Results from this trial are expected later this summer.
INCB28050: JAK Inhibitor Compound for Rheumatoid Arthritis and Other Inflammatory Conditions
- Initiated a six-month double-blind placebo-controlled dose-ranging Phase II trial that is scheduled to include 100 patients with active rheumatoid arthritis who have had inadequate response to currently available disease modifying therapies.
11beta-HSD1 Inhibitor Program
INCB13739: Type 2 Diabetes
- Presented clinical results at the American Diabetes Association 69th Scientific Sessions from a 3-month placebo-controlled, dose-ranging Phase IIb trial involving over 300 patients with type 2 diabetes which demonstrated that treatment with once-daily doses of INCB13739 significantly improved glycemic control, as measured by hemoglobin A1c, insulin sensitivity and total-cholesterol levels.
Sheddase Inhibitor Program
INCB7839: Breast Cancer
- Continued enrollment of a Phase II trial in combination with Herceptin(R) in breast cancer patients. We expect to present results from this trial at the San Antonio Breast Cancer Symposium in December 2009.
Second Quarter 2009 Financial Results
Cash Position
As of June 30, 2009, cash, short-term and long-term marketable securities totaled $147.5 million, compared to $217.8 million as of December 31, 2008.
During the six months ended June 30, 2009, we used $70.3 million in cash and marketable securities. Cash use guidance of $122 to $128 million for 2009 remains unchanged.
Revenues
Total revenues for the quarter ended June 30, 2009 were $0.8 million as compared to $0.6 million for the same period in 2008. Total revenues for the six months ended June 30, 2009 were $1.5 million, as compared to $1.9 million for the same period in 2008.
Net Loss
The net loss for the quarter ended June 30, 2009 was $40.0 million, or $0.41 per share, as compared to $45.6 million, or $0.54 per share, for the same period in 2008.
The net loss for the six months ended June 30, 2009 was $80.1 million, or $0.82 per share, as compared to $85.7 million or $1.01 per share, for the same period in 2008.
Included in the net loss for the quarter and the six months ended June 30, 2009 were $2.5 million and $5.9 million, respectively, of non-cash expense related to the impact of expensing share-based payments, including employee stock options, as compared to $3.9 million and $7.3 million, respectively, for the same periods in 2008.
Operating Expenses
Research and development expenses for the quarter ended June 30, 2009 were $29.0 million, as compared to $38.1 million for the same period last year. Research and development expenses for the six months ended June 30, 2009 were $58.6 million, as compared to $71.1 million for the same period last year. The decrease in research and development expenses was due to prioritization of our pipeline to focus on products we believe have a greater likelihood of creating near-term value. We expect our research and development expenses to vary from quarter to quarter, primarily due to the timing of our clinical development activities.
Included in research and development expenses for the quarter and the six months ended June 30, 2009 were $1.8 million and $4.2 million, respectively, of non-cash expense related to the impact of expensing share-based payments, including employee stock options, as compared to $2.9 million and $5.3 million, respectively, for the same periods in 2008.
Selling, general and administrative expenses for the quarter and the six months ended June 30, 2009 were $4.1 million and $8.9 million, respectively, as compared to $4.1 million and $8.5 million, respectively, for the same periods in 2008. Increased selling, general and administrative expenses for the six months ended June 30, 2009 reflected our initial sales and marketing preparations for the potential commercialization of INCB18424 for myeloproliferative disorders. Also included in selling, general and administrative expenses for the quarter and the six months ended June 30, 2009 were $0.7 million and $1.7 million, respectively, of non-cash expense related to the impact of expensing share-based payments, including employee stock options, as compared to $1.0 million and $2.0 million, respectively, for the same periods in 2008.
mfg ipollit
Incyte Reports Second Quarter 2009 Financial Results and Provides Update on Drug Development Programs
On Thursday July 30, 2009, 7:00 am EDT
WILMINGTON, Del.--(BUSINESS WIRE)--Incyte Corporation (Nasdaq:INCY - News) today reported second quarter 2009 financial results and provided an update on its highest priority clinical programs
"With the Phase III registration trials for our lead JAK1/JAK2 inhibitor, INCB18424, now underway in both the U.S. and Europe; the launch of a six-month Phase II trial for our second JAK1/JAK2 inhibitor, INCB28050, in rheumatoid arthritis; the release of positive Phase IIb results for our 11beta-HSD1 inhibitor for type 2 diabetes; and the expected release of Phase IIb results for topical INCB18424 in psoriasis later this summer, we have made substantial progress building our pipeline. I believe we are in a strong position to create and capture value from these programs both on our own and through strategic partnerships," stated Paul A. Friedman, M.D., Incyte's President and Chief Executive Officer.
Below is a summary of recent developments for our most advanced product candidates:
Janus Kinase (JAK) Inhibitor Program
INCB18424: (oral formulation) Myelofibrosis, Polycythemia Vera and Essential Thrombocythemia
- Agreement reached with the U.S. Food and Drug Administration (FDA) for a Special Protocol Assessment (SPA) for INCB18424 as a treatment in myelofibrosis (MF).
- Announcement that the Phase III trial under the SPA, COMFORT-I, is expected to begin patient enrollment in August and involve 240 patients with primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (PPV-MF) and post essential thrombocythemia myelofibrosis (PET-MF). COMFORT-I is scheduled to include over 90 clinical sites in the U.S., Canada and Australia.
- Initiation of a second Phase III trial, COMFORT-II, in Europe began enrolling patients in July and is expected to enroll 150 patients in approximately 70 clinical sites.
INCB18424: (topical formulation) Psoriasis and Other Inflammatory Conditions of the Skin
- Completed a three-month multiple-dose Phase IIb trial in patients with mild to moderate psoriasis. Results from this trial are expected later this summer.
INCB28050: JAK Inhibitor Compound for Rheumatoid Arthritis and Other Inflammatory Conditions
- Initiated a six-month double-blind placebo-controlled dose-ranging Phase II trial that is scheduled to include 100 patients with active rheumatoid arthritis who have had inadequate response to currently available disease modifying therapies.
11beta-HSD1 Inhibitor Program
INCB13739: Type 2 Diabetes
- Presented clinical results at the American Diabetes Association 69th Scientific Sessions from a 3-month placebo-controlled, dose-ranging Phase IIb trial involving over 300 patients with type 2 diabetes which demonstrated that treatment with once-daily doses of INCB13739 significantly improved glycemic control, as measured by hemoglobin A1c, insulin sensitivity and total-cholesterol levels.
Sheddase Inhibitor Program
INCB7839: Breast Cancer
- Continued enrollment of a Phase II trial in combination with Herceptin(R) in breast cancer patients. We expect to present results from this trial at the San Antonio Breast Cancer Symposium in December 2009.
Second Quarter 2009 Financial Results
Cash Position
As of June 30, 2009, cash, short-term and long-term marketable securities totaled $147.5 million, compared to $217.8 million as of December 31, 2008.
During the six months ended June 30, 2009, we used $70.3 million in cash and marketable securities. Cash use guidance of $122 to $128 million for 2009 remains unchanged.
Revenues
Total revenues for the quarter ended June 30, 2009 were $0.8 million as compared to $0.6 million for the same period in 2008. Total revenues for the six months ended June 30, 2009 were $1.5 million, as compared to $1.9 million for the same period in 2008.
Net Loss
The net loss for the quarter ended June 30, 2009 was $40.0 million, or $0.41 per share, as compared to $45.6 million, or $0.54 per share, for the same period in 2008.
The net loss for the six months ended June 30, 2009 was $80.1 million, or $0.82 per share, as compared to $85.7 million or $1.01 per share, for the same period in 2008.
Included in the net loss for the quarter and the six months ended June 30, 2009 were $2.5 million and $5.9 million, respectively, of non-cash expense related to the impact of expensing share-based payments, including employee stock options, as compared to $3.9 million and $7.3 million, respectively, for the same periods in 2008.
Operating Expenses
Research and development expenses for the quarter ended June 30, 2009 were $29.0 million, as compared to $38.1 million for the same period last year. Research and development expenses for the six months ended June 30, 2009 were $58.6 million, as compared to $71.1 million for the same period last year. The decrease in research and development expenses was due to prioritization of our pipeline to focus on products we believe have a greater likelihood of creating near-term value. We expect our research and development expenses to vary from quarter to quarter, primarily due to the timing of our clinical development activities.
Included in research and development expenses for the quarter and the six months ended June 30, 2009 were $1.8 million and $4.2 million, respectively, of non-cash expense related to the impact of expensing share-based payments, including employee stock options, as compared to $2.9 million and $5.3 million, respectively, for the same periods in 2008.
Selling, general and administrative expenses for the quarter and the six months ended June 30, 2009 were $4.1 million and $8.9 million, respectively, as compared to $4.1 million and $8.5 million, respectively, for the same periods in 2008. Increased selling, general and administrative expenses for the six months ended June 30, 2009 reflected our initial sales and marketing preparations for the potential commercialization of INCB18424 for myeloproliferative disorders. Also included in selling, general and administrative expenses for the quarter and the six months ended June 30, 2009 were $0.7 million and $1.7 million, respectively, of non-cash expense related to the impact of expensing share-based payments, including employee stock options, as compared to $1.0 million and $2.0 million, respectively, for the same periods in 2008.
mfg ipollit
Micromet... etwas seltsam trotz KE-Ankündigung heute deutlich im Plus
Micromet Announces Pricing of Public Offering of Common Stock
BETHESDA, Md., July 30 /PRNewswire-FirstCall/ -- Micromet, Inc. (Nasdaq: MITI - News), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced the pricing of its public offering of 14,000,000 shares of its common stock at a public offering price of $5.00 per share. All of the shares are being offered by Micromet. The gross proceeds to Micromet, before expenses, from the sale of the shares are expected to be approximately $70 million. The closing of the offering is expected to take place on August 4, 2009. The underwriters have a 30-day option to purchase up to an additional 2,100,000 shares of common stock to cover over-allotments, if any. Piper Jaffray & Co. is acting as the sole book running manager with RBC Capital Markets and Merriman Curhan Ford as co-managers in this offering.

mfg ipollit
Micromet Announces Pricing of Public Offering of Common Stock
BETHESDA, Md., July 30 /PRNewswire-FirstCall/ -- Micromet, Inc. (Nasdaq: MITI - News), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced the pricing of its public offering of 14,000,000 shares of its common stock at a public offering price of $5.00 per share. All of the shares are being offered by Micromet. The gross proceeds to Micromet, before expenses, from the sale of the shares are expected to be approximately $70 million. The closing of the offering is expected to take place on August 4, 2009. The underwriters have a 30-day option to purchase up to an additional 2,100,000 shares of common stock to cover over-allotments, if any. Piper Jaffray & Co. is acting as the sole book running manager with RBC Capital Markets and Merriman Curhan Ford as co-managers in this offering.
mfg ipollit
Antwort auf Beitrag Nr.: 37.665.320 von ipollit am 28.07.09 22:18:49
Hi ipollit, vielen Dank für Deine Erläuterungen zu dem Komplex Regeneron/Medarex/Novartis/CAPS.
Du schreibst dann zu Regeneron noch: "Die Phantasie entsteht allerdings mehr durch VEGF-Trap (das analoge zum Avastin-AK) und VEGF-Trap Eye (was Lucentis wäre)... zu beiden gibt es nächstes Jahr wichtige PIII-Daten."
Avastin und Lucentis sind ja ganz große Namen... Hast Du - aufgrund von P II-Daten, ein Feeling, was die Chancen angeht?
Zu Incyte: "Planmäßig" wäre der Cash zum Jahresende auf ca. 90 Mio. USD abgeschmolzen - bei einem Cashburn von deutlich über 100 Mio. pro Jahr. Hier stellt sich die Frage der Finanzierung mit relativ hoher Dringlichkeit imho. Ob man INCB18424 bald verpartnern kann? Oder wird man eine KE versuchen?
Sobald ich meine restlichen Medarex BMY in den Rachen geschmissen haben werde (Mitte August vermutlich), muss ich entscheiden, wie es weitergeht. Regenereon und Incyte sind in der engeren Wahl.
Gruß.
Hi ipollit, vielen Dank für Deine Erläuterungen zu dem Komplex Regeneron/Medarex/Novartis/CAPS.
Du schreibst dann zu Regeneron noch: "Die Phantasie entsteht allerdings mehr durch VEGF-Trap (das analoge zum Avastin-AK) und VEGF-Trap Eye (was Lucentis wäre)... zu beiden gibt es nächstes Jahr wichtige PIII-Daten."
Avastin und Lucentis sind ja ganz große Namen... Hast Du - aufgrund von P II-Daten, ein Feeling, was die Chancen angeht?
Zu Incyte: "Planmäßig" wäre der Cash zum Jahresende auf ca. 90 Mio. USD abgeschmolzen - bei einem Cashburn von deutlich über 100 Mio. pro Jahr. Hier stellt sich die Frage der Finanzierung mit relativ hoher Dringlichkeit imho. Ob man INCB18424 bald verpartnern kann? Oder wird man eine KE versuchen?
Sobald ich meine restlichen Medarex BMY in den Rachen geschmissen haben werde (Mitte August vermutlich), muss ich entscheiden, wie es weitergeht. Regenereon und Incyte sind in der engeren Wahl.
Gruß.
Antwort auf Beitrag Nr.: 37.683.135 von ipollit am 31.07.09 00:05:00MITI... ein Gerücht besagt, dass Micromet wieder die vollen Rechte an Blinatumomab halten könnte.
http://www.thestreet.com/story/10559063/2/biotech-stock-mail…
Last March, AstraZeneca returned North American rights for blinatumomab to Micromet, although AstraZeneca continued to manufacture the drug for Microment and retained a one-time option to reacquire rights to the drug upon its first regulatory approval.
However, Micromet is reportedly buying back AstraZeneca's remaining option on blinatumomab for $6 million, according to an investor who was briefed by Micromet during meetings this week tied to the follow-on offering. Furthermore, AstraZeneca will no longer manufacture the drug for Micromet, which is now contemplating plans to build its own facility to make the drug.
I asked Chrisian Itin, Micromet's CEO, about any changes to the company's relationship with AstraZeneca during a call Thursday morning. He wouldn't answer the question, stating only that as of now, the arrangement with AstraZeneca remains as it was.
It's hard to say what this all means. If blinatumomab were a great drug in the making, why did AstraZeneca relinquish North American rights last March?
Perhaps AstraZeneca's pipeline priorities changed and it could no longer devote resources to the drug. That's entirely possible, but then why would AstraZeneca also decide to accept just $6 million to further give up an option to reacquire the rights to the drug if it did turn out to be a big moneymaker sometime in the future?
If Micromet does buy out AstraZeneca completely, the company will own unencumbered worldwide rights to blinatumomab, which is being developed right now in two hematologic cancer indications. The company also has other drugs in earlier stages of development, some partnerned with other companies, and a platform to generate new antibodies.
mfg ipollit
http://www.thestreet.com/story/10559063/2/biotech-stock-mail…
Last March, AstraZeneca returned North American rights for blinatumomab to Micromet, although AstraZeneca continued to manufacture the drug for Microment and retained a one-time option to reacquire rights to the drug upon its first regulatory approval.
However, Micromet is reportedly buying back AstraZeneca's remaining option on blinatumomab for $6 million, according to an investor who was briefed by Micromet during meetings this week tied to the follow-on offering. Furthermore, AstraZeneca will no longer manufacture the drug for Micromet, which is now contemplating plans to build its own facility to make the drug.
I asked Chrisian Itin, Micromet's CEO, about any changes to the company's relationship with AstraZeneca during a call Thursday morning. He wouldn't answer the question, stating only that as of now, the arrangement with AstraZeneca remains as it was.
It's hard to say what this all means. If blinatumomab were a great drug in the making, why did AstraZeneca relinquish North American rights last March?
Perhaps AstraZeneca's pipeline priorities changed and it could no longer devote resources to the drug. That's entirely possible, but then why would AstraZeneca also decide to accept just $6 million to further give up an option to reacquire the rights to the drug if it did turn out to be a big moneymaker sometime in the future?
If Micromet does buy out AstraZeneca completely, the company will own unencumbered worldwide rights to blinatumomab, which is being developed right now in two hematologic cancer indications. The company also has other drugs in earlier stages of development, some partnerned with other companies, and a platform to generate new antibodies.
mfg ipollit
Antwort auf Beitrag Nr.: 37.685.236 von SLGramann am 31.07.09 11:20:23"Planmäßig" wäre der Cash zum Jahresende auf ca. 90 Mio. USD abgeschmolzen...
ja... das größere Problem ist noch, dass INCY über 300 Mio USD Schulden hat, die abgebaut werden müssen. Dafür sind wohl KEs nötig, wobei ein Teil durch Verpartnerungs-Deals aufgefangen werden soll.
Chancen für REGN kann ich nicht recht beurteilen... z.B. soll VEGF-Trap Eye vergleichbar mit Lucentis sein, nur dass die Wirkung länger anhält und damit weniger Eingriffe ins Auge erfolgen müssen. Mit einer MK von 1,7 Mrd USD ist REGN auch nicht mehr so billig.
mfg ipollit
ja... das größere Problem ist noch, dass INCY über 300 Mio USD Schulden hat, die abgebaut werden müssen. Dafür sind wohl KEs nötig, wobei ein Teil durch Verpartnerungs-Deals aufgefangen werden soll.
Chancen für REGN kann ich nicht recht beurteilen... z.B. soll VEGF-Trap Eye vergleichbar mit Lucentis sein, nur dass die Wirkung länger anhält und damit weniger Eingriffe ins Auge erfolgen müssen. Mit einer MK von 1,7 Mrd USD ist REGN auch nicht mehr so billig.
mfg ipollit
habe meine ARRY-Position etwas aufgestockt... hier sollten im Laufe der nächsten Monate einige Daten kommen, die sich vielleicht positiv auswirken.

mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 37.683.121 von ipollit am 30.07.09 23:57:29INCY...
eine Zusammenfassung/Kommentare zur CC von... http://siliconinvestor.advfn.com/readmsg.aspx?msgid=25826603
CC-transcript... http://seekingalpha.com/article/152573-incyte-corporation-q2…
here are the salient points:
(i) opening remarks
1) We think it will take about six months to enroll each of these registration studies assuming that the results from the trials are positive. It means we could file the NDA for 18424's use in myelofibrosis in late '10 or early '11. And similarly, our regulatory filing in Europe is likely to occur in the second half of 2011.
2) And while it remains our intent to keep North American rights for 424 for oncology, we believe a strong rest of world partner could help us rapidly optimize the myeloproliferative disease opportunity as well as improve our financial position
3) For 424's use in the other two myeloproliferative diseases, polycythemia vera and essential thrombocythemia, we have about 70 patients enrolled in an ongoing Phase II trial; this includes a total of six sites in the US and Europe. We're quite encouraged by the results we've seen in this trial, both in terms of safety and efficacy, and we intend to present these data as well as an update on 424 in MF at ASH.
4) We recently completed the three-month Phase IIb trial for topical 18424 in psoriasis and we also look forward to sharing those results from this study sometime in September, so a month, month and a half from now
5) For the JAK inflammation program we believe that we could form a broad global licensing deal where we could opt to do a deal in which certain indications we retained and/or shared and downstream economics
6) We ended the second quarter with $147.5 million in cash and investments
(ii) q&a
1) Do you need COMFORT-II to be complete in order to file in the US? - - No, we don't. We will do an analysis of the data as it stands at the time we plan to file COMFORT-I. And I think that's been agreed to.
2) The design of the Phase II trial for 050 in RA, just curious your thoughts on including biologic failures I guess in light of what we saw from Rigel last week, just maybe any thoughts you guys have there and why that's included in the design? - - Well, I mean if you look at -- there's -- if you look at what happens to the lipids with Actemra, with a Pfizer drug, and from what we anticipate and see with our drug, you see the effect of blocking interleukin-6 signal.
If you look at the Rigel comp results you don't see that affect. So we don't know how the Rigel drug works frankly, but despite the fact that you can show in cell free system that it's fairly potent against certain enzymes that could be key, it's also very highly protein bound and when you do sell assays with it you get very dramatic shifts in activity. In other words, you've got to go to much higher concentrations to see anything inside the cell.
If you put all that together and you look at the results with Actemra and you look at our early results with 424 and you look at the results that have been presented by Pfizer, people who are biologic failures do pretty well on these new agents if you interfere with signaling through some key pathways. And it's our belief at this point, and I think it's probably going to turn out to be true, that the Rigel molecule does not impact certain key pathways as Actemra, Pfizer JAK inhibitor and our compound do.
3) now that you settled on a 35% reduction in spleen size, will you show us the Phase II data cut by that metric? - - Yes, at ASH
4) I just had a question on COMFORT-II and the entry criteria and how it compares to the Phase II program. Are you are assuming you're going to see a very similar patient population, particularly (inaudible) interested in baseline spleen size? - - Yes, the patent populations are going to be quite similar in all of our studies
5) Wanted just to get some comments on whether there are any functional endpoints at all in COMFORT-I or COMFORT-II. If so what are they? - - So obviously we're measuring symptoms with the MFSAF and there are also some other questions that we're asking. But in terms of actual measurements like six-minute walk test, we're not doing those in either COMFORT-I or COMFORT-II. We will, however, be presenting -- we intend to present at ASH some more detailed results on some of the functional endpoints such as the six-minute walk test
6) I just wanted to turn to the psoriasis program; we've got the data coming up later in the summer. Perhaps can you just give us a sense of what takes [spec] and put into context what you would consider to be promising data from that program? - -
Sure, I'll be happy to. I'm not going to go into quantitative numbers per se, but let me refresh a little bit of what we're doing and what some of the historical data is. So first of all, it's a 200 patient study, we're looking at vehicle and then three different once-a-day strengths of 424 applied topically. And the endpoints are based on both percentage of patients who have a 50% reduction in total lesion score as well as the percentage of patients who achieved clear or almost clear.
Recently approved topicals have used a primary endpoint in the registration studies that present clear or almost clear. And drugs -- recently approved drugs have gotten approved with about 20% clear or almost clear. So we would like to see at least being as good as recently approved drugs and hopefully better
(iii) closing remark
The key is at tolerated doses and 15 BID we get maybe 50% of patients who get a durable, very durable greater than 35% decrease in spleen size. Control groups, that simply is not going to happen -- an occasional patient, but it's going to be a rare event. And so unless we get some off the wall safety issue you have to remember we've had people now on this drug for over two years, some of them on a higher dose than the doses that we are using in COMFORT-I and COMFORT-II, doses that have demonstrated this greater than 35% durable reduction in spleen size, that these studies should have an extremely high probability of being positive
*******
mfg ipollit
eine Zusammenfassung/Kommentare zur CC von... http://siliconinvestor.advfn.com/readmsg.aspx?msgid=25826603
CC-transcript... http://seekingalpha.com/article/152573-incyte-corporation-q2…
here are the salient points:
(i) opening remarks
1) We think it will take about six months to enroll each of these registration studies assuming that the results from the trials are positive. It means we could file the NDA for 18424's use in myelofibrosis in late '10 or early '11. And similarly, our regulatory filing in Europe is likely to occur in the second half of 2011.
2) And while it remains our intent to keep North American rights for 424 for oncology, we believe a strong rest of world partner could help us rapidly optimize the myeloproliferative disease opportunity as well as improve our financial position
3) For 424's use in the other two myeloproliferative diseases, polycythemia vera and essential thrombocythemia, we have about 70 patients enrolled in an ongoing Phase II trial; this includes a total of six sites in the US and Europe. We're quite encouraged by the results we've seen in this trial, both in terms of safety and efficacy, and we intend to present these data as well as an update on 424 in MF at ASH.
4) We recently completed the three-month Phase IIb trial for topical 18424 in psoriasis and we also look forward to sharing those results from this study sometime in September, so a month, month and a half from now
5) For the JAK inflammation program we believe that we could form a broad global licensing deal where we could opt to do a deal in which certain indications we retained and/or shared and downstream economics
6) We ended the second quarter with $147.5 million in cash and investments
(ii) q&a
1) Do you need COMFORT-II to be complete in order to file in the US? - - No, we don't. We will do an analysis of the data as it stands at the time we plan to file COMFORT-I. And I think that's been agreed to.
2) The design of the Phase II trial for 050 in RA, just curious your thoughts on including biologic failures I guess in light of what we saw from Rigel last week, just maybe any thoughts you guys have there and why that's included in the design? - - Well, I mean if you look at -- there's -- if you look at what happens to the lipids with Actemra, with a Pfizer drug, and from what we anticipate and see with our drug, you see the effect of blocking interleukin-6 signal.
If you look at the Rigel comp results you don't see that affect. So we don't know how the Rigel drug works frankly, but despite the fact that you can show in cell free system that it's fairly potent against certain enzymes that could be key, it's also very highly protein bound and when you do sell assays with it you get very dramatic shifts in activity. In other words, you've got to go to much higher concentrations to see anything inside the cell.
If you put all that together and you look at the results with Actemra and you look at our early results with 424 and you look at the results that have been presented by Pfizer, people who are biologic failures do pretty well on these new agents if you interfere with signaling through some key pathways. And it's our belief at this point, and I think it's probably going to turn out to be true, that the Rigel molecule does not impact certain key pathways as Actemra, Pfizer JAK inhibitor and our compound do.
3) now that you settled on a 35% reduction in spleen size, will you show us the Phase II data cut by that metric? - - Yes, at ASH
4) I just had a question on COMFORT-II and the entry criteria and how it compares to the Phase II program. Are you are assuming you're going to see a very similar patient population, particularly (inaudible) interested in baseline spleen size? - - Yes, the patent populations are going to be quite similar in all of our studies
5) Wanted just to get some comments on whether there are any functional endpoints at all in COMFORT-I or COMFORT-II. If so what are they? - - So obviously we're measuring symptoms with the MFSAF and there are also some other questions that we're asking. But in terms of actual measurements like six-minute walk test, we're not doing those in either COMFORT-I or COMFORT-II. We will, however, be presenting -- we intend to present at ASH some more detailed results on some of the functional endpoints such as the six-minute walk test
6) I just wanted to turn to the psoriasis program; we've got the data coming up later in the summer. Perhaps can you just give us a sense of what takes [spec] and put into context what you would consider to be promising data from that program? - -
Sure, I'll be happy to. I'm not going to go into quantitative numbers per se, but let me refresh a little bit of what we're doing and what some of the historical data is. So first of all, it's a 200 patient study, we're looking at vehicle and then three different once-a-day strengths of 424 applied topically. And the endpoints are based on both percentage of patients who have a 50% reduction in total lesion score as well as the percentage of patients who achieved clear or almost clear.
Recently approved topicals have used a primary endpoint in the registration studies that present clear or almost clear. And drugs -- recently approved drugs have gotten approved with about 20% clear or almost clear. So we would like to see at least being as good as recently approved drugs and hopefully better
(iii) closing remark
The key is at tolerated doses and 15 BID we get maybe 50% of patients who get a durable, very durable greater than 35% decrease in spleen size. Control groups, that simply is not going to happen -- an occasional patient, but it's going to be a rare event. And so unless we get some off the wall safety issue you have to remember we've had people now on this drug for over two years, some of them on a higher dose than the doses that we are using in COMFORT-I and COMFORT-II, doses that have demonstrated this greater than 35% durable reduction in spleen size, that these studies should have an extremely high probability of being positive
*******
mfg ipollit
Antwort auf Beitrag Nr.: 37.701.934 von ipollit am 03.08.09 19:48:26noch zu Array...
http://www.biohealthinvestor.com/2009/07/why-array-biopharma…
Why Array Biopharma’s MEK inhibitor pipeline may pay off (ARRY)
July 16, 2009
It is very early in the game, but Array Biopharma Inc. (Nasdaq: ARRY) may now have two MEK inhibitor drug candidates that may each eventually find markets in both both cancer and rheumatoid arthritis — ARRY-162 and ARRY-300.
The company announced this morning that it has filed an investigational new drug application for ARRY-162, and expects to start a Phase 1 cancer trial.
Array Biopharma also is in the midst of an ongoing Phase II trial for ARRY-162 in 200 patients with rheumatoid arthritis. The data in that trial is expected to be released in September, an event that’s a likely catalyst for the company’s stock.
Earlier this week, the company announced additional data for ARRY-162 in rheumatoid arthritis, saying that a four-week study of ARRY-162 in patients with stable RA receiving continued doses, showed that ARRY-162 was well tolerated and with no patients that discontinued the study due to an adverse event.
While ARRY-162 has been the headline-grabber, Array has another MEK inhibitor in ARRY-300 that also is a potential treatment for rheumatoid arthritis, and potentially cancer. The company began a Phase I trial for ARRY-300 early this year in RA. Back in January, Array’s Chief Scientific Officer Kevin Koch said, “We believe ARRY-300 is a best-in-class MEK inhibitor with superior pharmacologic properties.”
MEK is an enzyme that regulates the biosynthesis of inflammation. It also plays a key position in the signaling pathway that has been implicated in the development and progression of cancers. Array scientists have discovered potent MEK inhibitors that interfere with these biosynthetic processes.
Array has a total of five MEK inhibitors in total in its pipeline. And ARRY-162, and possibly ARRY-300 may have potential to treat multiple conditions.
Over time, thosemultiple MEK candidates and multiple potential markts may become valuable when the company seeks development partnerships. — Mike Tarsala
******
http://www.biohealthinvestor.com/2009/07/array-biopharma-tri…
Array BioPharma trial failure highlights another anti-inflammatory (ARRY)
July 9, 2009
Read the Array BioPharma Inc. (Nasdaq: ARRY) press release carefully, and while the headline says its inflammation drug candidate ARRY-797 was well tolerated in two Phase I clinical trials, it becomes clear near the very bottom of the document that it failed the efficacy portion of the test.
The company now says it will discontinue development of ARRY-797 in chronic inflammatory diseases and will cease enrollment in enrollment of new patients in its current Phase II trial of the candidate.
If it’s any consolation, halting a trial with 170 patients is likely to save the company some dough. And this particular candidate was not very far along, so the Street likely didn’t even build in expectations for it.
The attention may now turn to Array’s ARRY-162, another anti-inflammatory that showed much more promising efficacy in its Phase 1 trial, and could eventually become a drug to help treat osteoarthritis and rheumatoid arthritis. Results from a Phase II study of that candidate is expected in the third quarter. — Mike Tarsala
*********
ps. ein interessanter Biotech-Blog: www.biohealthinvestor.com/
mfg ipollit
http://www.biohealthinvestor.com/2009/07/why-array-biopharma…
Why Array Biopharma’s MEK inhibitor pipeline may pay off (ARRY)
July 16, 2009
It is very early in the game, but Array Biopharma Inc. (Nasdaq: ARRY) may now have two MEK inhibitor drug candidates that may each eventually find markets in both both cancer and rheumatoid arthritis — ARRY-162 and ARRY-300.
The company announced this morning that it has filed an investigational new drug application for ARRY-162, and expects to start a Phase 1 cancer trial.
Array Biopharma also is in the midst of an ongoing Phase II trial for ARRY-162 in 200 patients with rheumatoid arthritis. The data in that trial is expected to be released in September, an event that’s a likely catalyst for the company’s stock.
Earlier this week, the company announced additional data for ARRY-162 in rheumatoid arthritis, saying that a four-week study of ARRY-162 in patients with stable RA receiving continued doses, showed that ARRY-162 was well tolerated and with no patients that discontinued the study due to an adverse event.
While ARRY-162 has been the headline-grabber, Array has another MEK inhibitor in ARRY-300 that also is a potential treatment for rheumatoid arthritis, and potentially cancer. The company began a Phase I trial for ARRY-300 early this year in RA. Back in January, Array’s Chief Scientific Officer Kevin Koch said, “We believe ARRY-300 is a best-in-class MEK inhibitor with superior pharmacologic properties.”
MEK is an enzyme that regulates the biosynthesis of inflammation. It also plays a key position in the signaling pathway that has been implicated in the development and progression of cancers. Array scientists have discovered potent MEK inhibitors that interfere with these biosynthetic processes.
Array has a total of five MEK inhibitors in total in its pipeline. And ARRY-162, and possibly ARRY-300 may have potential to treat multiple conditions.
Over time, thosemultiple MEK candidates and multiple potential markts may become valuable when the company seeks development partnerships. — Mike Tarsala
******
http://www.biohealthinvestor.com/2009/07/array-biopharma-tri…
Array BioPharma trial failure highlights another anti-inflammatory (ARRY)
July 9, 2009
Read the Array BioPharma Inc. (Nasdaq: ARRY) press release carefully, and while the headline says its inflammation drug candidate ARRY-797 was well tolerated in two Phase I clinical trials, it becomes clear near the very bottom of the document that it failed the efficacy portion of the test.
The company now says it will discontinue development of ARRY-797 in chronic inflammatory diseases and will cease enrollment in enrollment of new patients in its current Phase II trial of the candidate.
If it’s any consolation, halting a trial with 170 patients is likely to save the company some dough. And this particular candidate was not very far along, so the Street likely didn’t even build in expectations for it.
The attention may now turn to Array’s ARRY-162, another anti-inflammatory that showed much more promising efficacy in its Phase 1 trial, and could eventually become a drug to help treat osteoarthritis and rheumatoid arthritis. Results from a Phase II study of that candidate is expected in the third quarter. — Mike Tarsala
*********
ps. ein interessanter Biotech-Blog: www.biohealthinvestor.com/
mfg ipollit
ONXX...
Die Nexavar-Umsätze entwickeln sich mit 200 Mio USD (+19%) in Q2 weiter positiv. Für das Gesamtjahr werden 850 Mio USD erwartet, so das Nexavar bald die 1 Mrd USD Schwelle übersteigen sollte.
Überraschenderweise soll nun 300 Mio USD zusätzliches Cash aufgenommen, obwohl Onyx bereits profitabel ist und über ein sehr konfortables Cash-Polster von fast netto 500 Mio USD verfügt. Über den Grund, wofür Onyx 800 Mio USD Cash benötigt, wurden keine Angaben gemacht. Ein Problem ist die nach Nexavar schwache Pipeline... es gibt zurzeit nichts in PII/PIII außer Nexavar. Somit könnte Onyx versuchen, ein zugelassenes oder PIII-Produkt einzukaufen oder gleich ein ganzes Unternehmen zu übernehmen. Die Sorge, dass sich Onyx verkalkuliert und irgendeinen Schrott für viel Geld einkauft, hat den Kurs deutlich gedrückt. Ich bin gespannt, was Onyx mit dem Geld vorhat. Merkwürdig ist zudem, dass gerade jetzt die KE durchgeführt wird, obwohl angeblich sehr positive Brustkrebs-Daten zu Nexavar in den nächsten Monaten veröffentlicht werden sollen... falls die Daten wirklich so positiv sind, dann könnte ONXX sehr viel leichter an die 300 Mio USD kommen.
*********

Onyx Pharma slides on guidance, stock issue
Onyx shares fall as analysts point to Nexavar sales guidance and unexpected capital raise
On Wednesday August 5, 2009, 12:53 pm EDT
NEW YORK (AP) -- Onyx Pharmaceuticals Inc.'s shares skidded Wednesday after the cancer drug maker said it will issue stock and convertible notes, and that sales of its drug Nexavar probably will not reach the high end of its forecasts for the year.
Shares of the Emeryville, Calif., company lost $5.58, or 15.3 percent, to $30.97 in heavy afternoon trading. The slide came despite the company Tuesday posting second-quarter results that surpassed Wall Street expectations.
Onyx maintained its forecast of $850 million to $875 million in Nexavar revenue, but said sales probably will not reach the upper end of that range due to marketing and reimbursement timing in other countries, the timing of its launch as a liver cancer treatment in Japan, overall economic conditions and unfavorable exchange rates.
Most of the revenue on Nexavar sales goes to Onyx's partner Bayer Healthcare. Sales rose 19 percent to $201 million in the second quarter -- $60.2 million of which went to Onyx -- and sales have totaled $379.1 million in the first six months of the year.
Onyx also said it will raise money by selling up to 4.6 million shares of common stock and $200 million in convertible senior notes due 2016.
Thomas Weisel Partners analyst Stephen Willey said Onyx has a solid balance sheet and its raising money to finance a partnership or buyout.
"We believe today's financing sets the stage for a strategic late-stage acquisition or partnership within the oncology space," he said.
BMO Capital Markets analyst Jason Zhang said the timing of the fundraising was "curious," but said he thinks Onyx is either preparing to license a late-stage drug candidate from another company, or may just be looking to raise cash. In July, the company reported positive results from a mid stage study of Nexavar against metastatic breast cancer.
********
http://www.thestreet.com/_yahoo/story/10566124/1/onyx-fund-r…
Onyx Fund-Raising Sparks Cry for Shareholder Input
08/06/09 - 12:30 PM EDT
EMERYVILLE, Calif. (TheStreet) -- A large investor in Onyx Pharmaceuticals(ONXX Quote) wants shareholders to have the right to approve or veto any significant deal the company may seek to do in the future.
Samuel Isaly, portfolio manager at Orbimed Advisors, which owns about 4% of Onyx, requested greater shareholder control over Onyx's deal-making decisions after the company announced a new offering of stock and debt securities.
Isaly says he's worried that Onyx's desire to raise another $300 million or more -- on top of the $460 million already in the company's coffers -- might lead management into an expensive or highly dilutive deal that won't benefit shareholders.
"Onyx has said little about how they intend to use these proceeds. We're worried about the company embarking on a transaction of significant size in which the shareholder can't vote," says Isaly.
Asked to define "significant size," Isaly says he wants Onyx to rewrite its corporate by-laws to require a shareholder vote on any transaction greater than 25% of the company's market value.
Onyx's market capitalization is currently around $2 billion, so Isaly is asking for shareholder votes on deals priced at $500 million or higher.
Onyx executives could not be reached for comment.
Isaly says he hopes to steer Onyx clear of making the same mistake OSI Pharmaceuticals(OSIP Quote) made when it acquired Eyetech Pharmaceuticals in 2005 for about $1 billion. OSI engineered that deal in a way that prevented a shareholder vote, but the acquisition never delivered promised returns because Eyetech's eye drug failed commercially.
Onyx shares fell 15% to $31.18 Wednesday, the day after the company announced plans to sell 4 million shares of common stock and $200 million in convertible debt.
Onyx also reported a second-quarter profit Tuesday that beat the Street consensus estimate and guided to the lower end of the company's 2009 sales forecast for its cancer drug Nexavar.
Still, most of the questions on the company's Tuesday night conference call were about the financing plans.
"People are mad" about Onyx's fund-raising plans, said Cowen & Co. biotech analyst Phil Nadeau, who covers Onyx. Some investors, like Isaly, are worried about what Onyx might do with the money; others are concerned with the timing of the offering, he says.
"Why is Onyx raising money now shortly after announcing positive top-line data from the Nexavar breast cancer trial but before disclosing all the data from the study," Nadeau says.
Onyx CEO Tony Coles responded to this concern on Tuesday's conference call. "But let me reassure you that, as a former practicing physician, we're very confident and very comfortable with the statistical significance of these findings, and believe that when the full data set are shared with everyone, that you'll be as thrilled and as pleased as we are around these particular findings."
Nexavar is used to treat patients with kidney cancer and liver cancer. Onyx said 2009 sales would likely come in at the lower end of its $850 million to $875 million guidance range.
An expansion of Nexavar into breast cancer could bolster sales by hundreds of millions of dollars.
Nexavar is Onyx's only revenue-producing drug. The company has other drugs in the pipeline but all are in the early stages of testing. Nexavar is marketed worldwide through a joint venture between Onyx and the German drug maker Bayer.
mfg ipollit
Die Nexavar-Umsätze entwickeln sich mit 200 Mio USD (+19%) in Q2 weiter positiv. Für das Gesamtjahr werden 850 Mio USD erwartet, so das Nexavar bald die 1 Mrd USD Schwelle übersteigen sollte.
Überraschenderweise soll nun 300 Mio USD zusätzliches Cash aufgenommen, obwohl Onyx bereits profitabel ist und über ein sehr konfortables Cash-Polster von fast netto 500 Mio USD verfügt. Über den Grund, wofür Onyx 800 Mio USD Cash benötigt, wurden keine Angaben gemacht. Ein Problem ist die nach Nexavar schwache Pipeline... es gibt zurzeit nichts in PII/PIII außer Nexavar. Somit könnte Onyx versuchen, ein zugelassenes oder PIII-Produkt einzukaufen oder gleich ein ganzes Unternehmen zu übernehmen. Die Sorge, dass sich Onyx verkalkuliert und irgendeinen Schrott für viel Geld einkauft, hat den Kurs deutlich gedrückt. Ich bin gespannt, was Onyx mit dem Geld vorhat. Merkwürdig ist zudem, dass gerade jetzt die KE durchgeführt wird, obwohl angeblich sehr positive Brustkrebs-Daten zu Nexavar in den nächsten Monaten veröffentlicht werden sollen... falls die Daten wirklich so positiv sind, dann könnte ONXX sehr viel leichter an die 300 Mio USD kommen.
*********
Onyx Pharma slides on guidance, stock issue
Onyx shares fall as analysts point to Nexavar sales guidance and unexpected capital raise
On Wednesday August 5, 2009, 12:53 pm EDT
NEW YORK (AP) -- Onyx Pharmaceuticals Inc.'s shares skidded Wednesday after the cancer drug maker said it will issue stock and convertible notes, and that sales of its drug Nexavar probably will not reach the high end of its forecasts for the year.
Shares of the Emeryville, Calif., company lost $5.58, or 15.3 percent, to $30.97 in heavy afternoon trading. The slide came despite the company Tuesday posting second-quarter results that surpassed Wall Street expectations.
Onyx maintained its forecast of $850 million to $875 million in Nexavar revenue, but said sales probably will not reach the upper end of that range due to marketing and reimbursement timing in other countries, the timing of its launch as a liver cancer treatment in Japan, overall economic conditions and unfavorable exchange rates.
Most of the revenue on Nexavar sales goes to Onyx's partner Bayer Healthcare. Sales rose 19 percent to $201 million in the second quarter -- $60.2 million of which went to Onyx -- and sales have totaled $379.1 million in the first six months of the year.
Onyx also said it will raise money by selling up to 4.6 million shares of common stock and $200 million in convertible senior notes due 2016.
Thomas Weisel Partners analyst Stephen Willey said Onyx has a solid balance sheet and its raising money to finance a partnership or buyout.
"We believe today's financing sets the stage for a strategic late-stage acquisition or partnership within the oncology space," he said.
BMO Capital Markets analyst Jason Zhang said the timing of the fundraising was "curious," but said he thinks Onyx is either preparing to license a late-stage drug candidate from another company, or may just be looking to raise cash. In July, the company reported positive results from a mid stage study of Nexavar against metastatic breast cancer.
********
http://www.thestreet.com/_yahoo/story/10566124/1/onyx-fund-r…
Onyx Fund-Raising Sparks Cry for Shareholder Input
08/06/09 - 12:30 PM EDT
EMERYVILLE, Calif. (TheStreet) -- A large investor in Onyx Pharmaceuticals(ONXX Quote) wants shareholders to have the right to approve or veto any significant deal the company may seek to do in the future.
Samuel Isaly, portfolio manager at Orbimed Advisors, which owns about 4% of Onyx, requested greater shareholder control over Onyx's deal-making decisions after the company announced a new offering of stock and debt securities.
Isaly says he's worried that Onyx's desire to raise another $300 million or more -- on top of the $460 million already in the company's coffers -- might lead management into an expensive or highly dilutive deal that won't benefit shareholders.
"Onyx has said little about how they intend to use these proceeds. We're worried about the company embarking on a transaction of significant size in which the shareholder can't vote," says Isaly.
Asked to define "significant size," Isaly says he wants Onyx to rewrite its corporate by-laws to require a shareholder vote on any transaction greater than 25% of the company's market value.
Onyx's market capitalization is currently around $2 billion, so Isaly is asking for shareholder votes on deals priced at $500 million or higher.
Onyx executives could not be reached for comment.
Isaly says he hopes to steer Onyx clear of making the same mistake OSI Pharmaceuticals(OSIP Quote) made when it acquired Eyetech Pharmaceuticals in 2005 for about $1 billion. OSI engineered that deal in a way that prevented a shareholder vote, but the acquisition never delivered promised returns because Eyetech's eye drug failed commercially.
Onyx shares fell 15% to $31.18 Wednesday, the day after the company announced plans to sell 4 million shares of common stock and $200 million in convertible debt.
Onyx also reported a second-quarter profit Tuesday that beat the Street consensus estimate and guided to the lower end of the company's 2009 sales forecast for its cancer drug Nexavar.
Still, most of the questions on the company's Tuesday night conference call were about the financing plans.
"People are mad" about Onyx's fund-raising plans, said Cowen & Co. biotech analyst Phil Nadeau, who covers Onyx. Some investors, like Isaly, are worried about what Onyx might do with the money; others are concerned with the timing of the offering, he says.
"Why is Onyx raising money now shortly after announcing positive top-line data from the Nexavar breast cancer trial but before disclosing all the data from the study," Nadeau says.
Onyx CEO Tony Coles responded to this concern on Tuesday's conference call. "But let me reassure you that, as a former practicing physician, we're very confident and very comfortable with the statistical significance of these findings, and believe that when the full data set are shared with everyone, that you'll be as thrilled and as pleased as we are around these particular findings."
Nexavar is used to treat patients with kidney cancer and liver cancer. Onyx said 2009 sales would likely come in at the lower end of its $850 million to $875 million guidance range.
An expansion of Nexavar into breast cancer could bolster sales by hundreds of millions of dollars.
Nexavar is Onyx's only revenue-producing drug. The company has other drugs in the pipeline but all are in the early stages of testing. Nexavar is marketed worldwide through a joint venture between Onyx and the German drug maker Bayer.
mfg ipollit
VPHM...
eine positive Überraschung ist ausgeblieben: Vancocin wird bald generisch werden und Cinryze muss das entstehende Umsatzloch füllen. Da dies bereits seit langen erwartet wurde, hat der Kurs relativ wenig darauf reagiert.
http://www.reuters.com/article/marketsNews/idINN044488902009…
UPDATE 3-US panel backs plan for ViroPharma drug copies
Tue Aug 4, 2009 9:58pm EDT
By Lisa Richwine
SILVER SPRING, Md., Aug 4 (Reuters) - U.S. advisers on Tuesday backed Food and Drug Administration guidelines for testing less costly generic versions of the antibiotic Vancocin over the objections of manufacturer ViroPharma Inc (VPHM.O).
Analysts said the recommendation should lead to FDA approval in the coming months of generic competitors to ViroPharma's best-selling product used to fight intestinal infections.
The company's shares fell to $7.35 in after-hours trading, down from an earlier close of $7.78 on Nasdaq.
ViroPharma had argued to the panel that the FDA's proposed criteria for generic rivals were not rigorous enough to prove the copies would work as well as the brand-name product.
But panel members said they felt confident in the FDA's suggested test methods for generic pills with the same ingredients as Vancocin. The committee voted 16-0 to back the FDA plan for the drug, known generically as vancomycin.
Many of the outside experts said there was a public health need for cheaper versions of oral vancomycin to fight a growing number of intestinal infections with Clostridium difficile, a common and sometimes deadly cause of diarrhea.
Many hospitals and patients find brand-name Vancocin too expensive, panelists and generic drug industry representatives said. To cut costs, doctors often try an older generic drug that may not work as well, or prepare liquid formulations using a form of vancomycin meant for intravenous use.
"As a clinician, I really am in favor of moving forward on the generic for this drug," said Dr. Thomas Moore, a panelist and professor of medicine at the University of Kansas.
Given the rise in serious Clostridium difficile infections, "it seems more important than ever to increase access to oral vancomycin," Moore added.
Generic drugmakers including Mylan Inc (MYL.O) and Akorn Inc (AKRX.O) have applied for approval to sell copies of Vancocin.
Deutsche Bank analyst Robyn Karnauskas said investors expected the panel decision and are looking for future growth fueled by the company's rare disease drug, Cinryze.
ViroPharma's stock is "fairly valued without Vancocin at current levels" given strong Cinryze sales, Karnauskas said.
Ira Loss, an analyst with Washington Analysis, predicted the FDA would clear a copycat version by the end of the year.
ViroPharma had argued to the panel that the standards recommended by the FDA, which involve lab tests of how the drug dissolves in water, may not accurately reflect how the medicine would be absorbed in real patients with serious infections.
ViroPharma Chief Executive Vincent Milano said he was "very disappointed" by the panel vote. "I think this puts patients seriously at risk," he told reporters.
Milano said it was too early to project how much Vancocin's sales would fall if generic rivals reach the market and said Cinryze remained the "growth engine" for the company. Cinryze treats a rare swelling disease called hereditary angioedema.
Vancocin was ViroPharma's best-selling drug in 2008 with sales of $232 million. Last month, ViroPharma withdrew 2009 sales guidance for Vancocin while projecting Cinryze sales of $80 million to $95 million this year.

mfg ipollit
eine positive Überraschung ist ausgeblieben: Vancocin wird bald generisch werden und Cinryze muss das entstehende Umsatzloch füllen. Da dies bereits seit langen erwartet wurde, hat der Kurs relativ wenig darauf reagiert.
http://www.reuters.com/article/marketsNews/idINN044488902009…
UPDATE 3-US panel backs plan for ViroPharma drug copies
Tue Aug 4, 2009 9:58pm EDT
By Lisa Richwine
SILVER SPRING, Md., Aug 4 (Reuters) - U.S. advisers on Tuesday backed Food and Drug Administration guidelines for testing less costly generic versions of the antibiotic Vancocin over the objections of manufacturer ViroPharma Inc (VPHM.O).
Analysts said the recommendation should lead to FDA approval in the coming months of generic competitors to ViroPharma's best-selling product used to fight intestinal infections.
The company's shares fell to $7.35 in after-hours trading, down from an earlier close of $7.78 on Nasdaq.
ViroPharma had argued to the panel that the FDA's proposed criteria for generic rivals were not rigorous enough to prove the copies would work as well as the brand-name product.
But panel members said they felt confident in the FDA's suggested test methods for generic pills with the same ingredients as Vancocin. The committee voted 16-0 to back the FDA plan for the drug, known generically as vancomycin.
Many of the outside experts said there was a public health need for cheaper versions of oral vancomycin to fight a growing number of intestinal infections with Clostridium difficile, a common and sometimes deadly cause of diarrhea.
Many hospitals and patients find brand-name Vancocin too expensive, panelists and generic drug industry representatives said. To cut costs, doctors often try an older generic drug that may not work as well, or prepare liquid formulations using a form of vancomycin meant for intravenous use.
"As a clinician, I really am in favor of moving forward on the generic for this drug," said Dr. Thomas Moore, a panelist and professor of medicine at the University of Kansas.
Given the rise in serious Clostridium difficile infections, "it seems more important than ever to increase access to oral vancomycin," Moore added.
Generic drugmakers including Mylan Inc (MYL.O) and Akorn Inc (AKRX.O) have applied for approval to sell copies of Vancocin.
Deutsche Bank analyst Robyn Karnauskas said investors expected the panel decision and are looking for future growth fueled by the company's rare disease drug, Cinryze.
ViroPharma's stock is "fairly valued without Vancocin at current levels" given strong Cinryze sales, Karnauskas said.
Ira Loss, an analyst with Washington Analysis, predicted the FDA would clear a copycat version by the end of the year.
ViroPharma had argued to the panel that the standards recommended by the FDA, which involve lab tests of how the drug dissolves in water, may not accurately reflect how the medicine would be absorbed in real patients with serious infections.
ViroPharma Chief Executive Vincent Milano said he was "very disappointed" by the panel vote. "I think this puts patients seriously at risk," he told reporters.
Milano said it was too early to project how much Vancocin's sales would fall if generic rivals reach the market and said Cinryze remained the "growth engine" for the company. Cinryze treats a rare swelling disease called hereditary angioedema.
Vancocin was ViroPharma's best-selling drug in 2008 with sales of $232 million. Last month, ViroPharma withdrew 2009 sales guidance for Vancocin while projecting Cinryze sales of $80 million to $95 million this year.
mfg ipollit
Medigene... EndoTag-Gespräche dauern weiter an.
derstandard.at/fs/1246543971003/bMedigeneb-mit-weniger-Verlu…
Medigene mit weniger Verlust
07. August 2009, 10:29
Kostensenkungsmaßnahmen greifen
Frankfurt - Das deutsche Biotechunternehmen MediGene hat im ersten Halbjahr dank seines Krebsmittels Eligard mehr umgesetzt und seinen operativen Verlust verringert. "Einerseits steigen die Erlöse unserer Produkte auf dem Markt und andererseits greifen unsere Kostensenkungsmaßnahmen im Unternehmen", erklärte Finanzvorstand Thomas Klaue am Freitag im Bericht über die ersten sechs Monate 2009.
Bei der Partnersuche für sein Krebsmittel Endotag-1 kam das Unternehmen weiter voran. Die Verhandlungen sind Medigene zufolge inzwischen im fortgeschrittenem Stadium. Medigene sprach zuletzt mit großen Pharmakonzernen sowie auch mit Firmen mittlerer Größe. Früheren Angaben aus Kreisen zufolge hatte unter anderem der US-Konzern Pfizer ein Auge auf die Arznei geworfen.
Im ersten Halbjahr verringerte Medigene seinen Verlust im Jahresvergleich um 50 Prozent auf 8,3 Mio. Euro. Vor Zinsen, Steuern und Abschreibungen sank der operative Verlust (Ebitda) ebenfalls um 59 Prozent auf 6,8 Mio. Euro. Für das zweite Quartal stand ein operatives Minus von 4,9 Mio. Euro zu Buche - eine Verringerung von 45 Prozent. Medigene setzte im Zeitraum Jänner bis Juni 20 Mio. Euro um - das sind 45 Prozent mehr als vor einem Jahr. Dazu trugen hauptsächlich die Einnahmen aus dem Verkauf des Krebsmittels Eligard bei, das in Europa vom japanischen Pharmakonzern Astellas vermarktet wird.
Medigene hat sich in der Forschung und Entwicklung auf Medikamente gegen Krebs und Autoimmunerkrankungen konzentriert. Mit der Warzensalbe Veregen und dem Prostatakrebsmittel Eligard hat Medigene bereits zwei Medikamente auf dem Markt. (APA)
mfg ipollit
derstandard.at/fs/1246543971003/bMedigeneb-mit-weniger-Verlu…
Medigene mit weniger Verlust
07. August 2009, 10:29
Kostensenkungsmaßnahmen greifen
Frankfurt - Das deutsche Biotechunternehmen MediGene hat im ersten Halbjahr dank seines Krebsmittels Eligard mehr umgesetzt und seinen operativen Verlust verringert. "Einerseits steigen die Erlöse unserer Produkte auf dem Markt und andererseits greifen unsere Kostensenkungsmaßnahmen im Unternehmen", erklärte Finanzvorstand Thomas Klaue am Freitag im Bericht über die ersten sechs Monate 2009.
Bei der Partnersuche für sein Krebsmittel Endotag-1 kam das Unternehmen weiter voran. Die Verhandlungen sind Medigene zufolge inzwischen im fortgeschrittenem Stadium. Medigene sprach zuletzt mit großen Pharmakonzernen sowie auch mit Firmen mittlerer Größe. Früheren Angaben aus Kreisen zufolge hatte unter anderem der US-Konzern Pfizer ein Auge auf die Arznei geworfen.
Im ersten Halbjahr verringerte Medigene seinen Verlust im Jahresvergleich um 50 Prozent auf 8,3 Mio. Euro. Vor Zinsen, Steuern und Abschreibungen sank der operative Verlust (Ebitda) ebenfalls um 59 Prozent auf 6,8 Mio. Euro. Für das zweite Quartal stand ein operatives Minus von 4,9 Mio. Euro zu Buche - eine Verringerung von 45 Prozent. Medigene setzte im Zeitraum Jänner bis Juni 20 Mio. Euro um - das sind 45 Prozent mehr als vor einem Jahr. Dazu trugen hauptsächlich die Einnahmen aus dem Verkauf des Krebsmittels Eligard bei, das in Europa vom japanischen Pharmakonzern Astellas vermarktet wird.
Medigene hat sich in der Forschung und Entwicklung auf Medikamente gegen Krebs und Autoimmunerkrankungen konzentriert. Mit der Warzensalbe Veregen und dem Prostatakrebsmittel Eligard hat Medigene bereits zwei Medikamente auf dem Markt. (APA)
mfg ipollit
Zu Incyte:
Ohad Hammer hat den schon seit einiger Zeit angekündigten Beitrag zu INCY veröffentlicht.
A drug with an almost certain approval and immediate sales potential of hundreds of millions of dollars is an asset very few biotech companies possess. In that sense, Incyte (INCY), which is developing a breakthrough drug for blood disorders, represents a unique opportunity in an industry plagued by risk and uncertainty. Incyte is also unique in its problematic capital structure, which makes an otherwise simple investment decision into a tricky one.
Hier gehts weiter:
http://www.hammerstockblog.com/biotech-portfolio-updates-%E2…
Ohad Hammer hat den schon seit einiger Zeit angekündigten Beitrag zu INCY veröffentlicht.
A drug with an almost certain approval and immediate sales potential of hundreds of millions of dollars is an asset very few biotech companies possess. In that sense, Incyte (INCY), which is developing a breakthrough drug for blood disorders, represents a unique opportunity in an industry plagued by risk and uncertainty. Incyte is also unique in its problematic capital structure, which makes an otherwise simple investment decision into a tricky one.
Hier gehts weiter:
http://www.hammerstockblog.com/biotech-portfolio-updates-%E2…
Antwort auf Beitrag Nr.: 37.739.413 von SLGramann am 09.08.09 10:29:28
Im Actelion-Thread habe ich mir mal Actelion als Partner für Incyte gewünscht. Meiner Meinung nach müsste das aus mehreren Gründen eigentlich ganz gut passen.
Im Actelion-Thread habe ich mir mal Actelion als Partner für Incyte gewünscht.
Meiner Meinung nach müsste das aus mehreren Gründen eigentlich ganz gut passen.
Antwort auf Beitrag Nr.: 37.739.413 von SLGramann am 09.08.09 10:29:28@SLGramann... danke für den Hinweis!
http://www.hammerstockblog.com/biotech-portfolio-updates-%e2…
Ohad Hammer Biotech Blog - Incyte
A drug with an almost certain approval and immediate sales potential of hundreds of millions of dollars is an asset very few biotech companies possess. In that sense, Incyte (INCY), which is developing a breakthrough drug for blood disorders, represents a unique opportunity in an industry plagued by risk and uncertainty. Incyte is also unique in its problematic capital structure, which makes an otherwise simple investment decision into a tricky one.
Incyte’s lead drug is INCB18424 (aka 424), currently in two registration trials in myelofibrosis (MF). Myelofibrosis is a blood disorder in which the bone marrow becomes dysfunctional. MF Patients, who often have enlarged spleen and anemia, suffer from a myriad of symptoms including infections, chronic fatigue, fever and weight loss. On average, patients survive five years from diagnosis as a result of infection, bleeding and organ failure. There are currently no approved drugs for MF and most patients are treated with drugs that alleviate symptoms, but typically have little impact on the course of the disease.
Commercial opportunity
Quantifying the opportunity in myelofibrosis is difficult, as there are only rough estimates as to the actual number of patients who are living with MF. According to Incyte, there are approximately 14 thousand MF patients in the US. This figure is substantially lower than other sources such as the MPD foundation, which suggests a prevalence of 40 thousand patients in the US. Sticking with the company’s estimation, and assuming an average price of ~$15k per patient per year (which is consistent with other drugs for chronic diseases), the market potential for 424 as a MF drug is $210 million in the US. Together with Europe and Japan, the market potential in MF could reach $500M annually.
424 is also being evaluated in two phase II trials for additional indications. The first phase II study is in additional blood disorders that are similar to MF, but are more common, with a prevalence of ~175k patients in the US. The company will present updated data from this phase II trial at the ASH meeting this December, and according to management remarks during the recent quarterly call, results are encouraging. The drug is also in a phase II trial in psoriasis, where a topical version is used. Initial data read out from the trial is scheduled for September.
Incyte’s development strategy
424 belongs to a novel class of drugs called Jak inhibitors. These drugs inhibit the protein Jak, which is implicated in the development of many diseases, including autoimmune disorders and blood cancer. The most prominent Jak inhibitor in development is Pfizer’s (PFE) CP-690,550, currently in a pivotal study in Rheumatoid Arthritis (RA).
424 also has activity in RA, but Incyte chose MF as a primary indication. The market opportunity in RA is much greater, but it is also a crowded market characterized by a long and expensive development path. In contrast to RA, MF seems like an ideal indication for small companies such as Incyte, as it represents a substantial commercial opportunity coupled with a cheap and fast route to market. The allure of MF as an indication is further enhanced by the lack of approved drugs, which could make patients and physicians very receptive towards new treatment options. Lastly, there was a strong scientific rationale to start with MF, since 50% of MF patients carry a mutation in a gene for a specific Jak subtype called Jak2. 424 hits primarily Jak2 whereas Pfizer’s compound primarily inhibits a different subtype - Jak3.
So far, Incyte’s strategy proved successful. If everything goes according to plans, 424 will be the first ever Jak inhibitor to reach the market as well as the first ever approved drug for MF, thanks to unprecedented efficacy and a reasonable safety profile.
A fixed fight
Incyte is regarded as an attractive investment because it can offer investors what only a handful of biotech companies can: An extremely high likelihood of approval. Obviously, there is never a 100% guarantee in a clinical trial, but given the specific study design and the data 424 has generated to date, the drug’s approval is almost inevitable.
The company is enrolling patients in two registration trials, one in North America and the other in Europe. The two studies are similar in terms of patient population and endpoints with several minor differences. The most notable difference is treatment duration, as the US trial will evaluate spleen size after 24 weeks and the EU trial will evaluate spleen size after 48 weeks of treatment. As a result, Incyte hopes to file for FDA approval during the fourth quarter of 2010, followed by European filing in the second half of 2011.
Clinical benefit in MF can be measured based on three main criteria: reduction in spleen size, improvement in symptoms, and resolution of anemia. So far, 424 has been given to more than 200 patients at multiple doses and had a dramatic effect on spleen size, with a rapid and significant reduction. The drug also generated a dramatic improvement in disease symptoms, but unfortunately had little effect on the anemia associated with the disease. The most promising drug for MF related anemia is Celgene’s (CELG) pomalidomide, which showed very encouraging resolution of anemia. Consequently, there is a strong rationale for combining the two drugs.
The regulators on both sides of the Atlantic agreed to approve Incyte’s drug based on reduction in spleen size. The primary endpoint of the studies is the percentage of patients achieving a 35% reduction in spleen size following a predefined treatment period. It is important to note that the ultimate goal in myelofibrosis is increased survival, however, in order to prove that, very long and large studies are needed. Fortunately for Incyte, the FDA and the EMEA agreed to settle for short term endpoints and quality of life assessments.
As there are no approved drugs for myelofibrosis, 424 is being compared to placebo, which is always better than being tested against another drug with proven activity. Patients in the placebo arm may continue to receive other standard treatments, such as hydroxyurea and infusions, through the course of the studies. The same supportive treatments will also be given to patients in the 424 arm, so that any additive benefit of 424 would be easily observed.
In addition, based on the available data, 424’s ability to reduce spleen size was so profound and dramatic that the trial looks like a “fixed fight”. According to the company, 424 led to a durable 35% reduction in spleen size in about half of the patients. Patients in the placebo arm are very unlikely to experience such a dramatic benefit, even with supportive care.
The registration trials will also evaluate 424’s ability to alleviate disease symptoms, which will be assessed based on patient self reporting. Subjective metrics are always more prone to a “placebo effect”, as can be seen in Rheumatoid arthritis trials. Nevertheless, according to physicians, the placebo effect in MF is expected to be marginal, based on previous trials with ineffective drugs.
Competition
Incyte is not alone in the MF arena, as there are additional Jak inhibitors in development for this indication. The most advanced of Incyte’s competitors is TargeGen, a private company which is approximately 18 months behind Incyte. Other companies, including Astra Zeneca (AZN)and Cephalon (CEPH) are joining the race as well, but Incyte will probably enjoy a first mover advantage for at least a couple of years.
But Incyte’s lead is not only in the time to market but also in the clinical activity as well as in the safety profile of its drug. As MF is a chronic disease that requires prolonged treatment, MF drugs must be very well tolerated with minimal side effects. Developing a MF drug that is both safe and effective could be challenging, even for experts such as Exelixis (EXEL), which scrapped its Jak inhibitor earlier this year due to toxicity issues. Incyte’s compound has been given to over 200 patients, so its safety profile is established and seems very different from TargeGen’s and Cephalon’s (CEPH) compounds in terms of toxicity. While 424’s side effects include primarily bone marrow toxicity, the other two involve a high level of additional toxicities (especially gastrointestinal side effects), which might hamper their chances of approval and future market acceptance.
Debt overhang
Incyte has only one cardinal disadvantage - its cash balance. The company has a market cap of almost $550M, a cash position of $147.5M and debt of $420M in the form of convertible notes. Such an issue can weigh down on any company, regardless of how promising its pipeline is. In Incyte’s case, the debt could theoretically bring the company to the brink of bankruptcy in 2011, when the debt is due.
In order to solve the huge debt overhang, Incyte will probably have to orchestrate a complex move that includes licensing of several of its programs (including ex US rights for 424), debt recycling and fund-raising.
Business development activities
Incyte’s mission looks particularly challenging since each of the different variables in the equation have an effect on each other. For example, a licensing deal could lift the stock price and consequently allow the company to raise capital under better terms. On the other hand, the company will have a problem getting the licensing deals it wants with the current balance sheet as potential partners will try to take advantage of its delicate situation. The only way to improve capital structure prior to the licensing deals is selling equity, but the stock price will not fully reflect the large 424 licensing deal. Lastly, because there are several drugs the company could license, including a mid stage diabetes drug, a mid stage autoimmune program and a couple of early oncology agents, there is the issue of sequencing the licensing deals in an optimal manner.
It appears that Incyte decided to start with a licensing deal for 424, as the company hired Goldman Sachs in order to license the compound as early as possible. The deal is expected to include only oncology indications, as Incyte is trying to license another Jak program for inflammatory and autoimmune diseases.
Compounds like 424 are in high demand these days, as pharmaceutical companies are eager to find near term revenue sources. The drug has potential in multiple blood disorders as well as blood cancers, thus leaving a lot of upside for potential partners. From what we are hearing, Goldman’s bankers are working diligently with Incyte in order to finalize a deal already this quarter.
The MF program is Incyte’s only opportunity to create a recurring revenue stream in the near term, which is why the company insists on retaining the US commercialization rights for 424. This is similar to what Seattle Genetics (SGEN) is doing with its lead drug, SGN-35, which could become approved as early as the first half of 2011. Seattle Genetics also encountered some challenges in retaining the US rights for the drug, which forced it to do a secondary offering earlier this year. Today, Seattle Genetics enjoys a better cash position as well as a nice flow of future milestone payments from various partners, putting it in a favorable position to strike the deal it wants. Incyte’s advantage is the larger market potential 424 has, as opposed to the niche opportunity SGN-35 represents. Seattle Genetics is also expected to strike a lucrative licensing deal in the coming months.
Portfolio update and performance
Incyte represents a dilemma for investors. On the one hand, it has a promising phase III program with an extremely high likelihood of success and a revenue stream starting in 2011. On the other hand, the company is in a challenging financial position that requires a multi-step strategy for reducing debt in parallel to out-licensing activities.
According to the company’s management, the debt issue will be solved in the coming months, which implies the company is in advanced negotiations on multiple fronts. The recent flood of public offerings in the biotech arena reflects a revival in the capital markets, which could make Incyte’s mission easier.
We are initiating a position in the company since we believe it is clear that by this time next year, Incyte will be a much stronger company, with at least two corporate partnerships under its belt, lower debt and a higher number of outstanding shares. Until a deal is struck, the stock could fluctuate, but the deal could drive shares higher.

mfg ipollit
http://www.hammerstockblog.com/biotech-portfolio-updates-%e2…
Ohad Hammer Biotech Blog - Incyte
A drug with an almost certain approval and immediate sales potential of hundreds of millions of dollars is an asset very few biotech companies possess. In that sense, Incyte (INCY), which is developing a breakthrough drug for blood disorders, represents a unique opportunity in an industry plagued by risk and uncertainty. Incyte is also unique in its problematic capital structure, which makes an otherwise simple investment decision into a tricky one.
Incyte’s lead drug is INCB18424 (aka 424), currently in two registration trials in myelofibrosis (MF). Myelofibrosis is a blood disorder in which the bone marrow becomes dysfunctional. MF Patients, who often have enlarged spleen and anemia, suffer from a myriad of symptoms including infections, chronic fatigue, fever and weight loss. On average, patients survive five years from diagnosis as a result of infection, bleeding and organ failure. There are currently no approved drugs for MF and most patients are treated with drugs that alleviate symptoms, but typically have little impact on the course of the disease.
Commercial opportunity
Quantifying the opportunity in myelofibrosis is difficult, as there are only rough estimates as to the actual number of patients who are living with MF. According to Incyte, there are approximately 14 thousand MF patients in the US. This figure is substantially lower than other sources such as the MPD foundation, which suggests a prevalence of 40 thousand patients in the US. Sticking with the company’s estimation, and assuming an average price of ~$15k per patient per year (which is consistent with other drugs for chronic diseases), the market potential for 424 as a MF drug is $210 million in the US. Together with Europe and Japan, the market potential in MF could reach $500M annually.
424 is also being evaluated in two phase II trials for additional indications. The first phase II study is in additional blood disorders that are similar to MF, but are more common, with a prevalence of ~175k patients in the US. The company will present updated data from this phase II trial at the ASH meeting this December, and according to management remarks during the recent quarterly call, results are encouraging. The drug is also in a phase II trial in psoriasis, where a topical version is used. Initial data read out from the trial is scheduled for September.
Incyte’s development strategy
424 belongs to a novel class of drugs called Jak inhibitors. These drugs inhibit the protein Jak, which is implicated in the development of many diseases, including autoimmune disorders and blood cancer. The most prominent Jak inhibitor in development is Pfizer’s (PFE) CP-690,550, currently in a pivotal study in Rheumatoid Arthritis (RA).
424 also has activity in RA, but Incyte chose MF as a primary indication. The market opportunity in RA is much greater, but it is also a crowded market characterized by a long and expensive development path. In contrast to RA, MF seems like an ideal indication for small companies such as Incyte, as it represents a substantial commercial opportunity coupled with a cheap and fast route to market. The allure of MF as an indication is further enhanced by the lack of approved drugs, which could make patients and physicians very receptive towards new treatment options. Lastly, there was a strong scientific rationale to start with MF, since 50% of MF patients carry a mutation in a gene for a specific Jak subtype called Jak2. 424 hits primarily Jak2 whereas Pfizer’s compound primarily inhibits a different subtype - Jak3.
So far, Incyte’s strategy proved successful. If everything goes according to plans, 424 will be the first ever Jak inhibitor to reach the market as well as the first ever approved drug for MF, thanks to unprecedented efficacy and a reasonable safety profile.
A fixed fight
Incyte is regarded as an attractive investment because it can offer investors what only a handful of biotech companies can: An extremely high likelihood of approval. Obviously, there is never a 100% guarantee in a clinical trial, but given the specific study design and the data 424 has generated to date, the drug’s approval is almost inevitable.
The company is enrolling patients in two registration trials, one in North America and the other in Europe. The two studies are similar in terms of patient population and endpoints with several minor differences. The most notable difference is treatment duration, as the US trial will evaluate spleen size after 24 weeks and the EU trial will evaluate spleen size after 48 weeks of treatment. As a result, Incyte hopes to file for FDA approval during the fourth quarter of 2010, followed by European filing in the second half of 2011.
Clinical benefit in MF can be measured based on three main criteria: reduction in spleen size, improvement in symptoms, and resolution of anemia. So far, 424 has been given to more than 200 patients at multiple doses and had a dramatic effect on spleen size, with a rapid and significant reduction. The drug also generated a dramatic improvement in disease symptoms, but unfortunately had little effect on the anemia associated with the disease. The most promising drug for MF related anemia is Celgene’s (CELG) pomalidomide, which showed very encouraging resolution of anemia. Consequently, there is a strong rationale for combining the two drugs.
The regulators on both sides of the Atlantic agreed to approve Incyte’s drug based on reduction in spleen size. The primary endpoint of the studies is the percentage of patients achieving a 35% reduction in spleen size following a predefined treatment period. It is important to note that the ultimate goal in myelofibrosis is increased survival, however, in order to prove that, very long and large studies are needed. Fortunately for Incyte, the FDA and the EMEA agreed to settle for short term endpoints and quality of life assessments.
As there are no approved drugs for myelofibrosis, 424 is being compared to placebo, which is always better than being tested against another drug with proven activity. Patients in the placebo arm may continue to receive other standard treatments, such as hydroxyurea and infusions, through the course of the studies. The same supportive treatments will also be given to patients in the 424 arm, so that any additive benefit of 424 would be easily observed.
In addition, based on the available data, 424’s ability to reduce spleen size was so profound and dramatic that the trial looks like a “fixed fight”. According to the company, 424 led to a durable 35% reduction in spleen size in about half of the patients. Patients in the placebo arm are very unlikely to experience such a dramatic benefit, even with supportive care.
The registration trials will also evaluate 424’s ability to alleviate disease symptoms, which will be assessed based on patient self reporting. Subjective metrics are always more prone to a “placebo effect”, as can be seen in Rheumatoid arthritis trials. Nevertheless, according to physicians, the placebo effect in MF is expected to be marginal, based on previous trials with ineffective drugs.
Competition
Incyte is not alone in the MF arena, as there are additional Jak inhibitors in development for this indication. The most advanced of Incyte’s competitors is TargeGen, a private company which is approximately 18 months behind Incyte. Other companies, including Astra Zeneca (AZN)and Cephalon (CEPH) are joining the race as well, but Incyte will probably enjoy a first mover advantage for at least a couple of years.
But Incyte’s lead is not only in the time to market but also in the clinical activity as well as in the safety profile of its drug. As MF is a chronic disease that requires prolonged treatment, MF drugs must be very well tolerated with minimal side effects. Developing a MF drug that is both safe and effective could be challenging, even for experts such as Exelixis (EXEL), which scrapped its Jak inhibitor earlier this year due to toxicity issues. Incyte’s compound has been given to over 200 patients, so its safety profile is established and seems very different from TargeGen’s and Cephalon’s (CEPH) compounds in terms of toxicity. While 424’s side effects include primarily bone marrow toxicity, the other two involve a high level of additional toxicities (especially gastrointestinal side effects), which might hamper their chances of approval and future market acceptance.
Debt overhang
Incyte has only one cardinal disadvantage - its cash balance. The company has a market cap of almost $550M, a cash position of $147.5M and debt of $420M in the form of convertible notes. Such an issue can weigh down on any company, regardless of how promising its pipeline is. In Incyte’s case, the debt could theoretically bring the company to the brink of bankruptcy in 2011, when the debt is due.
In order to solve the huge debt overhang, Incyte will probably have to orchestrate a complex move that includes licensing of several of its programs (including ex US rights for 424), debt recycling and fund-raising.
Business development activities
Incyte’s mission looks particularly challenging since each of the different variables in the equation have an effect on each other. For example, a licensing deal could lift the stock price and consequently allow the company to raise capital under better terms. On the other hand, the company will have a problem getting the licensing deals it wants with the current balance sheet as potential partners will try to take advantage of its delicate situation. The only way to improve capital structure prior to the licensing deals is selling equity, but the stock price will not fully reflect the large 424 licensing deal. Lastly, because there are several drugs the company could license, including a mid stage diabetes drug, a mid stage autoimmune program and a couple of early oncology agents, there is the issue of sequencing the licensing deals in an optimal manner.
It appears that Incyte decided to start with a licensing deal for 424, as the company hired Goldman Sachs in order to license the compound as early as possible. The deal is expected to include only oncology indications, as Incyte is trying to license another Jak program for inflammatory and autoimmune diseases.
Compounds like 424 are in high demand these days, as pharmaceutical companies are eager to find near term revenue sources. The drug has potential in multiple blood disorders as well as blood cancers, thus leaving a lot of upside for potential partners. From what we are hearing, Goldman’s bankers are working diligently with Incyte in order to finalize a deal already this quarter.
The MF program is Incyte’s only opportunity to create a recurring revenue stream in the near term, which is why the company insists on retaining the US commercialization rights for 424. This is similar to what Seattle Genetics (SGEN) is doing with its lead drug, SGN-35, which could become approved as early as the first half of 2011. Seattle Genetics also encountered some challenges in retaining the US rights for the drug, which forced it to do a secondary offering earlier this year. Today, Seattle Genetics enjoys a better cash position as well as a nice flow of future milestone payments from various partners, putting it in a favorable position to strike the deal it wants. Incyte’s advantage is the larger market potential 424 has, as opposed to the niche opportunity SGN-35 represents. Seattle Genetics is also expected to strike a lucrative licensing deal in the coming months.
Portfolio update and performance
Incyte represents a dilemma for investors. On the one hand, it has a promising phase III program with an extremely high likelihood of success and a revenue stream starting in 2011. On the other hand, the company is in a challenging financial position that requires a multi-step strategy for reducing debt in parallel to out-licensing activities.
According to the company’s management, the debt issue will be solved in the coming months, which implies the company is in advanced negotiations on multiple fronts. The recent flood of public offerings in the biotech arena reflects a revival in the capital markets, which could make Incyte’s mission easier.
We are initiating a position in the company since we believe it is clear that by this time next year, Incyte will be a much stronger company, with at least two corporate partnerships under its belt, lower debt and a higher number of outstanding shares. Until a deal is struck, the stock could fluctuate, but the deal could drive shares higher.
mfg ipollit
Allos...
bezügl. des Zulassungsantrags von Pralatrexate gegen PTCL:
- Expertenrunde gibt am 2.9. eine Empfehlung ab
- FDA-Entscheidung ca. 24.9.
*******
Allos Therapeutics Announces FDA Advisory Committee to Review Pralatrexate for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma
On Monday August 10, 2009, 10:25 am EDT
WESTMINSTER, Colo.--(BUSINESS WIRE)--Allos Therapeutics, Inc. (Nasdaq: ALTH - News) announced today that the Company has received notification from the U.S. Food and Drug Administration’s (FDA) that the Oncologic Drugs Advisory Committee (ODAC) will hold a meeting on September 2, 2009, to review the Company’s New Drug Application (NDA) for pralatrexate for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
The Company submitted the NDA in March 2009. In May 2009, the FDA accepted the NDA for priority review and established a Prescription Drug User Fee Act date of September 24, 2009 for a decision regarding approval of the NDA.
About Pralatrexate
Pralatrexate is a selective antifolate designed to accumulate preferentially in cancer cells. Based on preclinical studies, the Company believes that pralatrexate selectively enters cells expressing RFC-1, a protein that is over expressed on certain cancer cells compared to normal cells. Once inside cancer cells, pralatrexate is efficiently polyglutamylated, which leads to high intracellular drug retention. Polyglutamylated pralatrexate essentially becomes “trapped” inside cancer cells, making it less susceptible to efflux-based drug resistance. Acting on the folate pathway, pralatrexate interferes with DNA synthesis and triggers cancer cell death.
About Peripheral T-cell Lymphoma
Peripheral T-cell lymphoma (PTCL) comprises a biologically diverse group of blood cancers that account for approximately 10% to 15% of all newly diagnosed cases of non-Hodgkin's lymphoma (NHL) in the U.S. The American Cancer Society estimates that approximately 66,000 new cases of NHL were diagnosed in the U.S. in 2008. The Company estimates the current annual incidence of PTCL in the U.S. to be approximately 5,600 patients. There are currently no pharmaceutical agents approved for use in the treatment of either first-line or relapsed or refractory PTCL. In addition to those PTCL patients who do not respond to first-line treatment, a significant number of first-line responders relapse or become refractory after treatment. According to published clinical data, patients with aggressive PTCL have an overall five-year survival rate of approximately 25% to 40%, depending on sub-type.
*********
Allos Therapeutics 2Q loss widens on costs
Allos Therapeutics' 2nd-quarter loss widens on costs as it prepares for praletrexate marketing
On Wednesday August 5, 2009, 7:16 am EDT
WESTMINSTER, Colo. (AP) -- Development-stage biotechnology company Allos Therapeutics Inc. said late Tuesday its second-quarter loss widened on higher costs as it prepared to market the cancer treatment praletrexate.
The company lost $16.8 million, or 19 cents per share, compared with a loss of $11.8 million, or 16 cents per share, during the same period a year prior. There was no revenue in either quarter.
Analysts polled by Thomson Reuters expected a loss of 17 cents per share.
Operating expenses rose 36 percent to $16.8 million on a mix of higher research and development costs and marketing, general and administrative costs.
The company's cancer treatment candidate pralatrexate is under review by the Food and Drug Administration, with a regulatory decision expected Sept. 24. The drug candidate is aimed at treating relapsed or refractory peripheral T-cell lymphoma, a type of cancer that attacks the body's immune system.
Allos had $105.2 million in cash, cash equivalents and investments in marketable securities at the end of the quarter. Looking ahead, the company said it now expects to spend between $65 million and $70 million, overall, in 2009 as it prepares for eventual commercialization of praletrexate.

mfg ipollit
bezügl. des Zulassungsantrags von Pralatrexate gegen PTCL:
- Expertenrunde gibt am 2.9. eine Empfehlung ab
- FDA-Entscheidung ca. 24.9.
*******
Allos Therapeutics Announces FDA Advisory Committee to Review Pralatrexate for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma
On Monday August 10, 2009, 10:25 am EDT
WESTMINSTER, Colo.--(BUSINESS WIRE)--Allos Therapeutics, Inc. (Nasdaq: ALTH - News) announced today that the Company has received notification from the U.S. Food and Drug Administration’s (FDA) that the Oncologic Drugs Advisory Committee (ODAC) will hold a meeting on September 2, 2009, to review the Company’s New Drug Application (NDA) for pralatrexate for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
The Company submitted the NDA in March 2009. In May 2009, the FDA accepted the NDA for priority review and established a Prescription Drug User Fee Act date of September 24, 2009 for a decision regarding approval of the NDA.
About Pralatrexate
Pralatrexate is a selective antifolate designed to accumulate preferentially in cancer cells. Based on preclinical studies, the Company believes that pralatrexate selectively enters cells expressing RFC-1, a protein that is over expressed on certain cancer cells compared to normal cells. Once inside cancer cells, pralatrexate is efficiently polyglutamylated, which leads to high intracellular drug retention. Polyglutamylated pralatrexate essentially becomes “trapped” inside cancer cells, making it less susceptible to efflux-based drug resistance. Acting on the folate pathway, pralatrexate interferes with DNA synthesis and triggers cancer cell death.
About Peripheral T-cell Lymphoma
Peripheral T-cell lymphoma (PTCL) comprises a biologically diverse group of blood cancers that account for approximately 10% to 15% of all newly diagnosed cases of non-Hodgkin's lymphoma (NHL) in the U.S. The American Cancer Society estimates that approximately 66,000 new cases of NHL were diagnosed in the U.S. in 2008. The Company estimates the current annual incidence of PTCL in the U.S. to be approximately 5,600 patients. There are currently no pharmaceutical agents approved for use in the treatment of either first-line or relapsed or refractory PTCL. In addition to those PTCL patients who do not respond to first-line treatment, a significant number of first-line responders relapse or become refractory after treatment. According to published clinical data, patients with aggressive PTCL have an overall five-year survival rate of approximately 25% to 40%, depending on sub-type.
*********
Allos Therapeutics 2Q loss widens on costs
Allos Therapeutics' 2nd-quarter loss widens on costs as it prepares for praletrexate marketing
On Wednesday August 5, 2009, 7:16 am EDT
WESTMINSTER, Colo. (AP) -- Development-stage biotechnology company Allos Therapeutics Inc. said late Tuesday its second-quarter loss widened on higher costs as it prepared to market the cancer treatment praletrexate.
The company lost $16.8 million, or 19 cents per share, compared with a loss of $11.8 million, or 16 cents per share, during the same period a year prior. There was no revenue in either quarter.
Analysts polled by Thomson Reuters expected a loss of 17 cents per share.
Operating expenses rose 36 percent to $16.8 million on a mix of higher research and development costs and marketing, general and administrative costs.
The company's cancer treatment candidate pralatrexate is under review by the Food and Drug Administration, with a regulatory decision expected Sept. 24. The drug candidate is aimed at treating relapsed or refractory peripheral T-cell lymphoma, a type of cancer that attacks the body's immune system.
Allos had $105.2 million in cash, cash equivalents and investments in marketable securities at the end of the quarter. Looking ahead, the company said it now expects to spend between $65 million and $70 million, overall, in 2009 as it prepares for eventual commercialization of praletrexate.
mfg ipollit
ARRY
positive PI-Ergebnisse von ARRY-403 bei T2 Diabetes... ARRY-403 soll eins der aussichtsreichsten Kandidaten in der Pipeline von Array sein.
Array BioPharma Reports Positive Results of Its Oral Glucokinase Activator in Type 2 Diabetes Patients
– GK Activator ARRY-403 Demonstrates Effective Glucose Control –
On Monday August 10, 2009, 4:30 pm EDT
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (Nasdaq: ARRY - News) today announced positive top-line data from a Phase 1 clinical trial in patients with Type 2 diabetes with its novel small molecule glucokinase activator (GKA), ARRY-403. The drug met its primary and secondary endpoints of safety, pharmacokinetics and glucose control.
ARRY-403 was evaluated in a Phase 1 single ascending dose study. The study included seven dose cohorts, with a total of 41 patients with Type 2 diabetes receiving either placebo or single doses of ARRY-403 ranging from 25 mg to 400 mg. ARRY-403 was well tolerated at all doses. ARRY-403 was rapidly absorbed, and exposure was dose-dependent. The pharmacokinetic profile is consistent with once daily therapeutic dosing. ARRY-403 provided dose-dependent reduction in glucose excursions in response to a standardized meal as well as reduction in 24-hour fasting blood glucose.
Array is initiating a multiple ascending dose study in patients with Type 2 diabetes to evaluate safety, exposure and glucose control over a 10-day period. The Company plans to report the combined Phase 1 results at an upcoming scientific meeting during the first half of 2010.
“These results are highly encouraging, validating this mechanism-of-action in Type 2 diabetic patients,” said Kevin Koch, Ph.D., President and Chief Scientific Officer. “Unlike current therapies, ARRY-403 enhances insulin production from the pancreas while simultaneously regulating glucose metabolism in the liver. Based on the pharmacokinetic and safety profile, coupled with effective glucose control, we believe ARRY-403 has the potential to be a first-line therapy with once-a-day dosing.”
mfg ipollit
positive PI-Ergebnisse von ARRY-403 bei T2 Diabetes... ARRY-403 soll eins der aussichtsreichsten Kandidaten in der Pipeline von Array sein.
Array BioPharma Reports Positive Results of Its Oral Glucokinase Activator in Type 2 Diabetes Patients
– GK Activator ARRY-403 Demonstrates Effective Glucose Control –
On Monday August 10, 2009, 4:30 pm EDT
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (Nasdaq: ARRY - News) today announced positive top-line data from a Phase 1 clinical trial in patients with Type 2 diabetes with its novel small molecule glucokinase activator (GKA), ARRY-403. The drug met its primary and secondary endpoints of safety, pharmacokinetics and glucose control.
ARRY-403 was evaluated in a Phase 1 single ascending dose study. The study included seven dose cohorts, with a total of 41 patients with Type 2 diabetes receiving either placebo or single doses of ARRY-403 ranging from 25 mg to 400 mg. ARRY-403 was well tolerated at all doses. ARRY-403 was rapidly absorbed, and exposure was dose-dependent. The pharmacokinetic profile is consistent with once daily therapeutic dosing. ARRY-403 provided dose-dependent reduction in glucose excursions in response to a standardized meal as well as reduction in 24-hour fasting blood glucose.
Array is initiating a multiple ascending dose study in patients with Type 2 diabetes to evaluate safety, exposure and glucose control over a 10-day period. The Company plans to report the combined Phase 1 results at an upcoming scientific meeting during the first half of 2010.
“These results are highly encouraging, validating this mechanism-of-action in Type 2 diabetic patients,” said Kevin Koch, Ph.D., President and Chief Scientific Officer. “Unlike current therapies, ARRY-403 enhances insulin production from the pancreas while simultaneously regulating glucose metabolism in the liver. Based on the pharmacokinetic and safety profile, coupled with effective glucose control, we believe ARRY-403 has the potential to be a first-line therapy with once-a-day dosing.”
mfg ipollit
Antwort auf Beitrag Nr.: 37.750.367 von ipollit am 11.08.09 00:50:32Array BioPharma Reports Financial Results for the Fourth Quarter and Full Year of Fiscal 2009
On Monday August 10, 2009, 4:34 pm EDT
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (NASDAQ: ARRY - News) today reported financial results for the fourth quarter and full year of fiscal 2009.
Array reported revenue of $5.5 million for the fourth quarter of fiscal 2009, compared to revenue of $6.1 million for the same period in fiscal 2008. Array invested $21.3 million in proprietary research and development for the quarter to advance its seven wholly-owned drugs in clinical development and select discovery programs. This compares to $28.4 million invested in research and development during the fourth quarter of fiscal 2008. Array reported a net loss of $26.7 million, or ($0.55) per share, for the fourth quarter, compared to a net loss of $32.4 million, or ($0.68) per share, for the fourth quarter in fiscal 2008. Array ended the fourth quarter of fiscal 2009 with $57.5 million in cash, cash equivalents and marketable securities. In addition, Array received $40.0 million in capital from Deerfield Private Design Fund, L.P. and Deerfield Private Design International, L.P. (collectively the “Deerfield Funds”) on August 5, 2009 under a loan facility agreement dated May 15, 2009.
Array reported revenue of $25.0 million for the fiscal year ended June 30, 2009, compared to revenue of $28.8 million for fiscal 2008. Net loss for the fiscal year ended June 30, 2009, was $127.8 million, or ($2.67) per share, compared to a net loss of $96.3 million, or ($2.04) per share, reported in fiscal 2008. Array invested $89.6 million in proprietary research and development for the year, compared to $90.3 million for fiscal 2008.
“We are pleased to report positive results from our Phase 1 clinical trial of ARRY-403 in patients with Type 2 diabetes,” said Robert E. Conway, Chief Executive Officer. “There are 24 million Type 2 diabetic patients in the U.S. with the incidence of the disease accelerating at an alarming rate. Many of these patients are unable to control their glucose levels with existing therapy and need better approaches to manage their disease. ARRY-403 provides a unique mechanism of action for controlling diabetes with the potential to address this critical unmet medical need. We have initiated an aggressive development program, while also seeking a partner to maximize ARRY-403’s benefit to patients.”
SUMMARY OF RECENT PROGRESS
Clinical trial completed in Type 2 diabetic patients:
ARRY-403 – GK activator for Type 2 diabetes: Array announced positive top-line data from a Phase 1 clinical trial in patients with Type 2 diabetes with its novel small molecule glucokinase activator (GKA), ARRY-403. The drug met its primary and secondary endpoints of safety, pharmacokinetics and glucose control. Additional data is available from Array BioPharma’s website at www.arraybiopharma.com
Six clinical programs advanced for the treatment of cancer:
ARRY-162 – MEK inhibitor for cancer: Array filed an investigational new drug application with the FDA and is now able to proceed with a Phase 1 clinical trial in cancer patients with its most advanced wholly-owned MEK inhibitor, ARRY-162. Recent publications have shown that the MEK pathway acts as a central axis in the proliferation of different tumors including melanoma, non-small cell lung, colorectal and pancreatic cancers. The Phase 1 dose escalation study is designed to evaluate safety, pharmacokinetics and pharmacodynamics of ARRY-162 following daily oral administration to patients with advanced solid tumors.
ARRY-520 – KSP inhibitor for AML & MM: Array continued a Phase 1 trial of ARRY-520, a novel KSP inhibitor, in patients with solid tumors and two Phase 1/2 trials in patients with acute myelogenous leukemia and multiple myeloma, respectively.
ARRY-614 - p38/Tie-2 Inhibitor for MDS: In March 2009, Array reported data from a Phase 1 clinical trial with ARRY-614 in a single and multiple dose-escalation study in healthy subjects. This study showed that ARRY-614 was well tolerated and exhibited dose-dependent increases in exposure and strong evidence of pharmacodynamic activity. Based on this data and strong biological support for testing a p38 inhibitor in myelodysplastic syndromes (MDS), Array is dosing patients with MDS in a Phase 1 trial with ARRY-614 to determine the safety, maximum tolerated dose, pharmacokinetics and preliminary estimates of efficacy of the compound in this patient population.
ARRY-543 - ErbB family inhibitor for solid tumors: Array completed enrollment in a Phase 1b trial with ARRY-543 in patients with ErbB2+ metastatic breast cancer and other ErbB-expressing tumors. Array continued a Phase 1 dose-escalation study with tablet formulation in patients with solid tumors and a Phase 1b trial in combination with Xeloda® (capecitabine) in patients with solid tumors. Array initiated dosing patients in two additional Phase 1b trials, in combination with Taxotere® (docetaxel) and Gemzar® (gemcitabine).
ARRY-380 - ErbB2 selective inhibitor for cancer: Patient recruitment in a Phase 1 trial with ARRY-380, an oral, selective ErbB2 (Her2) inhibitor, remains on track to complete enrollment this year. The trial is designed to evaluate the safety and pharmacokinetics of ARRY-380 in patients with advanced cancer and to establish the maximum tolerated dose.
ARRY-300 – MEK inhibitor: Array completed enrollment in a Phase 1 trial with ARRY-300, a targeted small molecule MEK inhibitor. The Phase 1 trial was a randomized, single-blind, placebo-controlled, single-ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of ARRY-300 in healthy volunteers. ARRY-300 will be a back-up for ARRY-162.
Two clinical program updates for the treatment of chronic inflammatory disease and pain:
ARRY-162 - MEK inhibitor for RA: Array completed a 12 week Phase 2, randomized, double-blind, placebo controlled global proof-of-concept trial with ARRY 162 in patients with active RA who were receiving stable doses of methotrexate for ≥ 6 weeks. Top-line results are expected to be available in September 2009.
ARRY-797 - p38 inhibitor for AS: Array announced top-line results from its Phase 1, seven-day, dose escalation trial up to 1,200 mg daily of ARRY-797 in healthy volunteers. In addition, the top-line results were announced in a second study, where ARRY-797 was evaluated in a 28-day Phase 1b trial in stable RA patients taking methotrexate. A preliminary analysis of both trials indicates that ARRY-797 was well-tolerated with a pharmacokinetic profile consistent with earlier studies. In the 28-day, three-arm RA study with a total of 28 patients, ARRY-797 showed inhibition of CRP levels (marker of inflammation) only during the first three weeks of dosing and a beneficial reduction in NTx levels (marker of bone remodeling) throughout the study. In addition, ARRY-797 showed a trend to improve the patients’ assessment of pain (VAS score) over the course of the study.
Array continues to conduct a full analysis of safety, pharmacokinetics and efficacy data from both studies and anticipates that complete results from the studies will be presented at a medical conference in 2010. Based on these preliminary results, Array has discontinued the enrollment of new patients in its clinical trial of ARRY-797 in ankylosing spondylitis. Array is evaluating alternative development paths in sub-chronic pain and supportive care indications.
Array’s Information Technology Platform recognized by CIO magazine: IDG's CIO magazine announced Array as a recipient of the 2009 CIO 100. The 22nd annual award program recognizes organizations around the world that exemplify the highest level of operational and strategic excellence in information technology.
Array received additional capital: Array received $40.0 million in additional capital from the Deerfield Funds on August 5, 2009 under a loan facility agreement dated May 15, 2009. The outstanding principal under the new loan is due by April 2014 and interest is payable monthly. Principal and interest can be repaid, at Array’s option, at any time with shares of Array common stock.
mfg ipollit
On Monday August 10, 2009, 4:34 pm EDT
BOULDER, Colo.--(BUSINESS WIRE)--Array BioPharma Inc. (NASDAQ: ARRY - News) today reported financial results for the fourth quarter and full year of fiscal 2009.
Array reported revenue of $5.5 million for the fourth quarter of fiscal 2009, compared to revenue of $6.1 million for the same period in fiscal 2008. Array invested $21.3 million in proprietary research and development for the quarter to advance its seven wholly-owned drugs in clinical development and select discovery programs. This compares to $28.4 million invested in research and development during the fourth quarter of fiscal 2008. Array reported a net loss of $26.7 million, or ($0.55) per share, for the fourth quarter, compared to a net loss of $32.4 million, or ($0.68) per share, for the fourth quarter in fiscal 2008. Array ended the fourth quarter of fiscal 2009 with $57.5 million in cash, cash equivalents and marketable securities. In addition, Array received $40.0 million in capital from Deerfield Private Design Fund, L.P. and Deerfield Private Design International, L.P. (collectively the “Deerfield Funds”) on August 5, 2009 under a loan facility agreement dated May 15, 2009.
Array reported revenue of $25.0 million for the fiscal year ended June 30, 2009, compared to revenue of $28.8 million for fiscal 2008. Net loss for the fiscal year ended June 30, 2009, was $127.8 million, or ($2.67) per share, compared to a net loss of $96.3 million, or ($2.04) per share, reported in fiscal 2008. Array invested $89.6 million in proprietary research and development for the year, compared to $90.3 million for fiscal 2008.
“We are pleased to report positive results from our Phase 1 clinical trial of ARRY-403 in patients with Type 2 diabetes,” said Robert E. Conway, Chief Executive Officer. “There are 24 million Type 2 diabetic patients in the U.S. with the incidence of the disease accelerating at an alarming rate. Many of these patients are unable to control their glucose levels with existing therapy and need better approaches to manage their disease. ARRY-403 provides a unique mechanism of action for controlling diabetes with the potential to address this critical unmet medical need. We have initiated an aggressive development program, while also seeking a partner to maximize ARRY-403’s benefit to patients.”
SUMMARY OF RECENT PROGRESS
Clinical trial completed in Type 2 diabetic patients:
ARRY-403 – GK activator for Type 2 diabetes: Array announced positive top-line data from a Phase 1 clinical trial in patients with Type 2 diabetes with its novel small molecule glucokinase activator (GKA), ARRY-403. The drug met its primary and secondary endpoints of safety, pharmacokinetics and glucose control. Additional data is available from Array BioPharma’s website at www.arraybiopharma.com
Six clinical programs advanced for the treatment of cancer:
ARRY-162 – MEK inhibitor for cancer: Array filed an investigational new drug application with the FDA and is now able to proceed with a Phase 1 clinical trial in cancer patients with its most advanced wholly-owned MEK inhibitor, ARRY-162. Recent publications have shown that the MEK pathway acts as a central axis in the proliferation of different tumors including melanoma, non-small cell lung, colorectal and pancreatic cancers. The Phase 1 dose escalation study is designed to evaluate safety, pharmacokinetics and pharmacodynamics of ARRY-162 following daily oral administration to patients with advanced solid tumors.
ARRY-520 – KSP inhibitor for AML & MM: Array continued a Phase 1 trial of ARRY-520, a novel KSP inhibitor, in patients with solid tumors and two Phase 1/2 trials in patients with acute myelogenous leukemia and multiple myeloma, respectively.
ARRY-614 - p38/Tie-2 Inhibitor for MDS: In March 2009, Array reported data from a Phase 1 clinical trial with ARRY-614 in a single and multiple dose-escalation study in healthy subjects. This study showed that ARRY-614 was well tolerated and exhibited dose-dependent increases in exposure and strong evidence of pharmacodynamic activity. Based on this data and strong biological support for testing a p38 inhibitor in myelodysplastic syndromes (MDS), Array is dosing patients with MDS in a Phase 1 trial with ARRY-614 to determine the safety, maximum tolerated dose, pharmacokinetics and preliminary estimates of efficacy of the compound in this patient population.
ARRY-543 - ErbB family inhibitor for solid tumors: Array completed enrollment in a Phase 1b trial with ARRY-543 in patients with ErbB2+ metastatic breast cancer and other ErbB-expressing tumors. Array continued a Phase 1 dose-escalation study with tablet formulation in patients with solid tumors and a Phase 1b trial in combination with Xeloda® (capecitabine) in patients with solid tumors. Array initiated dosing patients in two additional Phase 1b trials, in combination with Taxotere® (docetaxel) and Gemzar® (gemcitabine).
ARRY-380 - ErbB2 selective inhibitor for cancer: Patient recruitment in a Phase 1 trial with ARRY-380, an oral, selective ErbB2 (Her2) inhibitor, remains on track to complete enrollment this year. The trial is designed to evaluate the safety and pharmacokinetics of ARRY-380 in patients with advanced cancer and to establish the maximum tolerated dose.
ARRY-300 – MEK inhibitor: Array completed enrollment in a Phase 1 trial with ARRY-300, a targeted small molecule MEK inhibitor. The Phase 1 trial was a randomized, single-blind, placebo-controlled, single-ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of ARRY-300 in healthy volunteers. ARRY-300 will be a back-up for ARRY-162.
Two clinical program updates for the treatment of chronic inflammatory disease and pain:
ARRY-162 - MEK inhibitor for RA: Array completed a 12 week Phase 2, randomized, double-blind, placebo controlled global proof-of-concept trial with ARRY 162 in patients with active RA who were receiving stable doses of methotrexate for ≥ 6 weeks. Top-line results are expected to be available in September 2009.
ARRY-797 - p38 inhibitor for AS: Array announced top-line results from its Phase 1, seven-day, dose escalation trial up to 1,200 mg daily of ARRY-797 in healthy volunteers. In addition, the top-line results were announced in a second study, where ARRY-797 was evaluated in a 28-day Phase 1b trial in stable RA patients taking methotrexate. A preliminary analysis of both trials indicates that ARRY-797 was well-tolerated with a pharmacokinetic profile consistent with earlier studies. In the 28-day, three-arm RA study with a total of 28 patients, ARRY-797 showed inhibition of CRP levels (marker of inflammation) only during the first three weeks of dosing and a beneficial reduction in NTx levels (marker of bone remodeling) throughout the study. In addition, ARRY-797 showed a trend to improve the patients’ assessment of pain (VAS score) over the course of the study.
Array continues to conduct a full analysis of safety, pharmacokinetics and efficacy data from both studies and anticipates that complete results from the studies will be presented at a medical conference in 2010. Based on these preliminary results, Array has discontinued the enrollment of new patients in its clinical trial of ARRY-797 in ankylosing spondylitis. Array is evaluating alternative development paths in sub-chronic pain and supportive care indications.
Array’s Information Technology Platform recognized by CIO magazine: IDG's CIO magazine announced Array as a recipient of the 2009 CIO 100. The 22nd annual award program recognizes organizations around the world that exemplify the highest level of operational and strategic excellence in information technology.
Array received additional capital: Array received $40.0 million in additional capital from the Deerfield Funds on August 5, 2009 under a loan facility agreement dated May 15, 2009. The outstanding principal under the new loan is due by April 2014 and interest is payable monthly. Principal and interest can be repaid, at Array’s option, at any time with shares of Array common stock.
mfg ipollit
NicOx... hat das in der PII gescheiterte PF-03187207 von Pfizer zurückgekauft
http://www.reuters.com/article/rbssHealthcareNews/idUSL67511…
France's NicOx to buy back eye drug rights from Pfizer
Thu Aug 6, 2009 4:07am EDT
* Regains rights to PF-03187207 glaucoma drug candidate
* Gets access to certain Pfizer data on eye drug Xalatan
* Open to finding new partner for PF-03187207
* Shares rise as much as 6.7 percent shortly after open
PARIS, Aug 6 (Reuters) - French biotech firm NicOx (NCOX.PA) will buy back the rights to develop and market a drug that seeks to treat eye disease glaucoma from U.S. drugmaker Pfizer (PFE.N) and said it was open to finding another partner.
Last year, Pfizer decided not to take PF-03187207 into final Phase III clinical trials after a mid-stage study failed to meet its main goal, denting NicOx shares. NicOx had been working on developing eye drugs with Pfizer for over four years.
Shares in NicOx, which did not disclose the financial details of the deal, rose as much as 6.7 percent shortly after the stock market opened on Thursday. They added 4.8 percent to reach 9.94 euros by 0731 GMT, giving NicOx a value of 425 million euros.
Some analysts said the agreement was to be expected, with one Paris-based analyst calling NicOx shares "volatile".
"NicOx did not have much choice other than taking back the product," one Paris-based analyst, who declined to be identified, said. "It will not have cost them much and they could find a partner."
As well as PF-03187207, which has completed two mid-stage tests in patients with primary open-angle glaucoma and ocular hypertension, NicOx has won access to data on Pfizer's eye drug Xalatan. Pfizer said the research and development programme was no longer core to its strategy.
In addition, NicOx will regain rights to several new nitric oxide-donating compounds seeking to treat diabetic retinopathy and glaucoma, eye diseases which can cause blindness.
"We will evaluate opportunities for advancing PF-03187207 into Phase III (clinical trials), including possible third-party partnerships," Gavin Spencer, NicOx's vice president of business development, said in a statement on Thursday.
Glaucoma is a group of eye diseases which can lead to the loss of peripheral vision and eventually total blindness. Diabetic retinopathy represents the most common cause of blindness among adults.
NicOx is also looking for a partner to help it market its experimental anti-inflammatory drug naproxcinod in the United States but the biotech firm said in June completion of such talks could face a delay.
mfg ipollit
http://www.reuters.com/article/rbssHealthcareNews/idUSL67511…
France's NicOx to buy back eye drug rights from Pfizer
Thu Aug 6, 2009 4:07am EDT
* Regains rights to PF-03187207 glaucoma drug candidate
* Gets access to certain Pfizer data on eye drug Xalatan
* Open to finding new partner for PF-03187207
* Shares rise as much as 6.7 percent shortly after open
PARIS, Aug 6 (Reuters) - French biotech firm NicOx (NCOX.PA) will buy back the rights to develop and market a drug that seeks to treat eye disease glaucoma from U.S. drugmaker Pfizer (PFE.N) and said it was open to finding another partner.
Last year, Pfizer decided not to take PF-03187207 into final Phase III clinical trials after a mid-stage study failed to meet its main goal, denting NicOx shares. NicOx had been working on developing eye drugs with Pfizer for over four years.
Shares in NicOx, which did not disclose the financial details of the deal, rose as much as 6.7 percent shortly after the stock market opened on Thursday. They added 4.8 percent to reach 9.94 euros by 0731 GMT, giving NicOx a value of 425 million euros.
Some analysts said the agreement was to be expected, with one Paris-based analyst calling NicOx shares "volatile".
"NicOx did not have much choice other than taking back the product," one Paris-based analyst, who declined to be identified, said. "It will not have cost them much and they could find a partner."
As well as PF-03187207, which has completed two mid-stage tests in patients with primary open-angle glaucoma and ocular hypertension, NicOx has won access to data on Pfizer's eye drug Xalatan. Pfizer said the research and development programme was no longer core to its strategy.
In addition, NicOx will regain rights to several new nitric oxide-donating compounds seeking to treat diabetic retinopathy and glaucoma, eye diseases which can cause blindness.
"We will evaluate opportunities for advancing PF-03187207 into Phase III (clinical trials), including possible third-party partnerships," Gavin Spencer, NicOx's vice president of business development, said in a statement on Thursday.
Glaucoma is a group of eye diseases which can lead to the loss of peripheral vision and eventually total blindness. Diabetic retinopathy represents the most common cause of blindness among adults.
NicOx is also looking for a partner to help it market its experimental anti-inflammatory drug naproxcinod in the United States but the biotech firm said in June completion of such talks could face a delay.
mfg ipollit
SGEN... weitere 100 Mio USD KE
Seattle Genetics plans to sell shares
Seattle Genetics plans stock sale worth $106.6 million based on Monday's close
On Monday August 10, 2009, 6:12 pm EDT
BOTHELL, Wash. (AP) -- Biotechnology company Seattle Genetics Inc. said on Monday it will sell 9 million shares of common stock, with a 30-day option for underwriters to buy another 1.35 million shares.
Joint book-runners are JPMorgan and Goldman Sachs.
Based on Monday's closing price of $11.84, selling 9 million shares would raise almost $106.6 million. The company did not say what it will use the money for. Its shares fell 64 cents, or 5.4 percent, to $11.20 after the announcement was made.
**********
Seattle Genetics Achieves Milestone Under Antibody-Drug Conjugate Collaboration with MedImmune
On Monday August 10, 2009, 9:00 am EDT
BOTHELL, Wash.--(BUSINESS WIRE)--Seattle Genetics, Inc. (NASDAQ:SGEN - News), announced today that it has achieved a milestone under its antibody-drug conjugate (ADC) collaboration agreement with MedImmune, LLC, a wholly owned subsidiary of AstraZeneca. The milestone was triggered by MedImmune’s initiation of a phase I clinical trial of MEDI-547, its anti-EphA2 ADC for solid tumors.
“With multiple product candidates using our ADC technology now in clinical trials, and additional programs expected to advance into the clinic within the next 12 months, there is continued momentum by us and our collaborators in the development of ADCs for both hematologic malignancies and solid tumors,” said Eric L. Dobmeier, Chief Business Officer of Seattle Genetics. “We believe empowered antibodies are the next step in the evolution of antibody-based therapies because they can deliver targeted cell-killing without the widespread toxicity of conventional chemotherapy.”
ADCs utilize the targeting ability of monoclonal antibodies to deliver potent, cell-killing payloads to specific cells. Seattle Genetics’ technology employs synthetic, highly potent drugs attached to antibodies through proprietary linker systems. The linkers are designed to be stable in the bloodstream but to release the drug payload under specific conditions once inside target cells, thereby sparing non-target cells many of the toxic effects of traditional chemotherapy.
Under the ADC collaboration agreement, MedImmune has exclusive rights to use Seattle Genetics' ADC technology with monoclonal antibodies against two targets, including EphA2. Seattle Genetics received an upfront fee and MedImmune pays ongoing technology access and material supply fees. MedImmune has also agreed to make progress-dependent milestone payments and pay royalties on net sales of ADC products. MedImmune is responsible for research, product development, manufacturing and commercialization of any products resulting from the collaboration.
Seattle Genetics is advancing a proprietary pipeline of ADC programs, including brentuximab vedotin (SGN-35), which is in an ongoing pivotal trial for relapsed or refractory Hodgkin lymphoma and a phase II trial for systemic anaplastic large cell lymphoma. Seattle Genetics is also developing a number of preclinical ADCs, including SGN-75, which the company is advancing towards a planned 2009 investigational new drug submission for CD70-positive hematologic malignancies and solid tumors.

mfg ipollit
Seattle Genetics plans to sell shares
Seattle Genetics plans stock sale worth $106.6 million based on Monday's close
On Monday August 10, 2009, 6:12 pm EDT
BOTHELL, Wash. (AP) -- Biotechnology company Seattle Genetics Inc. said on Monday it will sell 9 million shares of common stock, with a 30-day option for underwriters to buy another 1.35 million shares.
Joint book-runners are JPMorgan and Goldman Sachs.
Based on Monday's closing price of $11.84, selling 9 million shares would raise almost $106.6 million. The company did not say what it will use the money for. Its shares fell 64 cents, or 5.4 percent, to $11.20 after the announcement was made.
**********
Seattle Genetics Achieves Milestone Under Antibody-Drug Conjugate Collaboration with MedImmune
On Monday August 10, 2009, 9:00 am EDT
BOTHELL, Wash.--(BUSINESS WIRE)--Seattle Genetics, Inc. (NASDAQ:SGEN - News), announced today that it has achieved a milestone under its antibody-drug conjugate (ADC) collaboration agreement with MedImmune, LLC, a wholly owned subsidiary of AstraZeneca. The milestone was triggered by MedImmune’s initiation of a phase I clinical trial of MEDI-547, its anti-EphA2 ADC for solid tumors.
“With multiple product candidates using our ADC technology now in clinical trials, and additional programs expected to advance into the clinic within the next 12 months, there is continued momentum by us and our collaborators in the development of ADCs for both hematologic malignancies and solid tumors,” said Eric L. Dobmeier, Chief Business Officer of Seattle Genetics. “We believe empowered antibodies are the next step in the evolution of antibody-based therapies because they can deliver targeted cell-killing without the widespread toxicity of conventional chemotherapy.”
ADCs utilize the targeting ability of monoclonal antibodies to deliver potent, cell-killing payloads to specific cells. Seattle Genetics’ technology employs synthetic, highly potent drugs attached to antibodies through proprietary linker systems. The linkers are designed to be stable in the bloodstream but to release the drug payload under specific conditions once inside target cells, thereby sparing non-target cells many of the toxic effects of traditional chemotherapy.
Under the ADC collaboration agreement, MedImmune has exclusive rights to use Seattle Genetics' ADC technology with monoclonal antibodies against two targets, including EphA2. Seattle Genetics received an upfront fee and MedImmune pays ongoing technology access and material supply fees. MedImmune has also agreed to make progress-dependent milestone payments and pay royalties on net sales of ADC products. MedImmune is responsible for research, product development, manufacturing and commercialization of any products resulting from the collaboration.
Seattle Genetics is advancing a proprietary pipeline of ADC programs, including brentuximab vedotin (SGN-35), which is in an ongoing pivotal trial for relapsed or refractory Hodgkin lymphoma and a phase II trial for systemic anaplastic large cell lymphoma. Seattle Genetics is also developing a number of preclinical ADCs, including SGN-75, which the company is advancing towards a planned 2009 investigational new drug submission for CD70-positive hematologic malignancies and solid tumors.
mfg ipollit
Evotec...
ganz klar ist es mir nicht, warum Evotec so steigt... eigentlich sehe ich im Moment keine großen Chancen, wenn man Evotec mehr auf Dienstleistungen ausrichtet. Inzwischen vielleicht schon etwas zu teuer.
http://www.handelsblatt.com/unternehmen/industrie/biotechfir…
Biotechfirma Evotec setzt auf indische Kostenstrukturen
10.08.2009
von Siegfried Hofmann
Mit der Hamburger Evotec AG hat sich jetzt erstmals ein deutsches Biotechunternehmen in Indien engagiert. Durch die Übernahme des Synthese-Spezialisten RSIPL will Evotec die Kapazitäten und seine Marktpräsenz in der Wirkstoff-Suche erweitern.
FRANKFURT. Für knapp drei Mio. Euro erwirbt Evotec eine 70-prozentige Beteiligung an der indischen RSIPL, einem Serviceunternehmen im Bereich der Chemiesynthese. Ziel ist es, mit Hilfe von RSIPL eine Marktposition als global führender Serviceanbieter in der Wirkstoff-Forschung für die Pharmabranche aufzubauen.
Die Akquisition ist damit Teil eines Strategiewandels, den der neue Firmenchef Werner Lanthaler vor einigen Monaten bei dem Hamburger Unternehmen einleitete. Lanthaler hat das Engagement von Evotec in der Entwicklung eigener Medikamente gekürzt und versucht statt dessen, das alte Kerngeschäft im Bereich der Wirkstoff-Suche für externe Partner, insbesondere große Pharmahersteller, auszubauen. Er glaubt, dass Evotec in diesem Bereich technologisch führend ist. Der Zukauf des indischen Unternehmens mit rund 160 Mitarbeitern soll nun dabei helfen, die Kapazitäten zu erweitern und die Effizienz zu steigern. "Wir kombinieren sozusagen deutsche Wertarbeit mit asiatischen Kostenstrukturen", so Lanthaler.
Bei der Wirkstoffsuche (drug discovery) geht es vor allem darum, große Sammlungen potenzieller Pharmamoleküle oder Molekül-Fragmente darauf zu testen, inwieweit sie an therapeutische relevante Zielmoleküle binden. Auch versucht man, schon in dieser Phase Hinweise auf toxische Eigenschaften zu erhalten. Dazu erwarb Evotec kürzlich eine in England und Singapur tätige Forschungseinheit für die Prüfung von Pharmasubstanzen an Zebrafischen.
Ausgangsbasis für das Hamburger Unternehmen sind selbst entwickelte Verfahren für das so genannte Screening von Substanzen. Darüber hinaus ist das Unternehmen auch in der Strukturanalyse und Optimierung von Wirkstoffkandidaten engagiert. Zu den wichtigsten Partnern und Kunden aus der Pharmabranche gehören Novartis, Roche und Boehringer Ingelheim. Servicegebühren und Erfolgsprovisionen (Meilensteinzahlungen) von diesen Partnern stellen den Löwenanteil der Evotec-Umsätze von 39 Mio Euro im vergangenen Jahr und 18,7 Mio Euro (plus 29 Prozent) im ersten Halbjahr 2009.
Als wenig erfolgreich entpuppte sich dagegen bisher der kostenträchtige Versuch, die Technologieplattform für die Entwicklung eigener Medikamente zu nutzen. Gleich zwei wichtige Projekte, ein potenzielles Schlafmittel und ein Wirkstoff zur Raucherentwöhnung, musste das Unternehmen in den vergangenen Monaten beerdigen, weil die klinischen Daten nicht stimmten und man keine Lizenznehmer aus der Pharmabranche finden konnte.
Lanthaler will die Medikamenten-Entwicklung zwar nicht aufgegeben, aber deutlich zurückfahren, um auf diese Weise das Risiko zu reduzieren und die Cash-Position (von derzeit knapp 73 Mio Euro) zu schonen. Gleichzeitig versucht er, neue Wachstumsperspektiven für das Servicegeschäft zu entwickeln.
Westliche Biotechfirmen sind in Indien bisher noch kaum engagiert, während große Pharmafirmen zum Teil schon größere Aktivitäten betreiben, unter anderem in Produktion und klinischer Entwicklung. Insbesondere als Lieferanten von patentfreien Wirkstoffen (Generika) sind indische Firmen bereits relativ stark im Geschäft. Seit einigen Jahren versuchen sie zudem, auch in der Pharmaforschung und Biotechnologie stärker Fuß zu fassen.

mfg ipollit
ganz klar ist es mir nicht, warum Evotec so steigt... eigentlich sehe ich im Moment keine großen Chancen, wenn man Evotec mehr auf Dienstleistungen ausrichtet. Inzwischen vielleicht schon etwas zu teuer.
http://www.handelsblatt.com/unternehmen/industrie/biotechfir…
Biotechfirma Evotec setzt auf indische Kostenstrukturen
10.08.2009
von Siegfried Hofmann
Mit der Hamburger Evotec AG hat sich jetzt erstmals ein deutsches Biotechunternehmen in Indien engagiert. Durch die Übernahme des Synthese-Spezialisten RSIPL will Evotec die Kapazitäten und seine Marktpräsenz in der Wirkstoff-Suche erweitern.
FRANKFURT. Für knapp drei Mio. Euro erwirbt Evotec eine 70-prozentige Beteiligung an der indischen RSIPL, einem Serviceunternehmen im Bereich der Chemiesynthese. Ziel ist es, mit Hilfe von RSIPL eine Marktposition als global führender Serviceanbieter in der Wirkstoff-Forschung für die Pharmabranche aufzubauen.
Die Akquisition ist damit Teil eines Strategiewandels, den der neue Firmenchef Werner Lanthaler vor einigen Monaten bei dem Hamburger Unternehmen einleitete. Lanthaler hat das Engagement von Evotec in der Entwicklung eigener Medikamente gekürzt und versucht statt dessen, das alte Kerngeschäft im Bereich der Wirkstoff-Suche für externe Partner, insbesondere große Pharmahersteller, auszubauen. Er glaubt, dass Evotec in diesem Bereich technologisch führend ist. Der Zukauf des indischen Unternehmens mit rund 160 Mitarbeitern soll nun dabei helfen, die Kapazitäten zu erweitern und die Effizienz zu steigern. "Wir kombinieren sozusagen deutsche Wertarbeit mit asiatischen Kostenstrukturen", so Lanthaler.
Bei der Wirkstoffsuche (drug discovery) geht es vor allem darum, große Sammlungen potenzieller Pharmamoleküle oder Molekül-Fragmente darauf zu testen, inwieweit sie an therapeutische relevante Zielmoleküle binden. Auch versucht man, schon in dieser Phase Hinweise auf toxische Eigenschaften zu erhalten. Dazu erwarb Evotec kürzlich eine in England und Singapur tätige Forschungseinheit für die Prüfung von Pharmasubstanzen an Zebrafischen.
Ausgangsbasis für das Hamburger Unternehmen sind selbst entwickelte Verfahren für das so genannte Screening von Substanzen. Darüber hinaus ist das Unternehmen auch in der Strukturanalyse und Optimierung von Wirkstoffkandidaten engagiert. Zu den wichtigsten Partnern und Kunden aus der Pharmabranche gehören Novartis, Roche und Boehringer Ingelheim. Servicegebühren und Erfolgsprovisionen (Meilensteinzahlungen) von diesen Partnern stellen den Löwenanteil der Evotec-Umsätze von 39 Mio Euro im vergangenen Jahr und 18,7 Mio Euro (plus 29 Prozent) im ersten Halbjahr 2009.
Als wenig erfolgreich entpuppte sich dagegen bisher der kostenträchtige Versuch, die Technologieplattform für die Entwicklung eigener Medikamente zu nutzen. Gleich zwei wichtige Projekte, ein potenzielles Schlafmittel und ein Wirkstoff zur Raucherentwöhnung, musste das Unternehmen in den vergangenen Monaten beerdigen, weil die klinischen Daten nicht stimmten und man keine Lizenznehmer aus der Pharmabranche finden konnte.
Lanthaler will die Medikamenten-Entwicklung zwar nicht aufgegeben, aber deutlich zurückfahren, um auf diese Weise das Risiko zu reduzieren und die Cash-Position (von derzeit knapp 73 Mio Euro) zu schonen. Gleichzeitig versucht er, neue Wachstumsperspektiven für das Servicegeschäft zu entwickeln.
Westliche Biotechfirmen sind in Indien bisher noch kaum engagiert, während große Pharmafirmen zum Teil schon größere Aktivitäten betreiben, unter anderem in Produktion und klinischer Entwicklung. Insbesondere als Lieferanten von patentfreien Wirkstoffen (Generika) sind indische Firmen bereits relativ stark im Geschäft. Seit einigen Jahren versuchen sie zudem, auch in der Pharmaforschung und Biotechnologie stärker Fuß zu fassen.
mfg ipollit
Antwort auf Beitrag Nr.: 37.750.410 von ipollit am 11.08.09 01:25:44aufgrund der fehlenden Phantasie habe ich mein Evotec-Position halbiert und in Erwartung guter NHL-Ergebnisse von Arzerra meine Genmab Position etwas erhöht, obwohl diese Aktie kaum Momentum besitzt... mal schauen, sollten die anstehenden Ergebnisse alle negativ ausfallen und Arzerra von der FDA keine Zulassung erhalten, dann ist es wohl keine gute Entscheidung gewesen. 
mfg ipollit

mfg ipollit
Antwort auf Beitrag Nr.: 37.752.261 von ipollit am 11.08.09 11:16:39
Ich habe gestern eine kleine Position in Incyte aufgebaut. Zu Genmab kann ich mich irgendwie nicht durchringen, aber das ist nur ein irrationales Gefühl.
Ich habe gestern eine kleine Position in Incyte aufgebaut. Zu Genmab kann ich mich irgendwie nicht durchringen, aber das ist nur ein irrationales Gefühl.
Antwort auf Beitrag Nr.: 37.752.809 von SLGramann am 11.08.09 12:16:12hoffe mal, dass die bei INCY den Schuldenberg in den Griff bekommen.
In Genmab bin ich eigentlich zu hoch investiert... recht viel Risiko, denn die Zukunft von Genmab ist noch recht unsicher. In den nächsten Monaten stehen wichtige Ereignisse an: angeblich diesen Monat PIII-Ergebnisse von Arzerra bei NHL, bei denen Rituxan nicht wirkt... also in der Anwendung nach Rituxan. Dies dürfte eine Blockbusterindikation sein. Ende Oktober entscheidet die FDA über die Zulassung von Arzerra bei CLL... von der Expertenrunde gab es eine positive Empfehlung, die FDA Unterlagen enthielten aber auch einige Kritikpunkte, weshalb die Zulassung zwar wahrscheinlich ist, aber noch Unsicherheiten bestehen. Ende des Jahres könnten die PIII-Daten zu Zalutumumab vorliegen... da wird sich zeigen, ob dieses Produkt ebenfalls großes Potential besitzt. In dem ein oder anderen Erfolgsfall sollte auch Übernahmefantasie durch GSK entstehen.

mfg ipollit
In Genmab bin ich eigentlich zu hoch investiert... recht viel Risiko, denn die Zukunft von Genmab ist noch recht unsicher. In den nächsten Monaten stehen wichtige Ereignisse an: angeblich diesen Monat PIII-Ergebnisse von Arzerra bei NHL, bei denen Rituxan nicht wirkt... also in der Anwendung nach Rituxan. Dies dürfte eine Blockbusterindikation sein. Ende Oktober entscheidet die FDA über die Zulassung von Arzerra bei CLL... von der Expertenrunde gab es eine positive Empfehlung, die FDA Unterlagen enthielten aber auch einige Kritikpunkte, weshalb die Zulassung zwar wahrscheinlich ist, aber noch Unsicherheiten bestehen. Ende des Jahres könnten die PIII-Daten zu Zalutumumab vorliegen... da wird sich zeigen, ob dieses Produkt ebenfalls großes Potential besitzt. In dem ein oder anderen Erfolgsfall sollte auch Übernahmefantasie durch GSK entstehen.
mfg ipollit
zu ISIS
http://seekingalpha.com/article/155356-isis-makes-anti-sense…
ISIS Makes (Anti) Sense (and Value)
August 11, 2009
Since bottoming near the end of 2008, ISIS Pharmaceuticals (ISIS) has been one of the hottest stocks in the first half of 2009.
...
What is Isis Pharmaceuticals?
Isis Pharmaceuticals is a drug discovery and development company which is the leading researcher in the field of antisense therapies. Antisense is a treatment for genetic disorders which focuses on the use of specially coded nucleic strands to bind to mRNA within a patient’s cells. For those of you who remember high school biology class, mRNA is the nucleic acid which carries coding information from the nucleus to the ribosomes for protein synthesis. A sense is a single sequence which codes for a particular protein. Antisense therapies focus on suppressing expression of a particular sense which effectively “turns off” particular genes.
Antisense drugs are a new class of pharmaceuticals and have the potential to change the way diseases are treated. Since these drugs are highly specific, targeting individual proteins being coded by RNA, they represent a large step towards more personalized medicine and theoretically should reduce unwanted side-effects in comparison to traditional protein or small-molecule pharmaceuticals. Moreover, since all antisense treatments utilize the same basic functionality – the delivery of an antisense molecule to target a specific RNA sequence – development times for new drugs can be shortened as early stage responses are more predictable.
Isis was the first biotech firm to bring an antisense therapy to market (Vitravene in 1998) and is a market leader in the manipulation and research of RNA.
Liquidity and Profitability
Isis reported $638 million in cash in its last filing and expects to burn roughly $90 million in cash over the course of the year. At current cash burn rates, the Company should be more than able to finance the development of its more promising drugs without needing to access capital markets over the next few years. A good thing given the current environment for both debt and equity capital.
Consensus estimates have the Company losing roughly 30 cents per share in 2010. The Company’s drug pipeline includes 19 drugs in development either by Isis or in partnership with another pharmaceutical, but its most promising drug, Mipomersen (a drug for the treatment of high cholesterol in development with Genzyme), is not likely to be filed with the FDA until late 2010.
Investment Thesis
As with many smaller biotech firms, Isis does not have a large portfolio of on patent, in-the-market drugs and the stock today trades on the expectation of profitability as opposed to real earnings power. Isis benefits, however, from consistent research and development revenues as a result of partnerships with larger pharmaceuticals. Antisense and gene therapies in general represent the next generation of pharmaceuticals and are of particular interest to big pharma which faces dwindling patent protections and uninspired drug pipelines.
Isis’ track record as the only biotech to bring an antisense drug to market as well as the current co-developer of a phase III cholesterol drug which could receive FDA approval by 2010 makes it a particularly coveted partner (and potential acquisition candidate). With the Company’s long term stock trend in tact and intermediate stock action showing resilience near its 50-day moving average, I’m ready to jump in on this stock as a speculative buy with a target in the low-$20s.

mfg ipollit
http://seekingalpha.com/article/155356-isis-makes-anti-sense…
ISIS Makes (Anti) Sense (and Value)
August 11, 2009
Since bottoming near the end of 2008, ISIS Pharmaceuticals (ISIS) has been one of the hottest stocks in the first half of 2009.
...
What is Isis Pharmaceuticals?
Isis Pharmaceuticals is a drug discovery and development company which is the leading researcher in the field of antisense therapies. Antisense is a treatment for genetic disorders which focuses on the use of specially coded nucleic strands to bind to mRNA within a patient’s cells. For those of you who remember high school biology class, mRNA is the nucleic acid which carries coding information from the nucleus to the ribosomes for protein synthesis. A sense is a single sequence which codes for a particular protein. Antisense therapies focus on suppressing expression of a particular sense which effectively “turns off” particular genes.
Antisense drugs are a new class of pharmaceuticals and have the potential to change the way diseases are treated. Since these drugs are highly specific, targeting individual proteins being coded by RNA, they represent a large step towards more personalized medicine and theoretically should reduce unwanted side-effects in comparison to traditional protein or small-molecule pharmaceuticals. Moreover, since all antisense treatments utilize the same basic functionality – the delivery of an antisense molecule to target a specific RNA sequence – development times for new drugs can be shortened as early stage responses are more predictable.
Isis was the first biotech firm to bring an antisense therapy to market (Vitravene in 1998) and is a market leader in the manipulation and research of RNA.
Liquidity and Profitability
Isis reported $638 million in cash in its last filing and expects to burn roughly $90 million in cash over the course of the year. At current cash burn rates, the Company should be more than able to finance the development of its more promising drugs without needing to access capital markets over the next few years. A good thing given the current environment for both debt and equity capital.
Consensus estimates have the Company losing roughly 30 cents per share in 2010. The Company’s drug pipeline includes 19 drugs in development either by Isis or in partnership with another pharmaceutical, but its most promising drug, Mipomersen (a drug for the treatment of high cholesterol in development with Genzyme), is not likely to be filed with the FDA until late 2010.
Investment Thesis
As with many smaller biotech firms, Isis does not have a large portfolio of on patent, in-the-market drugs and the stock today trades on the expectation of profitability as opposed to real earnings power. Isis benefits, however, from consistent research and development revenues as a result of partnerships with larger pharmaceuticals. Antisense and gene therapies in general represent the next generation of pharmaceuticals and are of particular interest to big pharma which faces dwindling patent protections and uninspired drug pipelines.
Isis’ track record as the only biotech to bring an antisense drug to market as well as the current co-developer of a phase III cholesterol drug which could receive FDA approval by 2010 makes it a particularly coveted partner (and potential acquisition candidate). With the Company’s long term stock trend in tact and intermediate stock action showing resilience near its 50-day moving average, I’m ready to jump in on this stock as a speculative buy with a target in the low-$20s.
mfg ipollit
Antwort auf Beitrag Nr.: 37.750.392 von ipollit am 11.08.09 01:11:33noch etwas zu SGEN...
http://www.biohealthinvestor.com/2009/08/seattle-genetics-bo…
Seattle Genetics’ Boosted Secondary… Takeover Defense?
August 12, 2009
Seattle Genetics, Inc. (NASDAQ: SGEN) is one of those companies in the world of biotech and biohealth which some have believed would be an ultimate takeover target. The issue is that some companies either do not have enough funding on their own to secure a deal under the terms they would prefer. Other companies want to grow and thrive on their own in the quest to become the next mega-blockbuster producer on the block. So the news of an offering of common stock from Seattle Genetics, Inc. would now put the company in the second category, or so it may seem.
Seattle Genetics priced a secondary offering of 11,000,000 shares of common stock at $10.75 per share. The gross proceeds from the sale are expected to be approximately $118.2 million and should close on or about August 17, 2009. The good news is that all of the shares in the offering are being sold by Seattle Genetics. The other bit of good news is that the demand must have been strong. The announcement was made Monday that it would sell 9 million shares, yet this pricing is for 11 million shares.
The company said that the use of funds will be used to fund R&D, including manufacturing activities and clinical trials for its proprietary product candidates, build-out of a commercial infrastructure and for general corporate purposes, including working capital.
The company also has a very high profile group of companies as the underwriters.
J.P. Morgan and Goldman Sachs are the joint book-running managers; Needham, Oppenheimer, RBC Capital Markets, and William Blair are all listed as the co-managers of the offering. The underwriters have a 30-day option to purchase up to 1,650,000 additional shares of common stock to cover over-allotments.
Back in July this stock was under $10.00. Then came several developments. It reported earnings, Bristol-Myers Squibb (NYSE: BMY) announced the acquisition of Medarex (NASDAQ: MEDX), then the company announced the initiation of Phase II trials of brentuximab vedotin (SGN-35) for lymphoma. Then this week came the announcement that Seattle Genetics had achieved a milestone under its antibody-drug conjugate (ADC) collaboration agreement with MedImmune, LLC, a wholly owned subsidiary of AstraZeneca, after MedImmune’s initiation of a phase I clinical trial of MEDI-547 for solid tumors.
Seattle Genetics has an exclusive worldwide collaboration agreement with Roche’s Genentech for the development and commercialization of dacetuzumab (SGN-40). Under the agreement, Genentech paid us $60 million upfront, and has agreed to pay potentially more than $800 million in milestones and escalating double-digit royalties starting in the mid-teens on annual product sales. In addition, Genentech funds research, development, manufacturing and commercialization costs. Seattle Genetics also has an option for co-promotion rights on dacetuzumab in the U.S.
The company has licensed its antibody-drug conjugate (ADC) technology to Genentech, Bayer (NYSE: BAY), CuraGen Corp. (NASDAQ: CRGN), Progenics Pharmaceuticals Inc. (NASDAQ: PGNX), Daiichi Sankyo, AstraZeneca’s (NYSE: AZN) MedImmune, and Takeda’s Millennium. These collaborations all involve upfront cash payments, milestones and royalties on net sales of products incorporating Seattle Genetics’ ADC technology. The licensees are responsible for development, manufacturing and commercialization of any ADC product candidates that result from the collaborations.
Another deal is in place as a co-development agreement with Agensys, a wholly-owned subsidiary of Astellas Pharma. In this the companies will jointly develop ADC products where Agensys provides proprietary targets and monoclonal antibodies to be utilized with Seattle Genetics’ proprietary ADC technology. The companies share research and development costs on up to two ADC products, and share equally in any profits.
Shares closed down over 7% at $10.99 in anticipation of the secondary offering and the stock’s 52-week range is $6.81 to $13.40. The company has been public since 2001 and has spent the bulk of the time from early 2007 to now trading in a range of $8.00 to $12.00. Its market before the effects of any secondary offering was $947 million after Tuesday’s drop. SGEN is also a stock with a high short interest: in mid-July it had over 5.5 million shares in the short interest, about 13.9 days worth of average trading volume.
This is still very much a research and development stage company. Analysts are only looking for about $38 million in revenues for 2009 and about $54 million in revenues in 2010. Until its products are closer to commercialization, losses are expected well beyond this year and next.
mfg ipollit
http://www.biohealthinvestor.com/2009/08/seattle-genetics-bo…
Seattle Genetics’ Boosted Secondary… Takeover Defense?
August 12, 2009
Seattle Genetics, Inc. (NASDAQ: SGEN) is one of those companies in the world of biotech and biohealth which some have believed would be an ultimate takeover target. The issue is that some companies either do not have enough funding on their own to secure a deal under the terms they would prefer. Other companies want to grow and thrive on their own in the quest to become the next mega-blockbuster producer on the block. So the news of an offering of common stock from Seattle Genetics, Inc. would now put the company in the second category, or so it may seem.
Seattle Genetics priced a secondary offering of 11,000,000 shares of common stock at $10.75 per share. The gross proceeds from the sale are expected to be approximately $118.2 million and should close on or about August 17, 2009. The good news is that all of the shares in the offering are being sold by Seattle Genetics. The other bit of good news is that the demand must have been strong. The announcement was made Monday that it would sell 9 million shares, yet this pricing is for 11 million shares.
The company said that the use of funds will be used to fund R&D, including manufacturing activities and clinical trials for its proprietary product candidates, build-out of a commercial infrastructure and for general corporate purposes, including working capital.
The company also has a very high profile group of companies as the underwriters.
J.P. Morgan and Goldman Sachs are the joint book-running managers; Needham, Oppenheimer, RBC Capital Markets, and William Blair are all listed as the co-managers of the offering. The underwriters have a 30-day option to purchase up to 1,650,000 additional shares of common stock to cover over-allotments.
Back in July this stock was under $10.00. Then came several developments. It reported earnings, Bristol-Myers Squibb (NYSE: BMY) announced the acquisition of Medarex (NASDAQ: MEDX), then the company announced the initiation of Phase II trials of brentuximab vedotin (SGN-35) for lymphoma. Then this week came the announcement that Seattle Genetics had achieved a milestone under its antibody-drug conjugate (ADC) collaboration agreement with MedImmune, LLC, a wholly owned subsidiary of AstraZeneca, after MedImmune’s initiation of a phase I clinical trial of MEDI-547 for solid tumors.
Seattle Genetics has an exclusive worldwide collaboration agreement with Roche’s Genentech for the development and commercialization of dacetuzumab (SGN-40). Under the agreement, Genentech paid us $60 million upfront, and has agreed to pay potentially more than $800 million in milestones and escalating double-digit royalties starting in the mid-teens on annual product sales. In addition, Genentech funds research, development, manufacturing and commercialization costs. Seattle Genetics also has an option for co-promotion rights on dacetuzumab in the U.S.
The company has licensed its antibody-drug conjugate (ADC) technology to Genentech, Bayer (NYSE: BAY), CuraGen Corp. (NASDAQ: CRGN), Progenics Pharmaceuticals Inc. (NASDAQ: PGNX), Daiichi Sankyo, AstraZeneca’s (NYSE: AZN) MedImmune, and Takeda’s Millennium. These collaborations all involve upfront cash payments, milestones and royalties on net sales of products incorporating Seattle Genetics’ ADC technology. The licensees are responsible for development, manufacturing and commercialization of any ADC product candidates that result from the collaborations.
Another deal is in place as a co-development agreement with Agensys, a wholly-owned subsidiary of Astellas Pharma. In this the companies will jointly develop ADC products where Agensys provides proprietary targets and monoclonal antibodies to be utilized with Seattle Genetics’ proprietary ADC technology. The companies share research and development costs on up to two ADC products, and share equally in any profits.
Shares closed down over 7% at $10.99 in anticipation of the secondary offering and the stock’s 52-week range is $6.81 to $13.40. The company has been public since 2001 and has spent the bulk of the time from early 2007 to now trading in a range of $8.00 to $12.00. Its market before the effects of any secondary offering was $947 million after Tuesday’s drop. SGEN is also a stock with a high short interest: in mid-July it had over 5.5 million shares in the short interest, about 13.9 days worth of average trading volume.
This is still very much a research and development stage company. Analysts are only looking for about $38 million in revenues for 2009 and about $54 million in revenues in 2010. Until its products are closer to commercialization, losses are expected well beyond this year and next.
mfg ipollit
Antwort auf Beitrag Nr.: 37.753.832 von ipollit am 11.08.09 14:07:31zu Genmab
PII 1st-line Ergebnisse in CLL in Kombi mit FC... da die Studie keinen Placebo-Arm enthält und relativ klein ist, sind die Ergebnisse schlecht einzuordnen. Die Vom Prinzip vergleichbare PIII-Studie CLL8 mit Rituxan erzielte weit höhere overall response rates mit 93% und ein CR von 45% im Vergleich zu Arzerra mit RR von bis zu 77% und einem CR von bis zu 50%. Der direkte Vergleich scheint aber verzerrt zu sein, da der Placebo-Arm (also nur der mit FC behandelte Arm) in CLL8 bereits ein RR von 85% und ein CR von 23% besitzt. Wären die Studien vergleichbar, dann sollte die RR mit 73%-77% für FC+Arzerra nicht unterhalb der RR nur FC in CLL8 mit 85% liegen. Die Patienten in der PII scheinen demnach schwerer behandelbar zu sein, wofür die erzielten CR-Raten dann wieder gut sind.
Genmab Announces Preliminary Top-Line Results for Arzerra(TM) in Front Line CLL
11.08.2009
Copenhagen, Denmark; August 11, 2009 - Genmab A/S (OMX: GEN) announced today
top-line results from the Phase II study of Arzerra(TM) (ofatumumab) in
combination with fludarabine and cyclophosphamide (FC) to treat chronic
lymphocytic leukemia (CLL) in previously untreated patients.
A total of 61 patients were treated in the study. Treatment response was
assessed using the 1996 National Cancer Institute Guidelines. The complete
remission rate was 32% in patients who received 500 mg of ofatumumab (n=31) and
50% in patients who received 1000 mg of ofatumumab (n=30). The overall response
rate was 77% in the 500 mg treatment group and 73% in the 1000 mg treatment
group.
There were no unexpected safety findings reported during treatment and within 30
days after last infusion. The most common adverse event reported was neutropenia
at 48%. Other common adverse events (greater that 15 percent) were nausea,
leukopenia, rash, vomiting, pyrexia, headache and thrombocytopenia. The number
of patients, who experienced adverse events, including serious adverse events,
was similar between the two dose groups. One death was reported and was judged
by the investigator as unrelated to ofatumumab.
‘We are pleased to see the positive results of this first study investigating
ofatumumab for the treatment of front line CLL,’ said Lisa N. Drakeman, Ph.D.,
Chief Executive Officer of Genmab. ‘We look forward to presenting the full data
at a future medical meeting.’
About the study
Patients in this open label study were randomized into two treatment groups.
Each patient was to receive one infusion of 300 mg of ofatumumab in combination
with FC followed by 5 monthly infusions of either 500 or 1000 mg of ofatumumab
in combination with FC. Disease status was measured every 4 weeks until week 24
and every 3 months thereafter until disease progression or 24 months. Treatment
response was assessed according to the 1996 National Cancer Institute Working
Group guidelines by an Independent endpoints Review Committee. Patients not
having progressed on their disease at 24 months will be followed for disease
progression at 6 month intervals until 60 months.
The objective of the study was to determine the efficacy of ofatumumab in
combination with FC in previously untreated CLL patients. The primary endpoint
was complete remission rate from start of treatment until 3 months after last
infusion.
***********
Arzerra: good results in 1st line CLL
- Positive results for Arzerra Phase II in 1st line CLL
Genmab reported positive top line data of the Phase II investigating Arzerra in combination with Fludarabine and cyclophosphamide (FC) in un-treated CLL
patients. The 61 CLL patients enrolled received one infusion of 300mg followed by 5 monthly infusions of either 500mg or 1,000mg of Arzerra, with FC. The primary endpoint was complete remission rate (CR) at 3 months after last infusion.
- Good response rate and complete response rate
CR was 32% and 50% in the 500mg and 1,000mg group, respectively, which we view as impressive in light of the 77% and 73% overall response rate in both groups, respectively, suggesting hard to treat patients. The clinical trial continues to assess patients and is expected to complete by the end of 2013 (clinicaltrial.gov).
- Comparison to Rituximab in 1st line CLL – CLL8 Phase III study
In the Phase III CLL8 study investigating Rituximab in 1st line CLL with or without FC, overall response rate was 93% and 85% in the Rituximab and placebo arm, respectively, and CR was 45% and 23%, respectively. Although the two clinical trials cannot be compared, we believe the CR achieved by Arzerra is rather
impressive in light of the lower Overall Response Rate, suggesting harder to treat patients in the Arzerra Phase II.
- Valuation
Our risk adjusted DCF valuation of Genmab remains DKr270.

mfg ipollit
PII 1st-line Ergebnisse in CLL in Kombi mit FC... da die Studie keinen Placebo-Arm enthält und relativ klein ist, sind die Ergebnisse schlecht einzuordnen. Die Vom Prinzip vergleichbare PIII-Studie CLL8 mit Rituxan erzielte weit höhere overall response rates mit 93% und ein CR von 45% im Vergleich zu Arzerra mit RR von bis zu 77% und einem CR von bis zu 50%. Der direkte Vergleich scheint aber verzerrt zu sein, da der Placebo-Arm (also nur der mit FC behandelte Arm) in CLL8 bereits ein RR von 85% und ein CR von 23% besitzt. Wären die Studien vergleichbar, dann sollte die RR mit 73%-77% für FC+Arzerra nicht unterhalb der RR nur FC in CLL8 mit 85% liegen. Die Patienten in der PII scheinen demnach schwerer behandelbar zu sein, wofür die erzielten CR-Raten dann wieder gut sind.
Genmab Announces Preliminary Top-Line Results for Arzerra(TM) in Front Line CLL
11.08.2009
Copenhagen, Denmark; August 11, 2009 - Genmab A/S (OMX: GEN) announced today
top-line results from the Phase II study of Arzerra(TM) (ofatumumab) in
combination with fludarabine and cyclophosphamide (FC) to treat chronic
lymphocytic leukemia (CLL) in previously untreated patients.
A total of 61 patients were treated in the study. Treatment response was
assessed using the 1996 National Cancer Institute Guidelines. The complete
remission rate was 32% in patients who received 500 mg of ofatumumab (n=31) and
50% in patients who received 1000 mg of ofatumumab (n=30). The overall response
rate was 77% in the 500 mg treatment group and 73% in the 1000 mg treatment
group.
There were no unexpected safety findings reported during treatment and within 30
days after last infusion. The most common adverse event reported was neutropenia
at 48%. Other common adverse events (greater that 15 percent) were nausea,
leukopenia, rash, vomiting, pyrexia, headache and thrombocytopenia. The number
of patients, who experienced adverse events, including serious adverse events,
was similar between the two dose groups. One death was reported and was judged
by the investigator as unrelated to ofatumumab.
‘We are pleased to see the positive results of this first study investigating
ofatumumab for the treatment of front line CLL,’ said Lisa N. Drakeman, Ph.D.,
Chief Executive Officer of Genmab. ‘We look forward to presenting the full data
at a future medical meeting.’
About the study
Patients in this open label study were randomized into two treatment groups.
Each patient was to receive one infusion of 300 mg of ofatumumab in combination
with FC followed by 5 monthly infusions of either 500 or 1000 mg of ofatumumab
in combination with FC. Disease status was measured every 4 weeks until week 24
and every 3 months thereafter until disease progression or 24 months. Treatment
response was assessed according to the 1996 National Cancer Institute Working
Group guidelines by an Independent endpoints Review Committee. Patients not
having progressed on their disease at 24 months will be followed for disease
progression at 6 month intervals until 60 months.
The objective of the study was to determine the efficacy of ofatumumab in
combination with FC in previously untreated CLL patients. The primary endpoint
was complete remission rate from start of treatment until 3 months after last
infusion.
***********
Arzerra: good results in 1st line CLL
- Positive results for Arzerra Phase II in 1st line CLL
Genmab reported positive top line data of the Phase II investigating Arzerra in combination with Fludarabine and cyclophosphamide (FC) in un-treated CLL
patients. The 61 CLL patients enrolled received one infusion of 300mg followed by 5 monthly infusions of either 500mg or 1,000mg of Arzerra, with FC. The primary endpoint was complete remission rate (CR) at 3 months after last infusion.
- Good response rate and complete response rate
CR was 32% and 50% in the 500mg and 1,000mg group, respectively, which we view as impressive in light of the 77% and 73% overall response rate in both groups, respectively, suggesting hard to treat patients. The clinical trial continues to assess patients and is expected to complete by the end of 2013 (clinicaltrial.gov).
- Comparison to Rituximab in 1st line CLL – CLL8 Phase III study
In the Phase III CLL8 study investigating Rituximab in 1st line CLL with or without FC, overall response rate was 93% and 85% in the Rituximab and placebo arm, respectively, and CR was 45% and 23%, respectively. Although the two clinical trials cannot be compared, we believe the CR achieved by Arzerra is rather
impressive in light of the lower Overall Response Rate, suggesting harder to treat patients in the Arzerra Phase II.
- Valuation
Our risk adjusted DCF valuation of Genmab remains DKr270.
mfg ipollit
zu Genmab
(ipollit, Du hast es sicher schon gelesen, Arzerra mit einem deutlichen Rückschlag bei NHL):
LONDON, Aug 18 (Reuters) - Disappointing clinical results sent shares in Danish biotechnology company Genmab (GEN.CO) tumbling 26 percent on Tuesday as investors questioned sales prospects for its key drug Arzerra in treating blood cancer.
Arzerra's failure to help patients with non-Hodgkin's lymphoma (NHL) as much as hoped will also hit Genmab's bottom line in 2009, since it will not now get expected milestone payments linked to the indication from partner GlaxoSmithKline (GSK.L).
http://www.reuters.com/article/rbssHealthcareNews/idUSLI5820…
(ipollit, Du hast es sicher schon gelesen, Arzerra mit einem deutlichen Rückschlag bei NHL):
LONDON, Aug 18 (Reuters) - Disappointing clinical results sent shares in Danish biotechnology company Genmab (GEN.CO) tumbling 26 percent on Tuesday as investors questioned sales prospects for its key drug Arzerra in treating blood cancer.
Arzerra's failure to help patients with non-Hodgkin's lymphoma (NHL) as much as hoped will also hit Genmab's bottom line in 2009, since it will not now get expected milestone payments linked to the indication from partner GlaxoSmithKline (GSK.L).
http://www.reuters.com/article/rbssHealthcareNews/idUSLI5820…
Antwort auf Beitrag Nr.: 37.806.162 von SLGramann am 18.08.09 22:41:22
(Link übernommen von PathFinder2 aus dem MorphoSys-Thread)
(Link übernommen von PathFinder2 aus dem MorphoSys-Thread)
zu Micromet
gibts einen neuen Beitrag von Ohad Hammer:
http://www.hammerstockblog.com/the-clock-is-ticking-on-micro…
gibts einen neuen Beitrag von Ohad Hammer:
http://www.hammerstockblog.com/the-clock-is-ticking-on-micro…
Zu Regeneron:
Ein (zunächst kleinerer?) Rückschlag bei VEGF Trap:
PARIS and TARRYTOWN, N.Y., Sept 11, 2009 /PRNewswire-FirstCall via COMTEX/ -- Sanofi-aventis (Euronext: SAN and NYSE: SNY) and Regeneron Pharmaceuticals, Inc. /quotes/comstock/15*!regn/quotes/nls/regn (REGN 21.88, -0.11, -0.50%) today announced the discontinuation of the Phase 3 trial that evaluated aflibercept (VEGF Trap) plus gemcitabine versus placebo plus gemcitabine for the first-line treatment of metastatic pancreatic cancer (VANILLA), based on the recommendations by an Independent Data Monitoring Committee (IDMC). As part of a planned interim efficacy analysis, the IDMC determined that the addition of aflibercept to gemcitabine would be unable to demonstrate a statistically significant improvement in the primary endpoint of overall survival compared to placebo plus gemcitabine in this study. The types and frequencies of adverse events reported on the combination arm with aflibercept were generally as anticipated.
With the closure of the study, a detailed analysis of the efficacy and safety results will be conducted by the companies and results will be presented at a future medical meeting. Sanofi-aventis and Regeneron have notified the study investigators and appropriate regulatory authorities of the decision to discontinue the study. Patients in the study will continue to be provided access to aflibercept at the determination of the study investigators in consultation with the patients.
Metastatic pancreatic cancer is among the most intractable cancers. Clinical development of new therapies, including anti-VEGF agents, has been generally characterized by a failure to achieve significant incremental clinical benefit over existing treatments.
"We are disappointed with the result of this study and we will continue our efforts to bring new and effective treatments for these patients," said Dr. Marc Cluzel, Senior Vice President, Research and Development sanofi-aventis. "We remain committed to the other ongoing Phase 3 trials of aflibercept in colorectal cancer, non-small cell lung cancer, and hormone-refractory metastatic prostate cancer."
Three Phase 3 studies continue, each of which is currently over 70 percent enrolled:
-- VELOUR study: 2nd-line metastatic colorectal cancer in combination with
fluorouracil, leucovorin, and irinotecan (FOLFIRI)
-- VITAL study: 2nd-line non-small cell lung cancer in combination with
docetaxel
-- VENICE study: 1st-line hormone-refractory metastatic prostate cancer in
combination with docetaxel and prednisone
Ein (zunächst kleinerer?) Rückschlag bei VEGF Trap:
PARIS and TARRYTOWN, N.Y., Sept 11, 2009 /PRNewswire-FirstCall via COMTEX/ -- Sanofi-aventis (Euronext: SAN and NYSE: SNY) and Regeneron Pharmaceuticals, Inc. /quotes/comstock/15*!regn/quotes/nls/regn (REGN 21.88, -0.11, -0.50%) today announced the discontinuation of the Phase 3 trial that evaluated aflibercept (VEGF Trap) plus gemcitabine versus placebo plus gemcitabine for the first-line treatment of metastatic pancreatic cancer (VANILLA), based on the recommendations by an Independent Data Monitoring Committee (IDMC). As part of a planned interim efficacy analysis, the IDMC determined that the addition of aflibercept to gemcitabine would be unable to demonstrate a statistically significant improvement in the primary endpoint of overall survival compared to placebo plus gemcitabine in this study. The types and frequencies of adverse events reported on the combination arm with aflibercept were generally as anticipated.
With the closure of the study, a detailed analysis of the efficacy and safety results will be conducted by the companies and results will be presented at a future medical meeting. Sanofi-aventis and Regeneron have notified the study investigators and appropriate regulatory authorities of the decision to discontinue the study. Patients in the study will continue to be provided access to aflibercept at the determination of the study investigators in consultation with the patients.
Metastatic pancreatic cancer is among the most intractable cancers. Clinical development of new therapies, including anti-VEGF agents, has been generally characterized by a failure to achieve significant incremental clinical benefit over existing treatments.
"We are disappointed with the result of this study and we will continue our efforts to bring new and effective treatments for these patients," said Dr. Marc Cluzel, Senior Vice President, Research and Development sanofi-aventis. "We remain committed to the other ongoing Phase 3 trials of aflibercept in colorectal cancer, non-small cell lung cancer, and hormone-refractory metastatic prostate cancer."
Three Phase 3 studies continue, each of which is currently over 70 percent enrolled:
-- VELOUR study: 2nd-line metastatic colorectal cancer in combination with
fluorouracil, leucovorin, and irinotecan (FOLFIRI)
-- VITAL study: 2nd-line non-small cell lung cancer in combination with
docetaxel
-- VENICE study: 1st-line hormone-refractory metastatic prostate cancer in
combination with docetaxel and prednisone
Zu Seattle Genetics
Hier gab es einen Klinikgang des Partners Bayer:
BOTHELL, Wash., Sep 08, 2009 (BUSINESS WIRE) -- Seattle Genetics, Inc. (Nasdaq: SGEN) announced today that it has achieved a milestone under its antibody-drug conjugate (ADC) collaboration with Bayer Schering Pharma AG, Germany. The milestone was triggered by Bayer's submission of an Investigational New Drug (IND) application with the U.S. Food and Drug Administration (FDA) for MN-IC, an ADC for solid tumors.
"We are pleased with the continued progress by our collaborators in advancing ADCs spanning a range of oncology indications and settings. MN-IC will be the third new ADC program utilizing our technology to advance into clinical trials during the past 12 months," said Eric L. Dobmeier, Chief Business Officer of Seattle Genetics. "In addition to generating revenue, these collaborator programs broaden the clinical experience with our ADC technology and build on our understanding of its potential in the treatment of serious diseases."
Under the terms of the ADC collaboration agreement, Bayer has rights to use Seattle Genetics' ADC technology with antibodies against MN. Bayer is responsible for research, product development, manufacturing and commercialization of all products under the collaboration. Seattle Genetics receives material supply and annual maintenance fees as well as research support payments for any assistance provided to Bayer in developing ADC products. The antibody component of the ADC targeted to MN is derived from HuCAL technology that Bayer licensed from MorphoSys AG.
Seattle Genetics is advancing a proprietary pipeline of ADC programs, including brentuximab vedotin (SGN-35), which is in an ongoing pivotal trial for relapsed or refractory Hodgkin lymphoma and a phase II trial for systemic anaplastic large cell lymphoma. Seattle Genetics is also developing a number of preclinical ADCs, including SGN-75, which the company is advancing towards a planned 2009 IND submission for CD70-positive hematologic malignancies and solid tumors. The company is also developing ASG-5ME in collaboration with Agensys, a subsidiary of Astellas Pharma. An IND submission of ASG-5ME for solid tumors is planned in the first half of 2010.
Hier gab es einen Klinikgang des Partners Bayer:
BOTHELL, Wash., Sep 08, 2009 (BUSINESS WIRE) -- Seattle Genetics, Inc. (Nasdaq: SGEN) announced today that it has achieved a milestone under its antibody-drug conjugate (ADC) collaboration with Bayer Schering Pharma AG, Germany. The milestone was triggered by Bayer's submission of an Investigational New Drug (IND) application with the U.S. Food and Drug Administration (FDA) for MN-IC, an ADC for solid tumors.
"We are pleased with the continued progress by our collaborators in advancing ADCs spanning a range of oncology indications and settings. MN-IC will be the third new ADC program utilizing our technology to advance into clinical trials during the past 12 months," said Eric L. Dobmeier, Chief Business Officer of Seattle Genetics. "In addition to generating revenue, these collaborator programs broaden the clinical experience with our ADC technology and build on our understanding of its potential in the treatment of serious diseases."
Under the terms of the ADC collaboration agreement, Bayer has rights to use Seattle Genetics' ADC technology with antibodies against MN. Bayer is responsible for research, product development, manufacturing and commercialization of all products under the collaboration. Seattle Genetics receives material supply and annual maintenance fees as well as research support payments for any assistance provided to Bayer in developing ADC products. The antibody component of the ADC targeted to MN is derived from HuCAL technology that Bayer licensed from MorphoSys AG.
Seattle Genetics is advancing a proprietary pipeline of ADC programs, including brentuximab vedotin (SGN-35), which is in an ongoing pivotal trial for relapsed or refractory Hodgkin lymphoma and a phase II trial for systemic anaplastic large cell lymphoma. Seattle Genetics is also developing a number of preclinical ADCs, including SGN-75, which the company is advancing towards a planned 2009 IND submission for CD70-positive hematologic malignancies and solid tumors. The company is also developing ASG-5ME in collaboration with Agensys, a subsidiary of Astellas Pharma. An IND submission of ASG-5ME for solid tumors is planned in the first half of 2010.
Beitrag zu dieser Diskussion schreiben
Zu dieser Diskussion können keine Beiträge mehr verfasst werden, da der letzte Beitrag vor mehr als zwei Jahren verfasst wurde und die Diskussion daraufhin archiviert wurde.
Bitte wenden Sie sich an feedback@wallstreet-online.de und erfragen Sie die Reaktivierung der Diskussion oder starten Sie eine neue Diskussion.
Meistdiskutiert
| Wertpapier | Beiträge | |
|---|---|---|
| 243 | ||
| 98 | ||
| 89 | ||
| 80 | ||
| 75 | ||
| 47 | ||
| 38 | ||
| 37 | ||
| 34 | ||
| 33 |
| Wertpapier | Beiträge | |
|---|---|---|
| 31 | ||
| 30 | ||
| 27 | ||
| 24 | ||
| 23 | ||
| 22 | ||
| 21 | ||
| 20 | ||
| 19 | ||
| 19 |



















